
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne step access to oxindole-based β-lactams through Ugi four-center three-component reaction†
Giulia Rainoldia,
Giordano Lesmaa,
Claudia Picozzi b,
Leonardo Lo Prestia and
Alessandra Silvani*a
b,
Leonardo Lo Prestia and
Alessandra Silvani*a
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, Milano, 20133, Italy. E-mail: alessandra.silvani@unimi.it
bDepartment of Food, Environmental and Nutritional Sciences (DeFENS), Division of Food Microbiology and Bioprocessing, Via Celoria 2, 20133 Milan, Italy
First published on 11th October 2018
Abstract
A multicomponent Ugi reaction involving isatin, isocyanide and β-amino acid components has been developed. The reactions proceeded smoothly to give β-lactam-containing 3,3-disubstituted oxindoles in only one step and generally high yields. When chiral, non racemic, β-amino acids were used, products were obtained as enantiomerically pure β-lactams diastereoisomers, whose relative stereochemistry was determined by X-ray analysis. For one compound, a weak antibacterial activity has been preliminarily highlighted.
Introduction
The β-lactam fragment constitutes a part of various naturally occurring compounds, a number of which exhibit pronounced biological activity. Despite many decades of clinical significance from the discovery of penicillin forward, it remains an interesting pharmacophore for medicinal chemistry and novel applications of β-lactam derivatives continue to be developed.1 On the other hand, 3-substituted-3-amino-2-oxindoles are also recurring core structures, that can be found in drug molecules and biologically active compounds acting on different targets2 (Fig. 1). Since the manifold biological activities of 3-substituted-3-amino-2-oxindoles are strongly influenced by the type of substitution at C3 position, the rapid construction of such privileged compounds, displaying complex and varied architectures, is a valuable way to contribute to drug discovery.3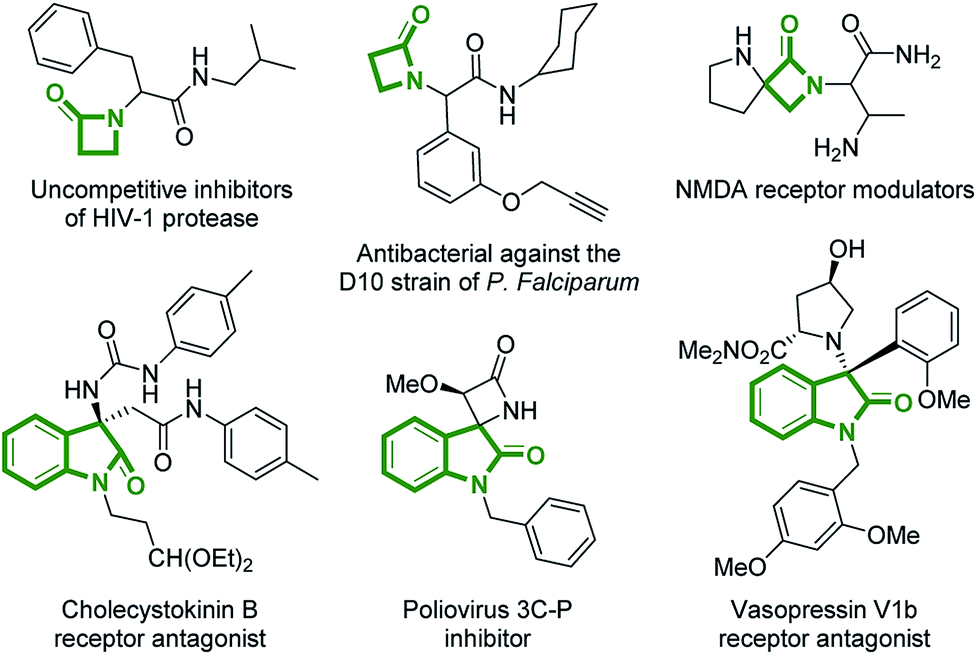 | ||
| Fig. 1 Examples of biologically relevant compounds containing β-lactam or 3-substituted-3-amino-2-oxindole fragments. | ||
As part of our interest in the synthesis of aminooxindoles and related spiro-compounds,4 we recently turned our attention to the molecular hybridization concept, a viable and effective approach envisioning the rational design of new functional compounds through the structural fusion of two pharmacophoric subunits from known structures into one chemical entity. Until now, several new chemical classes have been discovered by the combination of pharmacophoric moieties of known molecules, resulting often in novel and more potent hybrid derivatives.5 As, to the best of our knowledge, no methods have been reported for the preparation of oxindoles bearing a N-jointed β-lactam ring at the key C3 position, we envisioned the first construction of such hybrid molecules, selecting in particular, a peptidomimetic scaffold as the privileged target. New peptidomimetic small molecules are indeed desirable, particularly in the highly challenging fields of protein–protein interactions targeting and of antimicrobial drug discovery research.6 Among strategies to the β-lactam ring, Staudinger reaction involving [2 + 2] cycloaddition of ketenes and imines has been the most widely used protocol.7 Considering a multicomponent approach more suitable for the rapid generation of peptidomimetic backbones containing the β-lactam ring, we looked at the Ugi four-center three-component reaction (Ugi-4C-3CR),8 using easily accessible β-amino acids, isocyanides and isatins. Apart a single example reported,9 this is the first wide application of such reaction involving a ketone as the carbonyl component and following the complete Ugi pathway, including the final rearrangement, leading to β-lactam derivatives.
Herein, we report the high yield, single step synthesis of a variety of oxindole-based β-lactams, bearing a peptidomimetic backbone, some their post-transformations and they evaluation according to the Lipinski rule of five and against a set of bacterial strains.
