
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA graphene oxide polymer brush based cross-linked nanocomposite proton exchange membrane for direct methanol fuel cells†
Tianjian Yang ,
Zhongli Li,
Huilong Lyu,
Jianjun Zheng,
Jinglan Liu,
Fanna Liu,
Ziyong Zhang and
Huaxin Rao*
,
Zhongli Li,
Huilong Lyu,
Jianjun Zheng,
Jinglan Liu,
Fanna Liu,
Ziyong Zhang and
Huaxin Rao*
Department of Materials Science and Engineering, Jinan University, Guangzhou 510632, People's Republic of China. E-mail: traohx@jnu.edu.cn
First published on 26th April 2018
Abstract
Functional polymer brush modified graphene oxide (FPGO) with functional linear polysiloxane brushes was synthesized via surface precipitation polymerization (sol–gel) and chemical modification. Then, FPGO was covalently cross-linked to the sulfonated polysulfone (SPSU) matrix to obtain novel SPSU/FPGO cross-linked nanocomposite membranes. Meanwhile, SPSU/GO composite membranes and a pristine SPSU membrane were fabricated as control groups. Reduced agglomeration of the inorganic filler and better interfacial interaction, which are benefit to increase diffusion resistance of methanol and to generate continuous channels for fast proton transportation at elevated temperature, were observed in SPSU/FPGO cross-linked membranes. Moreover, the enhanced membrane stability (thermal, oxidative and dimensional stability) and good mechanical performance also guaranteed their proton conducting durability. It is noteworthy that the SPSU/FPGO-1 cross-linked membrane possesses the best comprehensive properties among all the prepared membranes and Nafion®117, it acquires the highest proton conductivity of 0.462 S cm−1 at 90 °C under hydrated conditions together with a low methanol permeability of 1.71 × 10−6 cm2 s−1 at 30 °C. The resulting high membrane selectivity displays the great potential of the SPSU/FPGO cross-linked membrane for DMFCs application.
1. Introduction
Direct methanol fuel cells (DMFCs) have been considered as prospective environment-friendly power sources which could lower our dependence on fossil fuel in the future. DMFCs convert the chemical energy of clean fuel (CH3OH) directly into electrical energy with many advantages such as high efficiency, compactness and low emission of pollution.1–4 The proton exchange membrane (PEM) plays a critical role in proton transportation from anode to cathode in the DMFCs. At present, Nafion, a perfluorosulfonic acid polymer membrane, is commonly used for proton conduction due to its good balance between the excellent chemical and electrochemical stabilities, good mechanical properties and high proton conductivity. However, its application in DMFCs is still limited due to its high cost, high fuel permeability and a confined operating temperature which is below 80 °C.5 Besides, because the transport mechanism and transport channels of protons and methanol molecules are similar, it is hard to simultaneously improve the membrane conductivity and decrease its methanol permeability when modifying PEMs.6One effective approach to improve the comprehensive properties of PEMs is to introduce inorganic nanofillers with excellent stability, high proton conductivity and methanol resistance into polyelectrolyte membrane matrix to obtain organic–inorganic hybrid PEMs. Graphene oxide (GO) has been considered as an attractive candidate, owing to its unique chemical structure with many oxygenated functional groups (carboxyl, hydroxyl etc.) for superior proton conductivity, methanol resistance, high stability and mechanical properties.7–9 Its large surface area with many polarized groups may help to construct continuous proton transport channels and resist the transportation of methanol, so a small amount of GO could be beneficial for improving the proton conducting behaviors and lower the methanol permeability.10–12
Polymer nanocomposite system is classified into two types: (i) hybrids with weak bonding (van der Waals force, ionic or hydrogen bonding) between inorganic filler and organic matrix, prepared by physical mixing of the two constituents; (ii) hybrids with strong bonds, prepared through mixing followed by covalent cross-linking. Direct physical mixing of the pristine GO and polyelectrolyte matrices often results in microscopic phase separation due to the low solubility of GO in organic solvents and low GO/polyelectrolyte interfacial compatibility, thereby limiting the maximum utilization of their nanostructures. Hence, additional functional groups (sulfonic acid and amine groups) or surfactant need to be introduced on the surface of nanofillers to further increase the organic–inorganic interaction.13–17 Recently, polymer brushes modified nanomaterials as PEMs has triggered more and more attentions.18–22 Compared with chemical modification using small molecules, polymer brushes offer the possibility of providing multiple selective functional groups on every soft polymer chains. Therefore, they can provide more effective opportunities to optimize the solubility of nanofiller in organic solvents, improve the nanofiller/polyelectrolyte interfacial compatibility by forming larger interfacial area with stronger interaction.23 L. Zhao et al.24 introduced sulfonated polymer brushes modified graphene oxide (SP–GO) into sulfonated poly(ether ether ketone) matrix. Interconnected ionic pathways were formed, leading to a 95.5% increase in proton conductivity of the membrane with 10 wt% of SP–GO under hydrated condition at 65 °C. Choi et al.25 incorporated GO into Nafion matrix, resulted in selective transport, a 40% decrease in methanol permeability and a maximum power density at 70 °C.
Compared with type (i) nanocomposite PEMs, covalently cross-linked nanocomposite membranes perform better mechanical property, markedly enhanced stability (dimensional, chemical and thermal stability) as well as higher methanol resistance for practical DMFCs applications.26–28 However, it's difficult to cross-link the widely used proton conducting polyelectrolytes onto GO surface through a mild reaction route without sacrificing too much ion exchange capacity (IEC) of the membrane. In addition, since PEMs with a very compact cross-linking network often performs significantly lowered proton conductivity due to their low water uptake (WU) and IEC, a moderately controlled cross-linking density is required.29 With a proper cross-linking density, the sacrifice of proton conductivity can be minimized,28,30–32 even enhanced proton conductivity compared with hybrid membrane without covalent cross-linking was observed, owing to the improved organic–inorganic interfacial compatibility, higher membrane water retention and continuous ionic exchange channels.33–35 With the help of functional polymer brushes modified GO (with reactive groups for cross-linking on the polymer brushes), a proper cross-linking density of the nanocomposite membrane can be realized by simply controlling the amount of incorporated polymer brushes modified GO fillers. Therefore, the trade-off between proton conductivity and methanol permeability of GO polymer brushes based cross-linked nanocomposite PEMs could be modulated for comprehensive DMFCs performance.
Aiming to enhance the comprehensive properties of PEMs including stability, proton conductivity and methanol resistance, we reported a novel hybrid PEM with cross-linked structure based on functional polysiloxane brushes modified GO (FPGO) and sulfonated polysulfone (SPSU). Herein, SPSU was chosen as polymer matrix for its excellent chemical and thermal stability, good mechanical property and excellent membrane-forming ability. The highly dispersible FPGO was synthesized by the following steps: surface precipitation polymerization (sol–gel) of vinylmethyldimethoxysilane (VMDMO) was adopted to synthesize novel polymer brushes modified graphene oxide (PGO), then 2,4,6,8-tetramethylcyclotetrasiloxane (DH4)/4-chlorostyrene cyclic cross-linker was connected onto the polysiloxane brushes to obtain FPGO. Subsequently, after the condensation reaction between terminal –OK groups of SPSU and 4-chlorostyrene groups of FPGO, SPSU/FPGO cross-linked membranes were fabricated by a facile solution casting method. The successful synthesis process were demonstrated by Fourier transform infrared spectroscopy (FTIR), dynamic light scattering (DLS), atomic force microscopy (AFM), solubility test, etc. Meanwhile, Nafion®117, pristine SPSU membrane and SPSU/GO composite membranes were set as control groups. Reduced agglomeration of GO and improved interfacial interaction was observed in SPSU/FPGO cross-linked membranes. In addition, thermal and dimensional stability of all the prepared membranes were analyzed. By measuring the WU, IEC, methanol permeability, proton conductivity and membrane selectivity, the effects of temperature, inorganic content and cross-linking density on the comprehensive membrane performance were evaluated.
2. Experimental
2.1. Materials
Sulfonated polysulfone (SPSU, Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, distribution index = 2, degree of sulfonation = 50%) was purchased from Shanghai Chunyi Materials Technology Co., Ltd., Shanghai, China. Nafion®117 (IEC = 0.909) was purchased from DuPont, Wilmington, USA. Few layers GO (>95 wt%, 0.5–5 μm2, 1–5 layers), which was prepared via modified Hummers method, was purchased from Suzhou Hengqiu Graphene Technology Co., Ltd., Suzhou, China. VMDMO and 4-chlorostyrene were purchased from Adamas, Emeryville, USA. 2,4,6,8-Tetramethylcyclotetrasiloxane (DH4) and chloroplatinic acid solution were obtained from the Research Center of Organic Silicone of Chengdu, China. N,N-Dimethylacetamide (DMAc), methanol, sodium hydroxide, absolute ethanol and phenolphthalein were obtained from Guangzhou Chemical Reagent Co., Ltd., Guangzhou, China.
000, distribution index = 2, degree of sulfonation = 50%) was purchased from Shanghai Chunyi Materials Technology Co., Ltd., Shanghai, China. Nafion®117 (IEC = 0.909) was purchased from DuPont, Wilmington, USA. Few layers GO (>95 wt%, 0.5–5 μm2, 1–5 layers), which was prepared via modified Hummers method, was purchased from Suzhou Hengqiu Graphene Technology Co., Ltd., Suzhou, China. VMDMO and 4-chlorostyrene were purchased from Adamas, Emeryville, USA. 2,4,6,8-Tetramethylcyclotetrasiloxane (DH4) and chloroplatinic acid solution were obtained from the Research Center of Organic Silicone of Chengdu, China. N,N-Dimethylacetamide (DMAc), methanol, sodium hydroxide, absolute ethanol and phenolphthalein were obtained from Guangzhou Chemical Reagent Co., Ltd., Guangzhou, China.
2.2. Synthesis of PGO and FPGO nanosheets
In order to obtain PGO (Scheme 1), VMDMO was adopted to modify the surface of GO nanosheets via surface precipitation polymerization. First, 400 mg of GO was dispersed in 800 mL of absolute ethanol by ultrasonication (SCIENTZ-IID ultrasonicator) for 3 h to get a homogeneous GO dispersion. Next, 3 g of VMDMO was dissolved into 50 mL of absolute ethanol and the resulted solution was slowly added into the GO dispersion. Then, 0.3 g of deionized water was added into the mixture dropwise to initiate the hydrolysis–condensation reaction of VMDMO on the surface of GO nanosheets. Surface precipitation polymerization was carried out at 65 °C under magnetic stirring for 24 h. Afterward, the dispersion was centrifuged and washed for 4 times with absolute ethanol to remove the unreacted traces of VMDMO. At last, PGO was obtained after being dried in a vacuum oven at 40 °C for 24 h.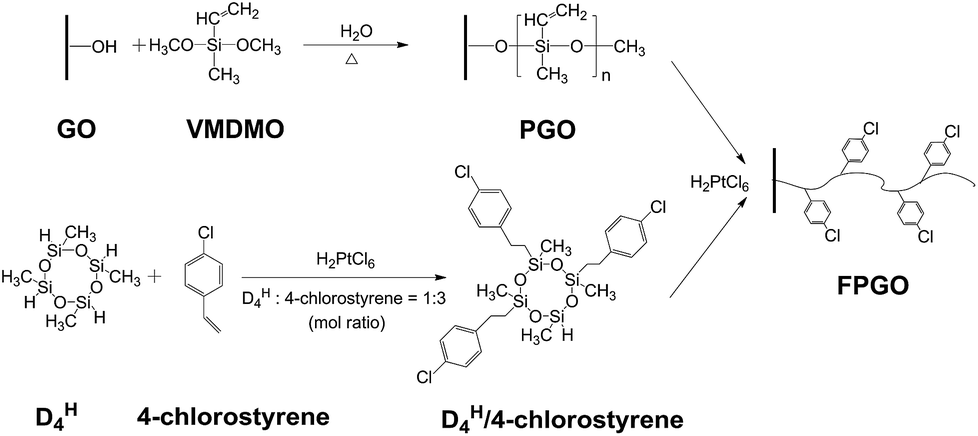 | ||
| Scheme 1 Synthesis procedure of PGO, DH4/4-chlorostyrene cyclic cross-linker and FPGO. | ||
To synthesize FPGO (Scheme 1), proper amount of chloroplatinic acid catalyst was added into the 60 mL of ethanol solution which contained 0.091 g of DH4 and 0.139 g of 4-chlorostyrene first. The hydrosilylation process between DH4 and 4-chlorostyrene was carried out at room temperature with constant magnetic stirring for 2 h. DH4/4-Chlorostyrene, a cyclic cross-linker which contains functional 4-chlorostyrene groups, was synthesized. After that, DH4/4-chlorostyrene cyclic cross-linker was connected to the polymer brushes of PGO via hydrosilylation reaction according to the following steps. First, 200 mg of PGO was dispersed in 400 mL of absolute ethanol by ultrasonication for 2 h to get a homogeneous PGO dispersion. Then, 10 mL of DH4/4-chlorostyrene cyclic cross-linker solution and proper amount of chloroplatinic acid were added into the PGO dispersion with rapid stirring. The hydrosilylation was carried out under ultrasonication for 1 h at room temperature. Afterward, the mixture was centrifuged and washed for 4 times with absolute ethanol to remove the unreacted traces of DH4/4-chlorostyrene cyclic cross-linker. Finally, the FPGO nanosheets, which contains functional 4-chlorostyrene groups for further cross-linking, were obtained after being dried in a vacuum oven at 40 °C for 24 h.
2.3. Preparation of SPSU/GO composite membranes
The SPSU/GO composite membranes were prepared via the solution casting method. A certain amount of GO was ultrasonicated in 10 g of DMAc for 2 h to prepare GO dispersion with various concentrations, respectively. Then, the GO dispersion was mixed with the 10 g SPSU solution (20 wt%) and kept stirring for 24 h at 35 °C. The mixture was again ultrasonicated for 1 h to attain better dispersion of GO within SPSU chains. The final casting solution was then cast on a smooth polyethylene terephthalate (PET) sheet in molds. The casting solution were dried at 60 °C for 12 h and 90 °C for 12 h, and then dried in a vacuum oven at 60 °C for 24 h to completely remove the solvent molecules. SPSU/GO composite membranes with various inorganic contents were fabricated. The obtained membranes were treated with 1 M HCl for 24 h to activate the sulfonic groups in the polymer matrix and the carboxyl groups in GO. Next, the treated membranes were washed using deionized water for many times until the washing solution is neutral. The prepared SPSU/GO composite membranes with different GO contents as 1 wt%, 2 wt% and 3 wt% were black, smooth, strong and homogeneous with thickness between 200 and 230 μm. They are coded as SPSU/GO-X, where X represented the weight percentage of GO in the composite membranes. All of the membranes were kept in deionized water before being used.2.4. Preparation of SPSU/FPGO cross-linked membranes
A certain amount of FPGO were ultrasonicated in 10 g of DMAc for 2 h to prepare the FPGO dispersion, respectively. Then, the obtained homogeneous FPGO dispersion was mixed with the 10 g of SPSU solution (20 wt%) and ultrasonicated for 1 h to attain better dispersion of FPGO with SPSU. Next, the mixture was added into a flask and keep the reaction temperature at 100 °C for 16 h under reflux condensation device with constant magnetic stirring. Through the condensation reaction between the –OK terminal groups of SPSU and the 4-chlorostyrene groups of FPGO, inorganic filler and polymer matrix can be cross-linked (Scheme 2). After cross-linking, the homogeneous and viscous solution was cast on a smooth polyethylene terephthalate (PET) sheet in the molds. The final sample was dried at 60 °C for 12 h and 90 °C for 12 h, then in a vacuum oven at 60 °C for 24 h to remove the residual solvents. The obtained SPSU/FPGO cross-linked membranes were then treated with 1 M HCl for 24 h to activate the sulfonic groups in the polymer matrix and the carboxyl groups in FPGO. The treated membranes were then washed with deionized water for many times until the washing solution is neutral. According to TG analysis of GO and FPGO, the GO content in FPGO is about 82 wt%. In order to make clear comparison with SPSU/GO composite membranes, filler contents of FPGO in SPSU/FPGO cross-linked membranes are also controlled at 1 wt%, 2 wt% and 3 wt%, respectively. The prepared cross-linked membranes were black, smooth, strong and homogeneous with thickness between 200 and 230 μm. They were kept in deionized water before being used. The membranes are coded as SPSU/FPGO-X, where X represented the weight percentage of FPGO in cross-linked membranes. | ||
| Scheme 2 Preparation procedure of SPSU/FPGO cross-linked membrane. | ||
2.5. Structure and morphology characterizations