Results and discussion
Initially, N-Bn isatin 1a, tert-butyl isocyanide 2a and β-alanine 3a were selected to optimize the conditions for the Ugi-4C-3CR (Table 1). We started our investigation considering aprotic solvents such as dichloromethane and toluene (entries 1–2), but the reaction was found to be sluggish. Working in MeOH (entry 3), the same reaction afforded the desired β-lactam 4a in low yield. When we switched to the more acidic trifluoroethanol (TFE), following our recent achievements in different Ugi-type reactions,10 both the reaction rate and yield definitely increased (entries 4–6). With satisfactory conditions in hand, a variety of substituted isatins 1a–j were next explored to investigate the carbonyl component scope of the Ugi-4C-3CR (Scheme 1). Working with tert-butyl isocyanide (2a) and β-alanine 3a, the protecting group on the oxindole nitrogen atom was found to have a moderate influence on the reaction, with compounds 4a–f obtained in substantial yields, up to 97%.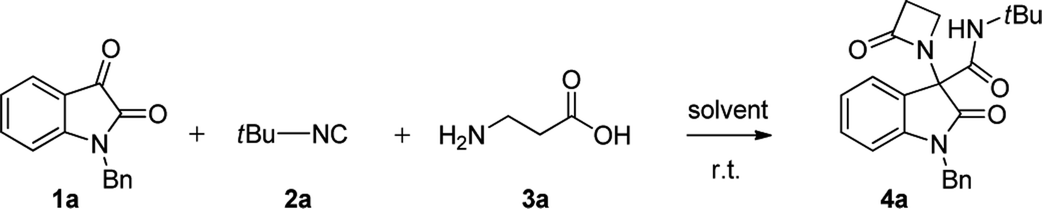
 | ||
| Scheme 1 Components scope of the Ugi-4C-3CRa. aReaction conditions: isatin 1 (0.5 mmol), isocyanide 2 (0.5 mmol) and β-amino acid 3 (0.5 mmol) in TFE (1 mL), stirred at room temperature. Isolated yields are reported. PBB = p-bromobenzyl, PNB = p-nitrobenzyl. | ||
Next, N-benzyl-isatins bearing various substituents on the aromatic ring were explored. Good yields of the corresponding β-lactams derivatives were obtained in the presence of a variety of substituents, including electron-donating group (4g) and halogen substituents at either the 5- or 6-position (4h and 4j), whereas the reaction proved to be moderately inhibited when a strongly electron-withdrawing group was present (4i).
Investigation of the isocyanide component (2a–e) scope was also conducted. Expected β-lactams 4k–n were readily obtained, both with aliphatic and heteroaromatic isocyanides, although in a generally bit lower yield.
Finally, two enantiomerically pure β-amino acids, namely (S)-3-amino-3-phenylpropanoic acid and (S)-3-amino-4-methoxy-4-oxobutanoic acid (3b, c), were tested in the reaction. Compounds 4o and 4p were easily obtained as mixtures of enantiomerically pure β-lactams diastereoisomers.
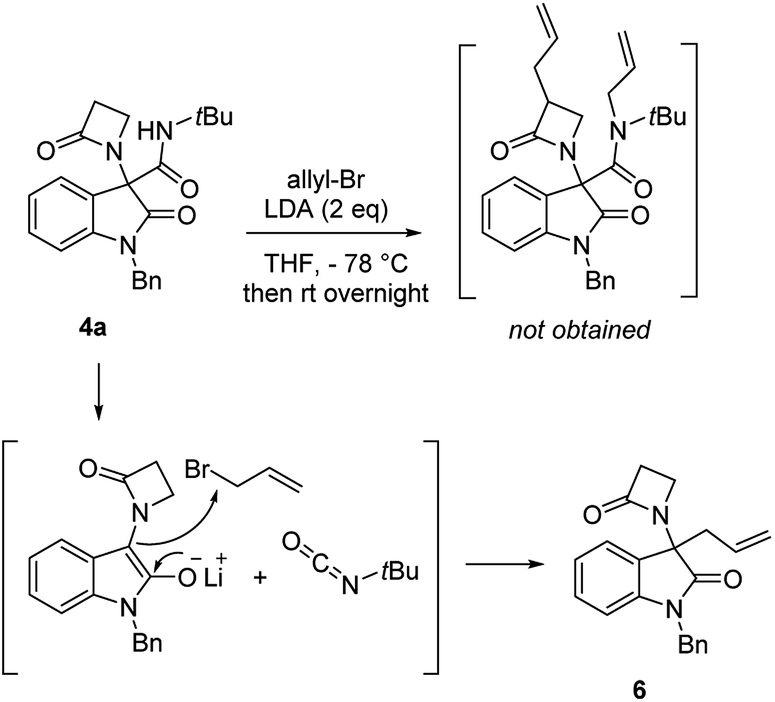
Chromatographic separation on compound 4o allowed to obtain crystals of diastereoisomers 4oa and 4ob, suitable for determination of relative (and absolute) stereochemistry. By means of X-ray analysis of the major diasteroisomer 4oa, given the fixed S configuration at the β-lactam stereocenter, the configuration at the oxindole ring stereocenter was found to be S (Fig. 2; see ESI† for full crystallographic details). Considering the quite similar chemical shifts trend in NMR spectra of 4o and 4p, the same S,S-configuration could be conceivably assigned also to the 4p major diastereoisomer. Having established the scope of the method, some post-transformations were performed on selected derivatives (Scheme 2). The reaction of β-lactam 4a with methyl iodide under basic conditions gave the alkylated amide 5 in high yield. Aiming to obtain the double functionalization on both the NH and at the α-position on the lactam moiety, compound 4a was also treated with two equivalents of LDA followed by the addition of allyl bromide.
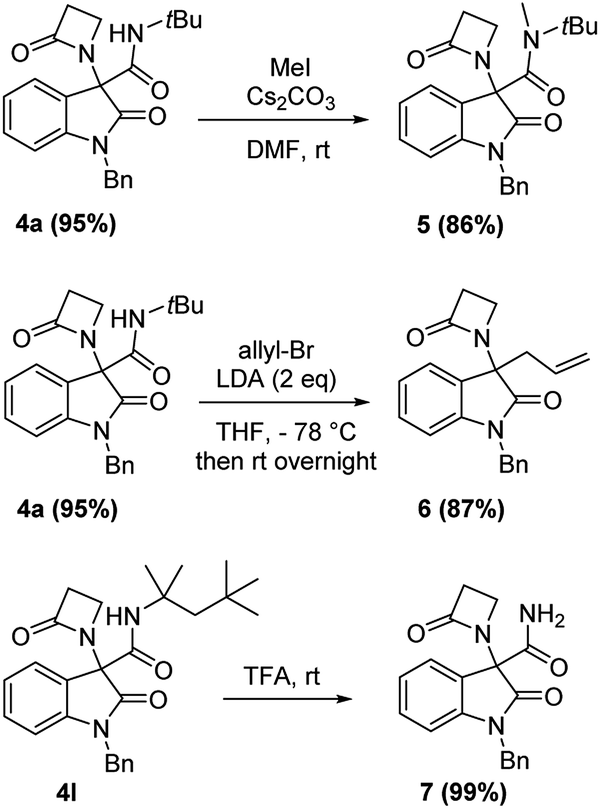 | ||
| Scheme 2 Post-transformation reactions performed on selected compounds. | ||
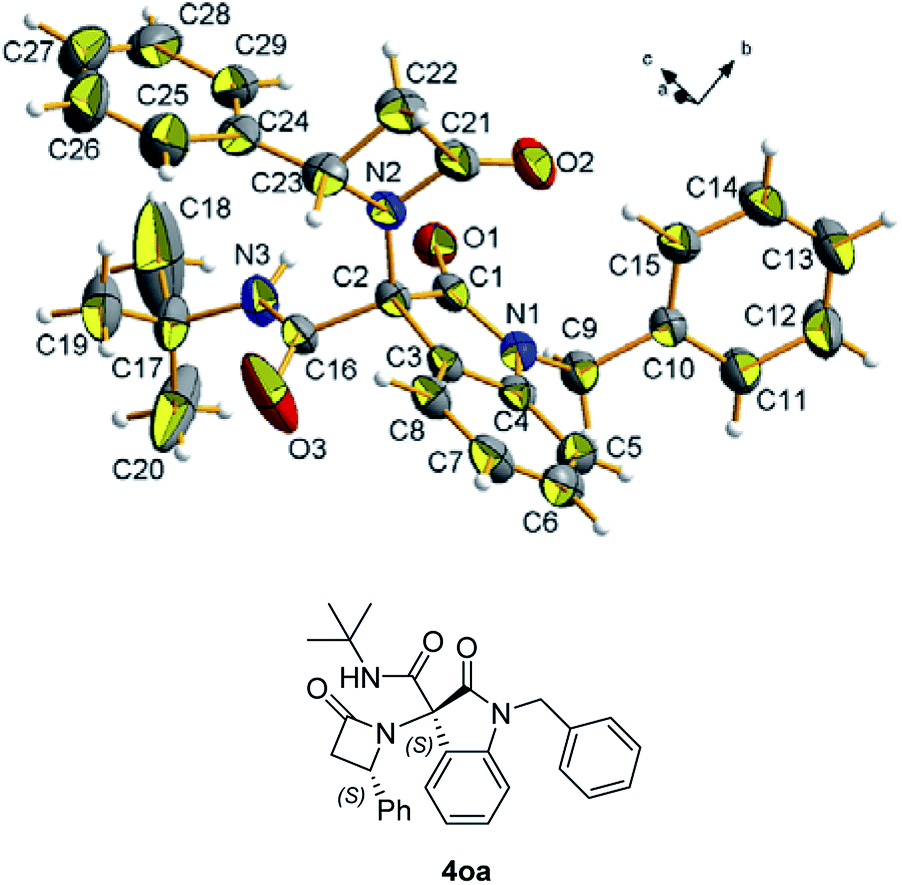 | ||
| Fig. 2 ORTEP view of compound 4oa with the atom-numbering scheme. The crystallographic reference system is also highlighted. Thermal ellipsoids of non-H atoms at RT were drawn at the 30% probability level. | ||
However, in this condition, only the unprecedented product 6 was obtained in good yield, likely deriving from a retro-condensation process, followed by irreversible allylation. This outcome discloses an effective deacylative alkylation strategy,11 that could be useful for the construction of a variety of 3,3-disubstituted 2-oxindoles. A proposed mechanism for this reaction is reported in Scheme 3. Finally, starting from compound 4l, the secondary amido group was easily dealkylated under acid conditions to give the primary amide 7 in quantitative yield, fully demonstrating the high versatility of 2-isocyano-2,4,4-trimethylpentane as cleavable isocyanide. To evaluate the suitability of the obtained compounds in drug discovery programs, their physicochemical properties have been calculated using DruLiTo12 (calculation details are provided in the ESI†). All compounds proved to be drug-like according to the rule of five (Ro5) proposed by Lipinski, having