FTIR were carried out using a Bruker Vertex 70 FTIR spectrometer in the range of 4000–400 cm−1. AFM images were taken to investigate the structure of the prepared polymer brushes in aqueous solution, using a multimode AFM (Bioscope Catalyst Nanoscope-V, Bruker, USA) with the automatic scanning mode. The measurement was performed with Tap150Al-G probe at the frequency of 150 kHz. The crystalline structure of the nanosheets and membranes were characterized by X-ray diffraction (XRD) using a X-ray diffractometer (MiniFlex 600, Rigaku, Japan) with Cu Kα radiation (λ = 0.15418 nm) in the 2θ range from 5° to 60°. The size and distribution of GO, PGO and FPGO polymer brushes were measured by DLS measurements using a Malvern Zetasizer Nano ZS, Britain. Cross-sections of the membranes were observed using field emission scanning electron microscope (ULTRA 55 FE-SEM, ZEISS, Germany). Vacuum-dried membrane samples were and freeze-fractured in liquid nitrogen and then sputtered with gold before being characterized by SEM. Field emission transmission electron microscope (JEM 2100 FE-TEM, JEOL, Japan) was utilized to investigate the nanostructure of ion clusters in nanocomposite membranes with an accelerating voltage of 200 kV. The membranes were stained with 0.5 M silver nitrate solution for 12 h and then vacuum-dried for 12 h before performing ultrathin section for TEM. Thermogravimetric analysis (TGA) was conducted at temperature range between 50 °C and 800 °C with a heating rate of 10 °C min−1 under nitrogen atmosphere using (TG 209 F3 Tarsus, NETZSCH, Germany). Determination methods of WU, area swelling and IEC of the prepared membranes were listed in ESI† in detail.2.6. Mechanical property
Tensile strength of various membranes was measured with a universal mechanical testing machine (Shimadzu AG-I, Japan) to investigate the change of mechanical properties upon the addition of GO or FPGO into the SPSU membranes. The membrane specimens were prepared to be 8 mm in width and 25 mm in length, and the testing elongation rate was 2 mm min−1. All the membranes were kept in DI water overnight at room temperature for testing, and water on the surface of membranes was removed using a filter paper before measurements. The final value of every group was the average of three measurements with an error within 5%.2.7. Oxidative stability
Each membrane sample was immersed in Fenton's reagent (2 ppm FeSO4 in 3% H2O2 solution) at 80 °C. Oxidative stability of all the prepared membranes was evaluated based on the weight change after treating in Fenton's reagent for 1 h, the time when membranes started to dissolve and the time for completely dissolving.2.8. Proton conductivity
Proton conductivity of membranes (2 cm × 1 cm) at 30, 50, 70 and 90 °C were measured using AC impedance spectroscopy of electrochemical workstation (Versa STAT3, AMETEK, USA) with an oscillating voltage of 20 mV over a frequency range of 10–106 Hz. The prepared membranes were fully hydrated in water for over 48 h prior to be tested. Proton conductivity measurement of fully hydrated membranes was carried out with the testing cell being immersed in deionized water bath. Proton conductivity (σ, S cm−1) of the membrane was the average value with an error within ±0.004 S cm−1 calculated by eqn (1):
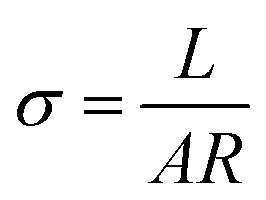 | (1) |
2.9. Methanol permeability and membrane selectivity
Methanol permeability of various membranes at 30 °C is carried out in a glass diffusion cell consisting of two compartments separated by a proton exchange membrane. Membranes are pre-hydrated for 24 h and then tightly clamped between the two diffusion compartments. Compartment A is filled with 50 wt% methanol aqueous solution and compartment B is filled with deionized water initially. Methanol concentration in compartment B is measured instantly during the diffusion process using a digital refractometer (ABBE WYA (2WAJ)). Finally, methanol permeability (P, cm2 s−1) was obtained with an error within 0.1 × 10−6 by eqn (2):
 | (2) |
 | (3) |
3. Results and discussion
3.1. Synthesis and characterization of GO and PGO nanosheets
As shown in Schemes 1 and 2, through hydrolysis–condensation reaction (sol–gel) between –OCH3 groups of VMDMO and –OH groups of the GO nanosheets, polysiloxane brushes were polymerized on the surface of GO nanosheets, leaving free vinyl groups on the PGO.To further confirm the existence of the polymer brushes on GO nanosheets, FTIR, AFM and TGA were performed. For the FTIR spectrum in Fig. 1a, the characteristic absorption peaks confirm the structure of GO. Peaks at 3450, 1724, 1628, 1396, 1220 and 1044 cm−1 correspond to the typical O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, C–OH, C–O–C and C–O groups of GO, respectively.36,37 For PGO, the characteristic peak at 1632 cm−1 corresponds to both CC bonds of GO substrate and the vinyl groups on polysiloxane brushes, the peak at 1220 cm−1 is assigned to C–O–C bonds of GO. The absorption bands at 1022 and 1127 cm−1 are assigned to Si–O–Si and Si–O–C bonds, respectively.38–40 And the peaks at 1452 and 1364 cm−1 are for the asymmetric and symmetric deformation vibrations of the methyl group in polysiloxane brushes. The above evidences confirm the successful reaction between –OH groups of GO nanosheets and –OCH3 groups of VMDMO polysiloxane brushes, leading to the successful surface precipitation polymerization. Some peaks can not be clearly described due to their overlapping, which is also reported in related literature.36
O, CC, C–OH, C–O–C and C–O groups of GO, respectively.36,37 For PGO, the characteristic peak at 1632 cm−1 corresponds to both CC bonds of GO substrate and the vinyl groups on polysiloxane brushes, the peak at 1220 cm−1 is assigned to C–O–C bonds of GO. The absorption bands at 1022 and 1127 cm−1 are assigned to Si–O–Si and Si–O–C bonds, respectively.38–40 And the peaks at 1452 and 1364 cm−1 are for the asymmetric and symmetric deformation vibrations of the methyl group in polysiloxane brushes. The above evidences confirm the successful reaction between –OH groups of GO nanosheets and –OCH3 groups of VMDMO polysiloxane brushes, leading to the successful surface precipitation polymerization. Some peaks can not be clearly described due to their overlapping, which is also reported in related literature.36
 | ||
| Fig. 1 (a) FTIR spectra of GO, PGO and FPGO, (b) TGA curves of GO and FPGO, (c) XRD patterns of GO and FPGO, (d) AFM images and height profiles of PGO. | ||
Morphology of GO and PGO were probed by AFM, and the images were showed in Fig. 1d and S1 (ESI†). By surface precipitation polymerization, PGO was successfully synthesized and showed a novel multi-layer sandwich structure. AFM investigation reveals the continuous and uniform layers of PGO. Due to the diffusion of VMDMO molecules into GO layers, polysiloxane polymerization reacted on both the upper and lower surfaces of each GO layer, leading to the growth of intercalated polysiloxane brushes on both sides. As shown in Fig. 1d, detailed thickness analysis on different areas of PGO nanosheets showed that the thickness of each PGO layer is approximately 4.7 nm. Meanwhile, the measured thickness of pristine single-layer GO is about 1.1 nm in Fig. S1 (ESI†), which is consistent with the thickness data of GO reported in the relevant literature.41 Thus, the thickness of the homogeneous polysiloxane brushes layers formed on upper and lower surface of GO nanosheets is expected to scale approximately as 1.8 nm.