a calculated octanol/water partition (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) lower than 3, therefore positioning themselves in the highly hydrophilic area (Fig. 3). Regarding the other Ro5 properties, all the synthesized compounds have hydrogen bonding donator groups (HBD) lower than five and hydrogen bonding acceptor groups (HBA) lower than ten (Fig. 4). Further, the presence of rotational bonds (RB) lower than ten and the polar surface area prediction (TPSA) lower than 140 Å2 (see ESI†) show that all compounds also meet the more recent Veber's rule13 on good oral bioavailability, making them definitively of potential interest from the pharmacological point of view.
P) lower than 3, therefore positioning themselves in the highly hydrophilic area (Fig. 3). Regarding the other Ro5 properties, all the synthesized compounds have hydrogen bonding donator groups (HBD) lower than five and hydrogen bonding acceptor groups (HBA) lower than ten (Fig. 4). Further, the presence of rotational bonds (RB) lower than ten and the polar surface area prediction (TPSA) lower than 140 Å2 (see ESI†) show that all compounds also meet the more recent Veber's rule13 on good oral bioavailability, making them definitively of potential interest from the pharmacological point of view.
 | ||
| Scheme 3 Proposed mechanism for compound 6. | ||
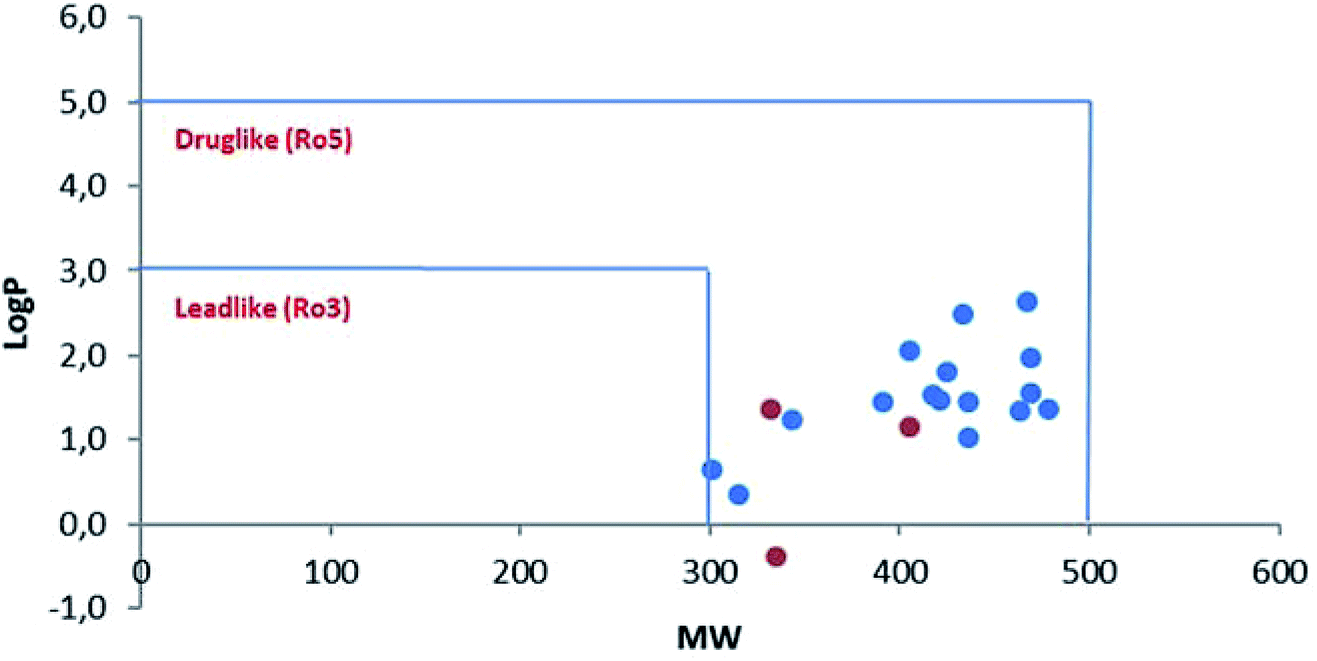 | ||
| Fig. 3 Drug- and lead-likeness (MW/logP) of all the products obtained in this work. Blue-spots: β-lactams derivatives 4a–p. Red-spots: post-transformation derivatives 5–7. | ||
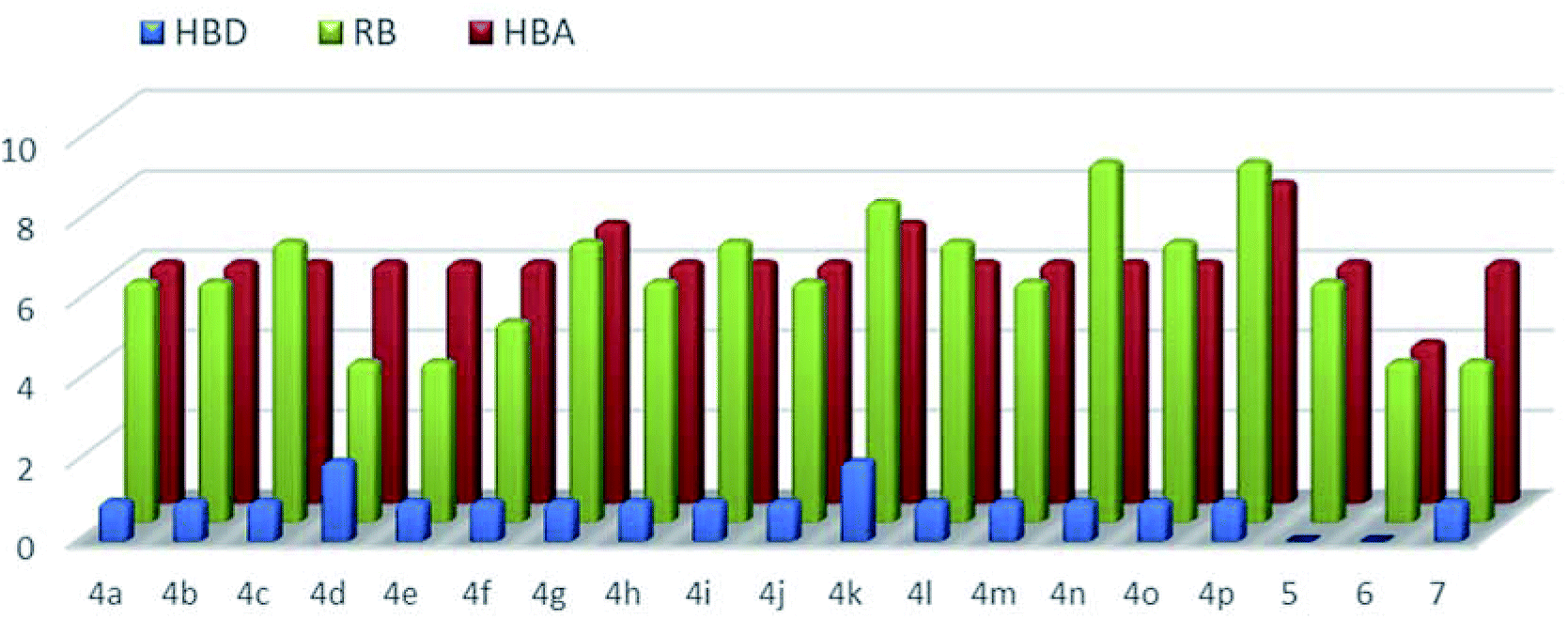 | ||
| Fig. 4 Calculated hydrogen bonding donators (HBD, blue-bars), hydrogen bonding acceptors (HBA, red-bars), and rotational bonds (RB, greenbars) for all the synthesized compounds. | ||
Lastly, a preliminary biological evaluation was also performed. The antibacterial activity was tested by the disc diffusion method14 using Gram-negative (Escherichia coli and Pseudomonas aeruginosa) and Gram-positive (Staphylococcus aureus and Streptococcus mutans) bacteria. The results show that one of the synthesised compounds, namely 4e, gives rise to a slight inhibition zone (9.5 ± 0.7 mm) on St. mutans at a concentration of 0.81 mM. None of the other compounds displayed activity at concentrations less than 1 mM.