3.2. Synthesis and characterization of FPGO nanosheets
DH4/4-Chlorostyrene cyclic cross-linker was synthesized via hydrosilylation using chloroplatinic acid solution as catalyst. The mole ratio of DH4 to 4-chlorostyrene was controlled at 1:3. In this way, the four Si–H bonds in each DH4 molecule can be mainly connected with three 4-chlorostyrene groups, leaving one Si–H in each cyclic cross-linker molecule to react with the vinyl groups on PGO via hydrosilylation to prepare FPGO. In this way, the agglomeration between nanosheets could be alleviated. Reaction route scheme of DH4/4-chlorostyrene cyclic cross-linker was shown in Scheme 1a.
For FTIR spectrum of FPGO in Fig. 1a, peaks at 1729 and 1628 cm−1 correspond to CO and CC bonds. The band at 804 cm−1 is attributed to the C–Cl stretching vibration,42 and the broad peak at 1074 cm−1 is assigned to Si–O–Si bond, which probably overlaps the absorption peaks at 1127 and 1044 cm−1 (νSi–O–C and νC–O).38 Moreover, no peak of Si–H bond is showed around 2160 cm−1. These evidences confirm that 4-chlorostyrene was successfully connected to DH4, and the cyclic cross-linker was successfully connected to the polysiloxane brushes of PGO by hydrosilylation.
Thermogravimetric analysis (TGA) and differential thermogravimetric analysis (DTG) was employed to access the relative composition of GO within the FPGO and its thermal stabilities. As shown in Fig. 1b and S2 (ESI†), pristine GO and FPGO were identically subjected to TGA for clear comparison. TGA curves suggest that GO and FPGO display three main stage weight loss, including the removal of moisture (mainly bonded water) from the hydrophilic groups as the first stage (50–150 °C), the degradation of oxygen containing functional groups as the second stage (150–220 °C) and the decomposition of GO backbone as the third stage (230–800 °C). The additional weight loss of FPGO at 150–760 °C largely corresponded to the decomposition of polysiloxane brushes, and this process could also prove that functional polymer brushes had been successfully grafted onto the GO nanosheets. The weight loss of GO and FPGO polymer brushes from 50 to 760 °C are 66.05 wt% and 55.37 wt%, respectively. According to reported data, degradation residual of linear polysiloxane at 800 °C in N2 atmosphere is around 6–8 wt%.43 Therefore, the weight percentage of polysiloxane brushes in FPGO nanosheets is calculated to be around 18 wt%. So the weight percentage of GO in FPGO is about 82 wt%.
As shown in Fig. 1c, XRD was performed to investigate the crystalline nanostructure of GO and FPGO. GO attains a characteristic diffraction band of (002) at 2θ = 8.9° corresponding to the interlayer spacing of adjacent GO sheets about 0.99 nm, which is close to the value in literature.44 This band for FPGO nanosheets is shifted to 10.5° after surface precipitation polymerization, indicating that the interlayer d-spacing of FPGO decreases to 0.84 nm. The slightly decreased d-spacing comes from the interaction between the adjacent polysiloxane brushes layers. Similar phenomenon has also been observed in other GO polymer brushes.24 Generally, GO and FPGO show very close XRD curves, which implies the crystalline structure of GO was not changed significantly.
The nanosize and distribution of GO, PGO and FPGO nanosheets were characterized using DLS and were shown in Fig. S3 (ESI†). By comparison, no obvious change of nanosize and distribution between GO (565 ± 45 nm), PGO (602 ± 58 nm) and FPGO (687 ± 54 nm) was found. Therefore, the agglomeration between PGO and FPGO nanosheets during the synthesis was successfully alleviated, which ensured their uniform dispersion in solvent and hybrid membranes.
As shown in Fig. S4a (ESI†), the GO dispersion in DMAc (on the left) prepared by ultrasonication is easy to agglomerate and then precipitate after being rested for 4 h. However, FPGO can be dispersed in DMAc uniformly without ultrasonication. Furthermore, the FPGO dispersion (on the right) keeps stable and homogeneous with no obvious agglomeration for at least 24 h. In this work, DMAc, a widely used solvent for PEMs fabrication, was chosen for the cross-linking reaction and the fabrication of hybrid membranes. Therefore, the aggregation of nano-filler in SPSU/FPGO cross-linked membranes could be effectively reduced to obtain uniformly cross-linked and homogeneous hybrid PEMs.
3.3. Preparation and characterization of the membranes
The synthesis route to SPSU/FPGO cross-linked membranes was shown in Scheme 1b. Through the condensation reaction between terminal –OK groups of SPSU and 4-chlorostyrene groups of FPGO, organic–inorganic cross-linked network structure can be obtained. The prepared SPSU/FPGO cross-linked membranes, whose FPGO content range is between 1 wt% to 3 wt%, were black, smooth, strong and homogeneous. Here, their cross-linking structure was confirmed by the solubility test. SPSU/GO-2 composite membrane and SPSU/FPGO-2 cross-linked membrane were immersed in 10 mL of DMAc at 80 °C for 1 h, respectively. As shown in Fig. S4b (ESI†), SPSU/GO-2 (on the left) dissolved more quickly than SPSU/FPGO-2 (on the right), making the color of the solution darker. While SPSU/FPGO-2 dissolved only partially in DMAc. Therefore, compared to SPSU/GO composite membrane, the cross-linking structure of SPSU/FPGO cross-linked membranes appeared to reduce their solubility, which also means better solvent resistance.Scanning electron microscopy (SEM) was used to investigate the internal microstructure of the pristine SPSU membrane, SPSU/GO-3 composite membrane and SPSU/FPGO-3 cross-linked membrane. Fig. 2a–d shows the images of the cross-section of different membranes, and all the membranes are dense and relatively homogeneous. The cross-section of the pristine SPSU membrane presents a smooth and uniform surface without any cracks, indicating fine quality. Compared with SPSU control membrane, the cross-section of SPSU/GO-3 composite membrane is rougher. From Fig. 2a and c, attributed to the hydrogen bonding between sulfonic acid groups of SPSU matrix and the polar groups (–O–, CO) of GO on the SPSU–GO interface, GO nanosheets exhibit good dispersion in the membrane. However, there are still some aggregations of GO nanosheets presented in the SPSU/GO-3 composite membrane, which are marked in Fig. 2b. These aggregations may result in a negative blocking effect on the proton conduction performance of composite membranes. For SPSU/FPGO-3 cross-linked membrane, its cross-section is rougher than that of SPSU/GO-3 composite membrane, but no obvious aggregations of inorganic filler are observed (Fig. 2c and d). Many smooth transition slopes are observed at the organic–inorganic interface of SPSU/FPGO cross-linked membrane. That indicates better interfacial adhesion between inorganic fillers and SPUS matrix, which is due to the better dispersion state of FPGO in DMAc and the uniform cross-linking structure between flexible FPGO polymer brushes and SPSU chains. Thereby, the interfacial compatibility between the inorganic fillers and matrix was efficiently improved, which may help to build more continuous proton transfer pathways at the interface.
 | ||
| Fig. 2 Cross-sectional SEM images of (a) pristine SPSU membrane, (b) SPSU/GO-3 composite membrane, (c) and (d) SPSU/FPGO-3 cross-linked membrane. | ||
The micro/nano phase segregation of SPSU/GO-1 and SPSU/FPGO-1 was investigated by TEM, as shown in Fig. S5 (ESI†). The bright area and dark area represent hydrophobic domains and hydrophilic ion cluster domains, respectively. For both SPSU/GO-1 and SPSU/FPGO-1 membranes, the bright and dark domains are uniformly dispersed, illustrating that the dispersion of inorganic fillers in polymeric matrices is uniform. So the ion clusters and proton conducting channels in SPSU/GO-1 and SPSU/FPGO-1 membranes are evenly formed to realize high membrane proton conductivity.
XRD was carried out to investigate the influence of incorporated inorganic nanosheets on the crystalline structure of prepared membranes, and the results were shown in Fig. 3a. It is found that all the membranes have the major diffraction band at 2θ = 10–35° corresponding to the crystalline domain of SPSU matrix, closing to the data in literature.45 For both SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes, intensities of their major crystalline bands are lower than that of pristine SPSU membrane. In addition, major peaks of this band for SPSU/GO and SPSU/FPGO membranes are shifted slightly to 2θ = 19.7° and 19.4°, respectively. The decrease of diffraction intensity and slightly increase of interplanar spacing in all the hybrid membranes is reasonably attributed to the mutual interaction between SPSU matrix and GO/FPGO fillers, which impede the ordered stacking of SPSU backbone and finally leading to lower crystallinity. Moreover, the intensity of diffraction bands of SPSU/FPGO membranes are relatively higher than that of SPSU/GO membranes. This phenomenon should be ascribed to the SEM observed smooth organic–inorganic polymer brushes interface in SPSU/FPGO membranes. Compared with GO nanosheets, there are less oxygen containing polarized groups presented on the surface of FPGO, so the interfacial interaction in SPSU/FPGO membranes is weaker than that in SPSU/GO membranes, leading to the lower inhibition on the crystallization of SPSU backbone.18 So the crystallinity in SPSU/FPGO membranes is higher than that of SPSU/GO membranes with the same inorganic content.
 | ||
| Fig. 3 (a) XRD patterns of pristine SPSU, SPSU/GO composite and SPSU/GO cross-linked membranes, (b) TGA curves, (c) DTG curves, (d) water uptake curves for various membranes at different temperature, (e) area swelling curves of SPSU and hybrid membranes at 70 °C. | ||
3.4. Mechanical properties
Table 1 presents the mechanical properties of the prepared membranes in wet state in terms of their stress–strain curves, fracture strain and tensile strength. Pristine SPSU membrane attains a good mechanical stability with a tensile strength of 32.77 MPa. Hybrid membranes show relatively lower tensile strength. When the inorganic content increased from 1 wt% to 3 wt%, their tensile strength decreased gradually. Among them, SPSU/FPGO-1 cross-linked membrane possesses the best tensile strength of 32.61 MPa, which is close to that of pristine SPSU membrane. By comparison, SPSU/FPGO membranes perform higher tensile strength than SPSU/GO membranes with the same inorganic content, and the increase in tensile strength between them are 10.3%, 24.5% and 50.0% when the inorganic content is 1 wt%, 2 wt% and 3 wt%, respectively. Higher FPGO contents lead to the higher cross-linking density, so the increasing amplitude is higher too. Tensile strength of as-synthesized hydrated membranes (20.41–32.77 MPa) is relatively lower than reported sulfonated polyimide based composite membranes.10 But it should be noted that all the prepared membranes perform higher tensile strength than our measured value of hydrated Nafion®117 (17.8 MPa) and reported sulfonated poly(arylene ether ketone) membranes (5.9–16.52 MPa),46 indicating that they satisfy the mechanical requirement of DMFCs.| Membrane | Oxidative stability | Mechanical properties | ||
|---|---|---|---|---|
| RWa (%) | Tb (h) | Tensile strength (MPa) | Elongation at break (%) | |
| a Retained weight of membranes after immersing in Fenton's reagent for 1 h at 80 °C.b The time for membranes began to dissolve. | ||||
| SPSU | 96.0 | 1.45 | 32.77 | 7.78 |
| SPSU/GO-1 | 96.6 | 2.2 | 29.57 | 6.99 |
| SPSU/GO-2 | 93.9 | 2.5 | 25.02 | 3.51 |
| SPSU/GO-3 | 95.2 | 3.5 | 20.41 | 2.87 |
| SPSU/FPGO-1 | 96.2 | 2.5 | 32.61 | 12.36 |
| SPSU/FPGO-2 | 94.3 | 3 | 31.15 | 5.06 |
| SPSU/FPGO-3 | 96.0 | 4 | 30.62 | 3.22 |
3.5. Thermal and oxidative stability
Thermal stability, which is closely related to the membrane structure, is an important prerequisite for PEMs. Degradation behaviors of pristine SPSU membrane, SPSU/GO-3 composite membrane and SPSU/FPGO-3 cross-linked membrane were evaluated by TGA and DTG in Fig. 3b and c. All the membranes exhibit analogous one minor and two major weight loss steps: (i) the first minor step is attributed to the evaporation of residual solvents (DMAc) and water around 50–250 °C; (ii) the second stage corresponds to the decomposition of sulfonic acid groups on SPSU chains around 280–425 °C; (iii) the third stage is the degradation of polymer backbones around 450–770 °C. Meanwhile, for the introduced GO in SPSU/GO composite membrane and SPSU/FPGO cross-linked membrane, a gradual decrease of mass is observed at 150–250 °C, possibly due to the degradation of its oxygen containing functional groups of the small introduced amount of GO. And the combustion of the residual char of GO happens at 300–800 °C. Thermal decomposition behavior of the embedded GO nanosheets in polymeric matrix is similar to previously reported values.10,24,47 Compared with pristine SPSU membrane, the incorporation of GO and FPGO fillers alters the decomposition behavior of the hybrid membranes. The degradation temperature of step (ii) (Td1) of pristine SPSU is 357 °C, while the Td1 of SPSU/GO-3 and SPSU/FPGO-3 membranes decreased by around 3 and 6 °C, respectively. The degradation temperature of step (ii) (Td2) of SPSU/GO-3 and SPSU/FPGO-3 decreased by 7 and 18 °C compared with SPSU (Td2 = 468 °C). In addition, the char yields of SPSU/GO composite membrane and SPSU/FPGO cross-linked membrane shows about 7.6 wt% higher than that of pristine SPSU control membrane. The above phenomena might be attributed to the incorporation of inorganic filler and the covalent cross-linking network, which impedes the degradation rate of SPSU backbone, leading to an improved thermal decomposition stability of the hybrid membranes. Results of the membrane structure obtained by TGA are well in agreement with that by FTIR and XRD. Generally, all the prepared membranes exhibited adequate thermal stability up to 280 °C, which is higher than the minimum decomposing temperature requirement for DMFCs.The degradation of polymer backbones of PEMs usually occur during fuel cell operations due to the attack of free radicals like HO˙ and HO2˙.28 Generally, a high degree of sulfonation leads to high membrane proton conductivity but low oxidative stability. Therefore, it's important to measure the lifetime of prepared pristine and hybrid PEMs under harsh oxidative fuel cell conditions where membranes were soaked in Fenton's reagent at 80 °C to accelerate the test. Oxidative stability of each samples was evaluated by comparing their dissolving behaviors (Table 1). After 1 h, SPSU/FPGO cross-linked membranes showed relatively lower weight loss than pristine SPSU membrane and SPSU/GO composite membranes with the same inorganic content. Then, compared to pristine SPSU membrane which began to dissolve within 1.45 h and completely dissolved within 2 h, SPSU/GO membranes and SPSU/FPGO membranes started to dissolve after 2.2–3.5 h and 2.5–4 h, respectively. And the elapsed time when the SPSU/FPGO membranes dissolved completely is also higher than that of SPSU and SPSU/GO membranes. With increasing inorganic contents, all the hybrid membranes performed better oxidative stability. Moderate membrane WU is beneficial to high proton conductivity while excess WU may consequently lead to severe membrane swelling and then weaken the membrane durability.48,49 As shown in Fig. 3d and Table S1 (ESI†), SPSU/FPGO membranes show lower WU than SPSU/GO membranes with the same inorganic contents at 70–90 °C. Furthermore, with the help of covalently cross-linked network which may slow down the oxidation process,29 SPSU/FPGO membranes showed better oxidative stability than SPSU/GO membranes.