Conclusions
In conclusion, we have achieved the one-step synthesis of new β-lactam-containing 3,3-disubstituted oxindole derivatives, relying on the Ugi four-center three-component reaction applied to isatin as the oxo component. The reaction conditions were optimized and adopted for a variety of substituted isatins and isocyanides, employing β-alanine or chiral, non racemic, β-amino acids as bifunctional components. Based on their calculated physicochemical properties, all compounds are eligible for being drug-like and therefore potentially suitable in drug discovery programs. Preliminary evaluation against selected bacterial strains highlighted compound 4e as the more promising for future tuning of functional groups on the β-lactam-oxindole scaffold.Experimental
General
All commercial materials (Aldrich, Fluka) were used without further purification. All solvents were of reagent grade or HPLC grade. All reactions were carried out under a nitrogen atmosphere unless otherwise noted. All reactions were monitored by thin layer chromatography (TLC) on precoated silica gel 60 F254; spots were visualized with UV light or by treatment with suitable TLC visualization reagents. Products were purified by flash chromatography (FC) on silica gel 60 (230–400 mesh). 1H NMR spectra and 13C NMR spectra were recorded on 300 and 400 MHz spectrometers. Chemical shifts are reported in parts per million relative to the residual solvent. 13C NMR spectra have been recorded using the APT pulse sequence. Multiplicities in 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br s = broad singlet. High-resolution MS spectra were recorded with a LCQ Fleet ion trap mass spectrometer, equipped with an ESI source. All the N-substituted isatins were synthesized according to reported literature.15:EtOAc, from 1.5:1 to 1:1.5; yield 95%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.6 Hz, 1H), 7.40–7.19 (m, 7H), 7.11 (t, J = 7.6 Hz, 1H), 6.73 (d, J = 7.6 Hz, 1H), 5.05 (d, J = 15.8 Hz, 1H), 4.90 (d, J = 15.8 Hz, 1H), 3.62–3.52 (m, 1H), 3.40–3.32 (m, 1H), 2.97–2.87 (m, 2H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.2, 167.7, 162.7, 143.0, 135.6, 130.7, 129.5 (2C), 128.5, 127.8 (2C), 127.3, 126.3, 124.3, 110.5, 68.9, 52.9, 45.0, 40.0, 36.7, 29.2 (3C); HRMS (ESI) calcd for C23H26N3O3+ [MH]+ 392.1969, found 392.1971.:EtOAc, from 1.5:1 to 1:1.5; yield 66%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.4 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.31 (br s, 1H), 7.29–7.19 (m, 3H), 7.10 (t, J = 7.4 Hz, 1H), 6.67 (d, J = 7.4 Hz, 1H), 5.00 (d, J = 15.9 Hz, 1H), 4.79 (d, J = 15.9 Hz, 1H), 3.63–3.53 (m, 1H), 3.43–3.31 (m, 1H), 2.98–2.86 (m, 2H), 1.38 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.5, 167.0, 161.9, 142.0, 134.0, 132.0 (2C), 130.1, 129.0 (2C), 126.6, 125.7, 123.8, 121.8, 109.7, 68.2, 52.3, 43.7, 39.4, 36.1, 28.5 (3C); HRMS (ESI) calcd for C23H24BrN3NaO3+ [MNa]+ 494.0893, found 494.0899.:EtOAc, from 1.5:1 to 1:1.5; yield 64%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.8 Hz, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.36 (br s, 1H), 7.25 (t, J = 7.8 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 6.63 (d, J = 7.8 Hz, 1H), 5.20 (d, J = 16.5 Hz, 1H), 4.93 (d, J = 16.5 Hz, 1H), 3.68–3.60 (m, 1H), 3.47–3.40 (m, 1H), 3.00–2.94 (m, 2H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.6, 167.0, 161.7, 147.6, 142.2, 141.7, 130.1, 128.0 (2C), 126.8, 125.7, 124.1 (3C), 109.4, 68.1, 52.4, 43.7, 39.5, 36.1, 28.5 (3C); HRMS (ESI) calcd for C23H24N4NaO5+ [MNa]+ 459.1639, found 459.1642.:EtOAc, from 1.5:1 to 1:1.5; yield 97%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 9.47 (br s, 1H), 7.63 (d, J = 7.5 Hz, 1H), 7.35 (br s, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.80 (d, J = 7.5 Hz, 1H), 3.65–3.50 (m, 1H), 3.45–3.30 (m, 1H), 2.99–2.81 (m, 2H), 1.36 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.8, 167.5, 162.3, 141.2, 130.1, 126.3, 126.0, 123.2, 111.1, 68.5, 52.2, 39.5, 35.8, 28.4 (3C); HRMS (ESI) calcd for C16H19N3NaO3+ [MNa]+ 324.1319, found 324.1325.:EtOAc, from 1.5:1 to 1:1.5; yield 91%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 7.5 Hz, 1H), 7.40–7.25 (m, 2H), 7.12 (td, J = 7.5, 0.9 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 3.64–3.45 (m, 1H), 3.41–3.29 (m, 1H), 3.24 (s, 3H), 2.95–2.80 (m, 2H), 1.35 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.3, 167.1, 162.1, 143.3, 130.1, 126.6, 125.3, 123.6, 108.9, 68.1, 52.2, 39.2, 36.0, 32.6, 28.4 (3C); HRMS (ESI) calcd for C17H21N3NaO3+ [MNa]+ 338.1475, found 338.1480.:EtOAc, from 1.5:1 to 1:1.5; yield 90%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.6 Hz, 1H), 7.42–7.24 (m, 2H), 7.11 (t, J = 7.6 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 4.59 (hept, J = 7.0 Hz, 1H), 3.62–3.46 (m, 1H), 3.38–3.22 (m, 1H), 3.00–2.75 (m, 2H), 1.52 (d, J = 7.0 Hz, 6H), 1.37 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 170.9, 166.9, 162.2, 142.2, 129.8, 126.9, 126.0, 123.0, 