3.6. Water uptake (WU), area swelling and ion exchange capacity (IEC)
WU and area swelling are important parameters for PEMs. WU plays a crucial role in proton conduction ability and transportation mechanism and area swelling directly determines the dimensional stability of PEMs.50 Since the excessive WU would lead to low membrane durability, a proper WU and area swelling level should be maintained to guarantee the overall performance for DMFCs.The absorbed water molecules, as the proton conductive medium, provide proton carriers for Vehicle mechanism and form hydrogen bond networks for Grotthuss-type proton transportation.2,51 Table S1 (ESI†) and Fig. 3d show the WU of different membranes at different temperatures. It is seen that the WU of all the hybrid membranes at 30 °C is higher than the WU of pristine SPSU without GO or FPGO. The increase is attributed to the addition of GO or FPGO, which contains many hydrophilic groups (–OH, –COOH and –O–). They provided additional water storage spaces at the SPSU matrix-filler interfacial domains. Meanwhile, the WU values of all the SPSU/FPGO cross-linked membranes at 30–90 °C are lower than that of SPSU/GO composite membrane with the same inorganic content. For example, compared to the pristine SPSU membrane, the increased proportion of WU is 50.7% for the SPSU/GO-1 at 30 °C, while the increased proportion of WU is 42.9% for the SPSU/FPGO-1 at 30 °C. This phenomenon is attributed to the compact polymeric network structure of the SPSU/FPGO cross-linked membranes and the increased organic–inorganic interfacial interaction due to covalently cross-linking.13,52 In this way, less water molecules were hold in the SPSU/FPGO membranes, leading to the lower WU compared with SPSU/GO membranes. For SPSU/GO composite membranes, due to the hydrogen bonds between oxygen functional groups of nanosheets and –SO3H groups of SPSU, the increase of WU is restricted as the temperature increased from 70 to 90 °C, leading to the lower WU than SPSU at 90 °C. And the phenomenon of sharp increased WU of pristine polymeric PEMs at elevated temperature is also observed in other works.24,53 Moreover, with the increasing contents of GO or FPGO fillers, WU values of hybrid membranes decrease at all the tested temperatures. For SPSU/FPGO cross-linked membranes, their polymer structure is compacted not only by hydrogen bonds but also by the covalent cross-linking bonds between FPGO and SPSU. Hence, with the increasing FPGO content, their WU values decrease much more.
As shown in Table S1 (ESI†), area swelling values of pristine SPSU membrane, SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes were measured. And the tested area swelling behavior is similar to the WU behavior of the membranes. It is reported that the area swelling of PEMs increases commonly when GO is incorporated as the inorganic filler,13,24 and similar phenomenon is also observed in our SPSU/GO composite membranes. At 30 °C, as shown in Fig. S6 (ESI†), area swelling of hybrid membranes is higher than that of pristine SPSU membrane due to the introduction of GO/FPGO inorganic fillers with many hydrophilic groups. For practical application as DMFCs, the area swelling of PEMs at relatively high temperature range (70–90 °C) is essential because it directly reflects the dimensional stability at operating temperature range, and the area swelling values of various membranes were shown in Fig. 3e. In this way, broader application of GO based PEMs might be limited due to the decreasing dimensional stability. However, through constructing polysiloxane brushes layer on GO surface to form compact polymeric network, and increase of polymer chain mobility and free volume when temperature increases can be restricted.26 Hence, at 70 °C, the cross-linked network in SPSU/FPGO prevents the temperature-WU-related structural changes in the hybrid membranes, resulting in lower area swelling values. Notably, the area swelling of SPSU/FPGO-1 membrane at 70 °C is even lower than that of pristine SPSU membrane, showing enhanced dimensional stability for DMFC applications.
IEC (mmol of sulfonic acid per g of the prepared membranes) indicates the number of ion exchangeable groups and available protons, so it determines the proton conductivity of membranes.54 IEC value of each sample was determined by titration method and the results are given in Table S1 (ESI†). The IEC of pristine SPSU control membrane is measured to be 1.44 mmol g−1, which is consistent to the theoretical value (1.46 mmol g−1). Previous research has suggested that, the IEC would be lowered by introducing organic or inorganic materials without sulfonic acid groups, which actually dilute the concentration of sulfonic acid groups in hybrid membranes.55 In this work, when different contents of GO and FPGO are added, the IEC values of SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes decreased too. For example, the IEC values of all the SPSU/GO composite membranes decreased from 1.44 to 1.23 mmol g−1 with increasing GO content, and the IEC values of all the SPSU/FPGO cross-linked membranes decreased from 1.44 to 1.21 mmol g−1. Moreover, the IEC values of SPSU/FPGO cross-linked membranes are lower than that of SPSU/GO-X composite membranes with the same inorganic filler content. The decreased IEC values are reasonably attributed to two factors: (i) the diluted concentration of sulfonic acid groups in the cross-linked membranes due to the introduction of functional polymer brushes; (ii) the more compact polymer structure of SPSU/FPGO cross-linked membranes which limits the amount of protons from being exchanged at room temperature.
3.7. Proton conductivity of the membranes
Proton conductivity is the most important property for PEMs, since it directly determines the operational voltage of the fuel cells. Proton conductivity values are closely related to temperature, inorganic content, WU and the cross-linking structure of the membranes. Generally, in order to describe proton transportation through the PEMs, two principle mechanisms were built. One is Vehicle mechanism, which assumes that free water molecules act like vehicles, protons are carried and transported by these vehicles by forming hydronium such as H3O+, H5O2+ and H9O4+, thus allowing the proton transportation through the membranes.56 The other is Grotthuss mechanism (hopping mechanism). For proton exchange membranes based on sulfonated polymer matrix, protons can hop from one proton-attracted group (H3O+·SO3−) to another by hydrogen bonds.57Fig. 4a–d and S7a–c (ESI†) show the proton conductivity of fully hydrated Nafion®117, pristine SPSU membrane, SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes measured at 30, 50, 70 and 90 °C. The obtained proton conductivity values of fully hydrated Nafion®117 are close to reported data.58 At low operating temperature range (30–50 °C), proton conductivity value of pristine SPSU membrane are close to Nafion®117. With 1 wt% of GO being incorporated into the membrane matrix, SPSU/GO-1 performs higher proton conductivity than Nafion®117 and SPSU. The enhancement is attributed to the following two reasons. First, the evenly distributed GO nanosheets possess high aspect ratio and many polarized functional groups, they create more facile interconnected channels for proton hopping and transfer, which is formed by ion clusters of the sulfonic acid groups and oxygenated groups.24,59 Second, from Table S1 (ESI†), SPSU/GO-1 membrane possesses higher WU at 30–50 °C than that of SPSU and Nafion®117. The increased WU may provide more proton conducting carriers and hydrogen networks, which could benefit the proton transportation.13,34 Thus, protons are able to transport more quickly through the SPSU/GO-1 membrane, leading to higher proton conductivity. However, when the GO content reached 2–3 wt%, proton conductivity of the SPSU/GO membranes decreased. The decrease is partially attributed to lower IEC caused by the dilution of GO on the concentration of sulfonic acid groups. Meanwhile, the blocking effect of GO nanosheets in nanocomposite membranes with 2–3 wt% of GO also plays a crucial role. As is illustrated in Section 3.3, the relatively high GO content and GO aggregation in SPSU/GO-3 membrane may generate blocking effect for proton transportation, which resulted in fewer, longer and narrower proton transfer pathways and reduced free volume of SPSU chains, so the protons were blocked by the 2D nanosheets during migration.16 The strong blocking effect in SPSU/GO-3 predominate from 30 °C to 90 °C, so the proton conductivity of SPSU/GO-3 composite membrane (0.103–0.354 S cm−1) is much lower than that of SPSU/GO-1 (0.142–0.462 S cm−1) and Nafion®117 (0.125–0.238 S cm−1).
 | ||
| Fig. 4 Temperature-dependent proton conductivity of (a) Nafion®117 and SPSU/GO composite membranes under hydrated condition, (b) Nafion®117 and SPSU/FPGO cross-linked membranes under hydrated condition, (c) and (d) Arrhenius plots of proton conductivity of pristine SPSU membrane, SPSU/GO membranes and SPSU/FPGO cross-linked membranes. | ||
As we can see in Fig. 4a and b, due to the enhanced mobility of protons/water molecules across the membrane, the increased free volume of the membranes and boosted mobility of protons at elevated temperature range,60,61 proton conductivity of every prepared membrane performs positive temperature-conductivity dependence. Moreover, attributed to their positive temperature-WU dependence, all the prepared membranes performed higher proton conductivity than Nafion®117 at 70–90 °C. For SPSU/GO-1, its proton conductivity at temperature range between 30 °C and 90 °C increased significantly (up to 0.416 S cm−1 at 90 °C). The enhancement is attributed to the evenly dispersed GO which facilitate proton transportation as well as its higher WU at 30–70 °C than that of pristine SPSU membrane.
As shown in Fig. 4a, b and S7 (ESI†), the proton conductivity of SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes with the same inorganic content are compared with pristine SPSU membrane. For SPSU/FPGO cross-linked membranes, they perform lower proton conductivity than SPSU/GO composite membranes at 30–50 °C. With covalently cross-linked network, the polymer structure of SPSU/FPGO membranes is more compact and chain mobility is restricted, leading to lower WU than SPSU/GO membranes. So they possess lower proton conductivity at 30–50 °C compared with the same inorganic contents. Interestingly, all the SPSU/FPGO cross-linked membranes shows higher conductivity than both pristine SPSU and SPSU/GO composite membranes at 70–90 °C, even if their WU and IEC values are lower. This might be attributed to the cross-linked network which provides a closer contact or connectivity between the proton conducting functional groups (carboxyl, hydroxyl, sulfonic acid groups etc.) in SPSU/FPGO membranes.34 In this way, sulfonic-riched domains tend to be induced on the surface of FPGO, promoting the formation of interconnected proton conducting channels at SPSU/FPGO interface.30,62,63 At 30–50 °C, due to the strong negative influence of lower WU values and lower IEC values on the proton conductivity of hybrid membranes, SPSU/FPGO cross-linked membranes show lower proton conductivity compared with SPSU/GO composite membranes. However, as the temperature increased to 70–90 °C, energy of thermal motion for polymer chains in SPSU/FPGO cross-linked membranes increases, resulting in the increasing chain conformation and flexibility. Therefore, more water molecules are absorbed in the SPSU/FPGO cross-linked membrane, providing more proton conductive carriers for both Grotthuss mechanism and Vehicle mechanism transportation at 70–90 °C. In this way, the interconnected proton conducting channels on the SPSU/FPGO interface facilitate high-efficiency proton transportation with the effectively improved proton conductivity at 70–90 °C. It is noteworthy that, even for the SPSU/FPGO-3 cross-linked membrane with the lowest IEC value and strong blocking effect negatively affecting its proton conductivity (Fig. S7c (ESI†)), it still shows a conductivity (0.354 S cm−1) which is higher than SPSU/GO-3 composite membrane (0.313 S cm−1) and is very close to pristine SPSU membrane (0.358 S cm−1) at 90 °C. Among all the tested membranes, the SPSU/FPGO-1 cross-linked membrane performs the best proton conduction behavior. Its proton conductivity reaches 0.4621 S cm−1 at 90 °C, which is 1.94-folds of the value of Nafion®117, 33% higher than pristine SPSU membrane and 14.5% higher than SPSU/GO-1 composite membrane. As is known, the operating temperature of DMFCs is generally above 80 °C, so the studied SPSU/FPGO-1 cross-linked membrane is effective as a practical PEM.
To further investigate the mechanism of proton transportation, proton conductivity values in Arrhenius plot of the PEMs were depicted in Fig. 4c and d, and the activation energy (Ea) values were calculated via Arrhenius equation. For Grotthuss mechanism, the activation energy for proton transportation is around 14–40 kJ mol−1,64 and the Ea obtained for all the prepared membranes in this work (14.99–19.19 kJ mol−1) are close to the above data. This would suggest that the proton transportation in all the membranes may occur through Grotthuss mechanism. The Ea data of 15.47 kJ mol−1 is observed for pristine SPSU membrane. After the introduction of GO, the Ea values of SPSU/GO composite membranes decrease (ranging from 14.99 to 15.22 kJ mol−1), indicating the creation of proton conduction channels with lower energy barrier along the surface of GO. However, with the increasing inorganic content to 2–3 wt%, the membrane IEC decreases and blocking effect also increases the resistance for proton transportation simultaneously, so SPSU/GO-2 and SPSU/GO-3 membranes show higher Ea values than the pristine SPSU membrane. For SPSU/FPGO cross-linked membranes, their Ea values (17.83–19.19 kJ mol−1) are little higher than that of pristine SPSU and SPSU/GO composite membranes with the same inorganic content, but falls more into the Ea scale of Grotthuss mechanism. The increased Ea mainly owes to the covalent cross-linking which makes the polymeric network compact, letting more protons migrate by Grotthuss hopping mechanism. Similar phenomenon was also reported in literature.18 Besides, with the FPGO content increases to 2–3 wt%, blocking effect increases and the polymer network is compacted more, leading to harder proton conduction in SPSU/FPGO cross-linked membranes with higher Ea values than SPSU/FPGO-1 membrane.
3.8. Methanol permeability and selectivity
In addition to their high proton conductivity, PEMs also act as essential methanol barriers in DMFCs to prevent the diffusion of methanol molecules from anode to cathode so as to improve the fuel cell efficiency. Herein, methanol permeability and selectivity values of Nafion®117, pristine SPSU, SPSU/GO composite membranes and SPSU/FPGO cross-linked membranes at 30 °C were measured and presented in Table 2. Compared with reported membranes with similar IEC, methanol permeability values of our prepared SPSU based nanocomposite membranes at 30 °C (1.96 × 10−6 to 1.46 × 10−6 cm2 s−1) are lower than sulfonated polyimide10 and close to sulfonated poly (arylene ether sulfone)65 and chitosan based nanocomposites.66 Moreover, both SPSU/GO and SPSU/FPGO membranes perform effectively reduced methanol permeability values compared with the measure values of Nafion®117 (2.53 × 10−6 cm2 s−1) and pristine SPSU membrane (2.34 × 10−6 cm2 s−1) at 30 °C. This could be attributed to the well-dispersed 2-D nanosheets of GO, which reduced the chain mobility and free volume of polymeric matrices and also formed twisted methanol diffusion paths in the nanocomposite membranes. With the increasing inorganic content from 1 wt% to 3 wt%, diffusion resistance of methanol molecules further increases, so the methanol transportation across the membranes is further restricted. Noteworthy, benefited from the covalently cross-linked structure which makes the polymeric network much more compact, SPSU/FPGO cross-linked membranes show even lower methanol permeability than SPSU/GO composite membranes with the same filler content. Benefiting from the highly increased diffusion resistance of methanol, selectivity values of hybrid membranes show a prominent increase compared with that of pristine SPSU membrane. Among them, SPSU/FPGO-1 cross-linked membrane posses the highest selectivity (8.28 × 104 S s cm−3 at 30 °C), which is 1.67-folds of the value higher than Nafion®117, further illustrating its excellent comprehensive transport properties for DMFCs applications. Herein, the trade-off transportation properties between proton conductivity and methanol permeability in the nanocomposite PEMs has been effectively modulated through constructing novel SPSU/FPGO cross-linked structure.| Membranes | Proton conductivity (S cm−1, 30 °C) | Methanol permeability (10−6 cm2 s−1, 30 °C) | Selectivity (104 S s cm−3, 30 °C) |
|---|---|---|---|
| Nafion®117 | 0.1250 | 2.53 | 4.94 |
| SPSU | 0.1301 | 2.34 | 5.56 |
| SPSU/GO-1 | 0.1521 | 1.96 | 7.76 |
| SPSU/GO-2 | 0.1279 | 1.65 | 7.75 |
| SPSU/GO-3 | 0.1134 | 1.52 | 7.46 |
| SPSU/FPGO-1 | 0.1415 | 1.71 | 8.28 |
| SPSU/FPGO-2 | 0.1173 | 1.62 | 7.24 |
| SPSU/FPGO-3 | 0.1031 | 1.46 | 7.06 |
4. Conclusions
To summarize, FPGO nanosheets with cross-linker bonded onto their polysiloxane brushes were synthesized via surface precipitation polymerization and hydrosilylation. By cross-linking FPGO into SPSU matrices and then fabricating the membrane through the facile solution casting method, novel GO polymer brushes based cross-linked nanocomposite PEMs (FPGO/SPSU) was successfully prepared. Compared with Nafion®117, pristine SPSU membrane, SPSU/GO composite membranes and other widely reported uncross-linked nanocomposite membranes, SPSU/FPGO cross-linked membranes showed comprehensive advantages for both membrane preparation process and DMFCs applications: (i) optimizing the dispersion property of GO based nano-filler in polar aprotic solvent (DMAc) to effectively reduce their aggregation in nanocomposite PEMs; (ii) improving thermal, oxidative and dimensional stability and mechanical property of nanocomposite membranes; (iii) enhancing proton conductivity at relatively high operating temperature range (70–90 °C); (iv) reducing the methanol permeability of PEMs. Particularly, SPSU/FPGO-1 cross-linked membrane acquires the best over-all properties including a low methanol permeability of 1.71 × 106 S s cm−3 at 30 °C and the highest proton conductivity of 0.462 S cm−1 at 90 °C under hydrated condition, which is 33% and 14.5% higher than the proton conductivity of pristine SPSU and SPSU/GO-1 composite membrane, respectively. Based on these results, we believe the SPSU/FPGO cross-linked membranes merit further research and might be considered as potential candidates for DMFCs application. This study might also provide an alternative method to build more proton transport channels on the surface of inorganic fillers via the cross-linking between polymer brushes and the sulfonated polymer matrix.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for the financial support of the National Natural Science Fund of China (31000446) and the Science and Technology Program of Guangzhou (201604020175), China.References
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- T. Ous and C. Arcoumanis, J. Power Sources, 2013, 240, 558–582 CrossRef CAS.