110.3, 67.9, 52.1, 44.9, 39.0, 35.9, 28.5 (3C), 19.2 (2C); HRMS (ESI) calcd for C19H25N3NaO3+ [MNa]+ 366.1788, found 366.1790.:EtOAc, from 1.5:1 to 1:1.5; yield 78%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.64–7.12 (m, 8H), 6.73 (dd, J = 8.6, 2.6 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 4.98 (d, J = 15.7 Hz, 1H), 4.85 (d, J = 15.8 Hz, 1H), 3.74 (s, 3H), 3.60–3.45 (m, 1H), 3.40–3.25 (m, 1H), 3.00–2.85 (m, 1H), 1.37 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.4, 167.0, 161.8, 156.5, 135.5, 135.0, 128.8 (2C), 127.8, 127.2 (2C), 126.6, 115.0, 113.4, 110.4, 68.4, 55.8, 52.2, 44.3, 39.3, 36.1, 28.5 (3C); HRMS (ESI) calcd for C24H27N3NaO4+ [MNa]+ 444.1894, found 444.1899.:EtOAc, from 1.5:1 to 1:1.5; yield 64%; yellow foamy solid; 1H NMR (300 MHz, CDCl3) d 7.83 (d, J = 1.0 Hz, 1H), 7.50–7.20 (m, 7H), 6.58 (d, J = 8.4 Hz, 1H), 5.04 (d, J = 15.8 Hz, 1H), 4.86 (d, J = 15.8 Hz, 1H), 3.71–3.50 (m, 1H), 3.50–3.30 (m, 1H), 3.16–2.83 (m, 2H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.3, 166.8, 161.0, 141.3, 134.4, 132.9, 129.8, 129.0 (2C), 128.0, 127.3, 127.1 (2C), 116.5, 111.0, 67.7, 52.4, 44.5, 39.5, 36.2, 28.5 (3C); HRMS (ESI) calcd for C23H24BrN3NaO3+ [MNa]+ 492.0893, found 492.0899.:EtOAc, from 1.5:1 to 1:1.5; yield 41%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 2.3 Hz, 1H), 8.18 (dd, J = 8.7, 2.3 Hz, 1H), 7.45–7.23 (m, 6H), 6.80 (d, J = 8.7 Hz, 1H), 5.14 (d, J = 15.9 Hz, 1H), 4.94 (d, J = 15.9 Hz, 1H), 3.73–3.62 (m, 1H), 3.55–3.48 (m, 1H), 3.07–2.97 (m, 2H), 1.42 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.2, 166.8, 160.1, 147.7, 144.2, 133.7, 129.1 (2C), 128.3, 127.1, 126.7 (2C), 126.3, 122.5, 109.7, 67.0, 52.7, 44.8, 39.7, 36.3, 28.5 (3C); HRMS (ESI) calcd for C23H24N4NaO5+ [MNa]+ 459.1639, found 459.1641.:EtOAc, from 1.5:1 to 1:1.5; yield 65%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 1H), 7.42–7.27 (m, 5H), 7.23 (br s, 1H), 7.09 (dd, J = 8.0, 1.8 Hz, 1H), 6.73 (d, J = 1.8 Hz, 1H), 5.00 (d, J = 15.8 Hz, 1H), 4.87 (d, J = 15.8 Hz, 1H), 3.64–3.54 (m, 1H), 3.42–3.35 (m, 1H), 3.25–2.75 (m, 2H), 1.39 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.7, 167.0, 161.5, 143.5, 136.0, 134.3, 129.0 (2C), 128.1, 127.7, 127.1 (2C), 123.9, 123.7, 110.6, 67.7, 52.4, 44.4, 39.5, 36.1, 28.5 (3C); HRMS (ESI) calcd for C23H24ClN3NaO3+ [MNa]+ 448.1398, found 448.1400.:EtOAc, from 1.5:1 to 1:1.5; yield 47%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 8.32 (br s, 1H), 7.79 (d, 7.9 Hz, 1H), 7.61 (d, 7.9 Hz, 1H), 7.50–6.90 (m, 12H), 6.71 (d, J = 7.9 Hz, 1H), 4.95 (d, J = 15.8 Hz, 1H), 4.87 (d, J = 15.8 Hz, 1H), 3.75–3.57 (m, 2H), 3.56–3.44 (m, 1H), 3.30–3.18 (m, 1H), 3.20–2.80 (m, 2H), 2.82 (t, J = 3.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 171.0, 167.4, 163.8, 142.5, 136.4, 135.0, 130.2, 128.9 (2C), 127.9, 127.1 (2C), 126.6, 125.4, 123.7, 122.5, 122.0, 119.4, 118.6, 112.2, 111.3, 109.9, 60.4, 44.3, 40.5, 39.5, 36.0, 29.7, 24.9; HRMS (ESI) calcd for C29H26N4NaO3+ [MNa]+ 501.1897, found 501.1900.:EtOAc, from 1.5:1 to 1:1.5; yield 43%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.8 Hz, 1H), 7.42–7.20 (m, 6H), 7.20–7.13 (m, 2H), 6.75 (d, J = 7.8 Hz, 1H), 5.00 (d, J = 15.7 Hz, 1H), 4.93 (d, J = 15.7 Hz, 1H), 3.75–3.50 (m, 1H), 3.50–3.25 (m, 1H), 3.02–2.85 (m, 2H), 1.86 (d, J = 15.0 Hz, 1H), 1.55 (d, J = 15.0 Hz, 1H), 1.46 (s, 3H), 1.45 (s, 3H), 0.94 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.0, 167.7, 162.4, 143.0, 135.6, 130.7, 129.5 (2C), 128.5, 127.9 (2C), 127.5, 126.2, 124.3, 110.4, 69.0, 56.9, 52.9, 45.0, 40.1, 36.8, 32.2, 32.0 (3C), 29.2 (2C); HRMS (ESI) calcd for C27H33N3NaO3+ [MNa]+ 470.2414, found 470.2419.:EtOAc, from 1.5:1 to 1:1.5; yield 48%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.4 Hz, 1H), 7.45–7.18 (m, 7H), 7.11 (t, J = 7.4 Hz, 1H), 6.73 (d, J = 7.4 Hz, 1H), 5.03–4.92 (m, 2H), 3.82 (m, br, 1H), 3.60 (q, J = 4.7 Hz, 1H), 3.36 (q, J = 4.7 Hz, 1H), 2.95 (t, br, J = 4.4 Hz, 2H), 1.98 (m, br, 1H), 1.86 (m, br, 1H), 1.79–1.65 (m, 2H), 1.60 (m, br, 1H), 1.44–1.11 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 171.3, 167.3, 162.6, 142.4, 134.9, 130.1, 128.9 (2C), 127.8, 127.1 (2C), 126.5, 125.6, 123.7, 109.9, 68.1, 49.0, 44.3, 39.4, 36.1, 32.6, 32.5, 25.5, 24.5 (2C); HRMS (ESI) calcd for C25H27N3NaO3+ [MNa]+ 440.1945, found 440.1943.:EtOAc, from 1.5:1 to 1:1.5; yield 62%; yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 7.3 Hz, 1H), 7.42 (br m, 1H), 7.40–7.20 (m, 6H), 7.11 (t, J = 7.3 Hz, 1H), 6.73 (d, J = 7.3 Hz, 1H), 5.05–4.91 (m, 2H), 3.60 (q, J = 4.7 Hz, 1H), 3.35–3.20 (m, 3H), 3.00–2.90 (m, 2H), 1.56 (quint, J = 7.1 Hz, 2H), 1.42–1.21 (m, 4H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.2, 167.4, 163.7, 142.5, 134.8, 130.2, 128.9 (2C), 127.9, 127.2 (2C), 126.6, 125.6, 123.7, 109.9, 68.2, 44.3, 40.2, 39.4, 36.1, 29.0, 28.9, 22.3, 14.0; HRMS (ESI) calcd for C24H27N3NaO3+ [MNa]+ 428.1945, found 428.1943.:EtOAc, from 1.5:1 to 1:1.5; yield 56%. Major diastereoisomer (S,S) (4oa): yellow foamy solid; [α]D = –179.1 (c. 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.4 Hz, 1H), 7.33–7.12 (m, 12H), 7.10 (t, J = 7.4 Hz, 1H), 6.51 (d, J = 7.4 Hz, 1H), 5.17 (dd, J = 5.5 and 2.4 Hz, 1H), 4.83 (d, J = 15.9 Hz, 1H), 4.47 (d, J = 15.9 Hz, 1H), 3.44 (dd, J = 15.0 and 5.5 Hz, 1H), 2.81 (dd, J = 15.0 and 2.4 Hz, 1H), 1.17 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.3, 168.8, 163.0, 143.0, 139.1, 135.4, 130.5, 129.5 (2C), 129.2 (2C), 129.1, 128.3, 127.8, 127.6 (2C), 127.5 (2C), 126.7, 124.3, 110.2, 69.5, 56.5, 52.7, 47.3, 44.8, 28.9 (3C); HRMS (ESI) calcd for C29H29N3NaO3+ [MNa]+ 490.2101, found 490.2105. Minor diastereoisomer (R,S) (4ob): yellow foamy solid; [α]D = –39.8 (c. 0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 7.8 Hz, 1H), 7.35–7.02 (m, 13H), 6.48 (d, J = 7.8 Hz, 1H), 4.82 (d, J = 15.8 Hz, 1H), 4.73 (dd, J = 5.5 and 2.5 Hz, 1H), 4.38 (d, J = 15.8 Hz, 1H), 3.43 (dd, J = 15.0 and 5.5 Hz, 1H), 2.93 (dd, J = 15.0 and 2.5 Hz, 1H), 1.34 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.5, 168.5, 166.9, 142.5, 137.6, 134.8, 130.0, 128.8 (2C), 128.6, 128.5 (2C), 127.7, 127.2, 127.1 (4C), 123.8, 123.3, 109.5, 69.5, 55.3, 52.2, 45.8, 44.0, 28.3 (3C); HRMS (ESI) calcd for C29H29N3NaO3+ [MNa]+ 490.2101, found 490.2108.:EtOAc, from 1.5:1 to 1:1.5; yield 82%; yellow foamy solid; 1H NMR (400 MHz, CDCl3, 7:3 mixture of diasteroisomers) δ 7.85 (d, J = 7.5 Hz, 0.7H), 7.44–7.20 (m, 6.6H), 7.12 (t, J = 7.6 Hz, 1H), 6.77 (d, J = 7.8 Hz, 0.7H), 6.73 (d, J = 7.9 Hz, 0.3H), 6.36 (s, br, 0.7H), 5.02 (d, J = 15.7 Hz, 0.7H), 4.93 (s, 0.6H), 4.85 (d, J = 15.7 Hz, 0.7H), 4.75 (dd, J = 6.0 and 2.7 Hz, 0.7H), 4.37 (dd, J = 5.7 and 2.3 Hz, 0.3H), 3.84 (s, 0.9H), 3.58 (s, 2.1H), 3.33 (dd, J = 14.7 and 6.1 Hz, 0.7H), 3.22 (dd, J = 14.6 and 5.8 Hz, 0.3H), 3.00–2.80 (m, 1H), 1.38 (s, 2.7H), 1.31 (s, 6.3H); 13C NMR (101 MHz, CDCl3) δ 171.7, 170.9 and 170.3 (1C), 166.9 and 166.6 (1C), 163.9 and 163.1 (1C), 143.4, 135.7, 130.9 and 130.8 (1C), 129.6 (2C), 128.5, 128.0 (2C), 127.7, 126.8 and 126.5 (1C), 124.4, 110.4 and 110.3 (1C), 69.6, 53.4 and 53.0 (1C), 52.9, 52.7 and 51.8 (1C), 45.0, 42.3 and 42.0 (1C), 29.1 (3C); HRMS (ESI) calcd for C25H27N3NaO5+ [MNa]+ 472.1843, found 472.1840.Post-transformation reactions
:EtOAc, from 1.5:1 to 1:1.5) affording the desired product 5 (86% yield) as a yellow foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.3 Hz, 1H), 7.39–7.26 (m, 5H), 7.22 (t, J = 7.5 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.73 (d, J = 7.8 Hz, 1H), 4.99 (d, J = 15.8 Hz, 1H), 4.90 (d, J = 15.7 Hz, 1H), 3.35 (m, br, 1H), 3.28–3.12 (m, 6H), 1.33 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.6, 168.0, 163.3, 142.3, 136.1, 130.0, 129.6 (2C), 128.4, 127.9 (2C), 127.5, 124.5, 124.0, 110.4, 69.8, 57.0, 44.6, 38.3, 36.8, 30.4 (3C), 22.4; HRMS (ESI) calcd for C24H27N3NaO3+ [MNa]+ 428.1945, found 428.1940.:EtOAc, from 1.5:1 to 1:1.5) obtaining compound 6 (87% yield) as a white foamy solid; 1H NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 7.2 and 1.0 Hz, 1H), 7.37–7.23 (m, 5H), 7.21 (td, J = 7.2 and 1.0 Hz, 1H), 7.07 (td, J = 7.2 and 1.0 Hz, 1H), 6.70 (d, J = 7.2 Hz, 1H), 5.41 (dddd, J = 16.5, 10.1, 8.2 and 6.4 Hz, 1H), 5.11 (dq, J = 16.5 and 1.4 Hz, 1H), 5.00 (d, br, J = 10.1 Hz, 1H), 4.95 (d, J = 15.7, 1H), 4.86 (d, J = 15.7, 1H), 3.36 (td, J = 5.5 and 3.0 Hz, 1H), 3.22 (td, J = 5.5 and 3.1 Hz, 1H), 3.20 (ddt, J = 13.3, 6.5 and 1.0 Hz, 1H), 3.05 (dd, J = 13.3, 8.2 Hz, 1H), 2.93 (ddd, J = 14.7, 5.8 and 3.1 Hz, 1H), 2.86 (ddd, J = 14.7, 5.8 and 3.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 173.7, 167.2, 142.3, 135.4, 130.1, 129.5, 128.8 (2C), 127.7, 127.3 (2C), 124.8, 123.1, 120.7, 115.2, 109.5, 65.1, 44.0, 39.2, 37.9, 36.1; HRMS (ESI) calcd for C21H20N2NaO2+ [MNa]+ 355.1417, found 355.1420.Conflicts of interest
There are no conflicts to declare.Notes and references
- Reviews: (a) A. V. Sklyarenko, M. A. El'darov, V. B. Kurochkina and S. V. Yarotsky, Enzymatic synthesis of β-lactam acids (review), Appl. Biochem. Microbiol., 2015, 51, 627–640 CrossRef CAS; (b) B. K. Banik, Novel synthesis of β-lactams and their biological evaluation, J. Indian Chem. Soc., 2014, 91, 1837–1860 CAS; (c) G. S. Singh, β-Lactams in the new millenium. Part-II: Cephems, oxacephems, penams and sulbactam, Mini-Rev. Med. Chem., 2004, 4, 93–109 CrossRef CAS PubMed . Selected examples: ; (d) T. Sperka, J. Pitlik, P. Bagossi and J. Tozser, Beta-lactam compounds as apparently uncompetitive inhibitors of HIV-1 protease, Bioorg. Med. Chem. Lett., 2005, 15, 3086–3090 CrossRef CAS PubMed; (e) M. A. L. Blackie, F. Tzu-Shean, P. J. Smith and K. Chibale, Synthesis of novel β-lactams and in vitro evaluation against the human malaria parasite Plasmodium falciparum, ARKIVOC, 2016, iii, 214–235 Search PubMed; (f) A. Khan, J. Moskal and P. Wood, NMDA receptor modulators and uses thereof for the treatment of cognitive and other conditions, PCT Int. Appl., WO 2010033757 A1 20100325, 2010.
- (a) T. Oost, G. Backfisch, S. Bhowmik, M. M. van Gaalen, H. Geneste, W. Hornberger, W. Lubisch, A. Netz, L. Unger and W. Wernet, Potent and selective oxindole-ased vasopressin 1b receptor antagonists with improved pharmacokinetic properties, Bioorg. Med. Chem. Lett., 2011, 21, 3828–3831 CrossRef CAS PubMed; (b) K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stukey, K. Krajewski, P. P. Roller and S. Wang, Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS PubMed; (c) T. Emura, T. Esaki, K. Tachibana and M. Shimizu, Efficient Asymmetric Synthesis of Novel Gastrin Receptor Antagonist AG-041R via Highly Stereoselective Alkylation of Oxindole Enolates, J. Org. Chem., 2006, 71, 8559–8564 CrossRef CAS PubMed.
- (a) J. –S. Yu, F. Zhou, Y. –L. Liu and J. Zhou, A Journey in the Catalytic Synthesis of 3-Substituted 3-Aminooxindoles, Synlett, 2015, 26, 2491–2504 CrossRef CAS; (b) J. Kaur, S. Singh Chimni, S. Mahajan and A. Kumar, Stereoselective synthesis of 3-amino-2-oxindoles from isatin imines: new scaffolds for bioactivity evaluation, RSC Adv., 2015, 5, 52481–52496 RSC; (c) P. Chauhan and S. Singh Chimni, Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetra-substituted stereocenter, Tetrahedron: Asymmetry, 2013, 24, 343–356 CrossRef CAS.
- (a) G. Rainoldi, M. Faltracco, L. Lo Presti, A. Silvani and G. Lesma, Highly diastereoselective entry into chiral spirooxindole-based 4-methyleneazetidines via formal [2 + 2] annulation reaction, Chem. Commun., 2016, 52, 11575–11578 RSC; (b) G. Rainoldi, A. Sacchetti, A. Silvani and G. Lesma, Organocatalytic vinylogous Mannich reaction of trimethylsiloxyfuran with isatin-derived benzhydryl-ketimines, Org. Biomol. Chem., 2016, 14, 7768–7776 RSC; (c) M. Stucchi, G. Lesma, F. Meneghetti, G. Rainoldi, A. Sacchetti and A. Silvani, Organocatalytic Asymmetric Biginelli-like Reaction Involving Isatin, J. Org. Chem., 2016, 81, 1877–1884 CrossRef CAS PubMed; (d) G. Lesma, F. Meneghetti, A. Sacchetti, M. Stucchi and A. Silvani, Asymmetric Ugi 3CR on isatin-derived ketimine: synthesis of chiral 3,3-disubstituted 3-aminooxindole derivatives, Beilstein J. Org. Chem., 2014, 10, 1383–1389 CrossRef PubMed; (e) A. Sacchetti, A. Silvani, F. G. Gatti, G. Lesma, T. Pilati and B. Trucchi, Addition of TMSCN to chiral ketimines derived from isatin. Synthesis of an oxindole-based peptidomimetic and a bioactive spirohydantoin, Org. Biomol. Chem., 2011, 9, 5515–5522 RSC; (f) G. Lesma, N. Landoni, T. Pilati, A. Sacchetti and A. Silvani, Grignard Addition to Imines Derived from Isatine: A Method for the Asymmetric Synthesis of Quaternary 3-Aminooxindoles, J. Org. Chem., 2009, 74, 4537–4541 CrossRef CAS PubMed.
- Design of Hybrid Molecules for Drug Development, ed. M. Decker, Elsevier Ed, 2017 Search PubMed.
- S. Lohan and G. T. Bisht, Recent approaches in design of peptidomimetics for antimicrobial drug discovery research, Mini-Rev. Med. Chem., 2013, 13, 1073–1088 CrossRef CAS PubMed.
- R. Tuba, Synthesis of β-actams by transition metal promoted Staudinger reactions: alternative synthetic approaches from transition metal enhanced organocatalysis to in situ, highly reactive intermediate synthesis and catalytic tandem reactions, Org. Biomol. Chem., 2013, 11, 5976–5988 RSC.
- Review: (a) S. S. van Berkel, B. G. M. Bögels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Recent Advances in Asymmetric Isocyanide-Based Multicomponent Reactions, Eur. J. Org. Chem., 2012, 3543–3559 CrossRef CAS . Selected examples: ; (b) C. C. Răzvan, L. Schaepkens van Riempst, P. Schuckman, E. Ruijter and R. V. A. Orru, Ugi Four-Center Three-Component Reaction as a Direct Approach to Racetams, Synthesis, 2017, 49, 1664–1674 Search PubMed; (c) T. M. Vishwanatha, N. Narendra and V. V. Sureshbabu, Synthesis of β-lactam peptidomimetics through Ugi MCR: first application of chiral Nβ-Fmoc amino alkyl isonitriles in MCRs, Tetrahedron Lett., 2011, 52, 5620–5624 CrossRef CAS.
- M. A. Mironov, M. I. Tokarev, M. N. Ivantsova and V. S. Mokrushin, Ugi reaction with isocyanoindoles, Russ. J. Org. Chem., 2004, 40, 847–853 CrossRef CAS.
- G. Rainoldi, F. Begnini, M. de Munnik, L. Lo Presti, C. M. L. Vande Velde, R. Orru, G. Lesma, E. Ruijter and A. Silvani, Sequential Multicomponent Strategy for the Diastereoselective Synthesis of Densely Functionalized Spirooxindole-Fused Thiazolidines, ACS Comb. Sci., 2018, 20, 98–105 CrossRef CAS PubMed.
- N. Kumar, M. Kanti Das, S. Ghosh and A. Bisai, Development of catalytic deacylative alkylations (DaA) of 3-acyl-2-oxindoles: total synthesis of meso-chimonanthine and related alkaloids, Chem. Commun., 2017, 53, 2170–2173 RSC.
- DruLiTo, https://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- E. Matuschek, D. F. J. Brown and G. Kahlmeter, Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories, Clin. Microbiol. Infect., 2014, 20, 255–266 CrossRef PubMed.
- (a) M. Ganga, N. Kashyap, K. Kumar, S. Goyal and V. A. Nair, Imidazolidinone based chiral auxiliary mediated acetate aldol reactions of isatin derivatives and stereoselective synthesis of 3-substituted-3-hydroxy-2-oxindoles, Tetrahedron Lett., 2015, 56, 7074–7081 CrossRef; (b) G. C. Senadi, H. W. Chandru, S. S. K. Boominathan and J. J. Wang, Palladium (0)-Catalyzed Single and Double Isonitrile Insertion: A Facile Synthesis of Benzofurans, Indoles, and Isatins, Chem.–Eur. J., 2015, 21, 998–1003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra for all new compounds, X-ray data, table of physicochemical properties. CCDC 1849730. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08165d |
| This journal is © The Royal Society of Chemistry 2018 |