- M. Noroozifar, Z. Yavari, M. Khorasani-Motlagh, T. Ghasemi, S. Rohani-Yazdia and M. Mohammadi, RSC Adv., 2016, 6, 563–574 RSC.
- Y. Yang, H. Gao and L. Zheng, RSC Adv., 2015, 5, 17683–17689 RSC.
- W. Jia, B. Tang and P. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 22620–22627 CAS.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS PubMed.
- A. Yu, I. Roes, A. Davies and Z. Chen, Appl. Phys. Lett., 2010, 96, 253105 CrossRef.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- C. Y. Tseng, Y. S. Ye, M. Y. Cheng, K. Y. Kao, W. C. Shen, J. Rick, J. C. Chen and B. J. Hwang, Adv. Energy Mater., 2011, 1, 1220–1224 CrossRef CAS.
- K. Feng, B. Tang and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 1481–1488 CAS.
- M. Vinothkannan, A. R. Kim, G. G. kumar and D. J. Yoo, RSC Adv., 2018, 8, 7494–7508 RSC.
- H. Beydaghi, M. Javanbakht and E. Kowsar, Polymer, 2016, 87, 26–37 CrossRef CAS.
- G. He, C. Chang, M. Xu, S. Hu, L. Li, J. Zhao, Z. Li, Z. Li, Y. Yin, M. Gang, H. Wu, X. Yang, M. D. Guiver and Z. Jiang, Adv. Funct. Mater., 2015, 25, 7502–7511 CrossRef.
- J. Zhang, S. Lu, H. Zhu, K. Chen, Y. Xiang, J. Liu, M. Forsyth and S. P. Jiang, RSC Adv., 2016, 6, 86575–86585 RSC.
- Y. Heo, H. Im and J. Kim, J. Membr. Sci., 2013, 425, 11–22 CrossRef.
- K. R. Reddy, H. M. Jeong, Y. Lee and A. V. Raghu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1477–1484 CrossRef CAS.
- J. Wang, H. Bai, J. Zhang, L. Zhao, P. Chen, Y. Li and J. Liu, J. Membr. Sci., 2017, 531, 47–58 CrossRef CAS.
- X. Qiu, M. Ueda, H. Hu, Y. Sui, X. Zhang and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 33049–33058 CAS.
- Y. He, J. Wang, H. Zhang, T. Zhang, B. Zhang, S. Cao and J. Liu, J. Mater. Chem. A, 2014, 2, 9548–9558 CAS.
- X. He, G. He, A. Zhao, F. Wang, X. Mao, Y. Yin, L. Cao, B. Zhang, H. Wu and Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 27676–27687 CAS.
- M. U. Khan, K. R. Reddy, T. Snguanwongchai, E. Haque and V. G. Gomes, Colloid Polym. Sci., 2016, 294, 1599–1610 CAS.
- Y. Li, G. He, S. Wang, S. Yu, F. Pan, H. Wu and Z. Jiang, J. Mater. Chem. A, 2013, 1, 10058–10077 CAS.
- L. Zhao, Y. Li, H. Zhang, W. Wu, J. Liu and J. Wang, J. Power Sources, 2015, 286, 445–457 CrossRef CAS.
- B. G. Choi, Y. S. Huh, Y. C. Park, D. H. Jung, W. H. Hong and H. Park, Carbon, 2012, 50, 5395–5402 CrossRef CAS.
- H. Pan, Y. Zhang, H. Pu and Z. Chang, J. Power Sources, 2014, 263, 195–202 CrossRef CAS.
- E. A. Mistri and S. Banerjee, RSC Adv., 2014, 4, 22398–22410 RSC.
- C. Gong, Y. Liang, Z. Qi, H. Li, Z. Wu, Z. Zhang, S. Zhang, X. Zhang and Y. Li, J. Membr. Sci., 2015, 476, 364–372 CrossRef CAS.
- N. Zhang, B. Wang, C. Zhao, Y. Zhang, F. Bu, Y. Cui, X. Li and H. Na, J. Power Sources, 2015, 275, 815–822 CrossRef CAS.
- Y. N. Chang, J. Y. Lai and Y. L. Liu, J. Membr. Sci., 2012, 403, 1–7 Search PubMed.
- K. Si, R. Wycisk, D. Dong, K. Cooper, M. Rodgers, P. Brooker, D. Slattery and M. Litt, Macromolecules, 2013, 46, 422–433 CrossRef CAS.
- P. Dai, Z. H. Mo, R. W. Xu, S. Zhang and Y. X. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20329–20341 CAS.
- K. J. Peng, J. Y. Lai and Y. L. Liu, J. Membr. Sci., 2016, 514, 86–94 CrossRef CAS.
- J. Ren, S. L. Zhang, Y. Liu, Y. Wang, J. H. Pang, Q. H. Wang and G. B. Wang, J. Membr. Sci., 2013, 434, 161–170 CrossRef CAS.
- C. H. Lee, H. B. Park, Y. S. Chung, Y. M. Lee and B. D. Freeman, Macromolecules, 2006, 39, 755–764 CrossRef CAS.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- S. Park, D. A. Dikin, S. T. Nguyen and R. S. Ruoff, J. Phys. Chem. C, 2009, 113, 15801–15804 CAS.
- L. Liu, C. Tong, Y. He, Y. Zhao and C. Lü, J. Membr. Sci., 2015, 487, 99–108 CrossRef CAS.
- C. Y. Lee, J. H. Bae, T. Y. Kim, S. H. Chang and S. Y. Kim, Composites, Part A, 2015, 75, 11–17 CrossRef CAS.
- S. F. Hou, S. J. Su, M. L. Kasner, P. Shah, K. Patel and C. J. Madarang, Chem. Phys. Lett., 2010, 501, 68–74 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- J. Wang, M. Ren, S. Wang and Y. Qu, Spectrochim. Acta, Part A, 2011, 78, 1126–1132 CrossRef PubMed.
- J. D. Jovanovic, M. N. Govedarica, P. R. Dvornic and I. G. Popovic, Polym. Degrad. Stab., 1998, 61, 87–93 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- Y. Devrima, S. Erkana, N. Baçb and I. Eroğlua, Int. J. Hydrogen Energy, 2009, 34, 3467–3475 CrossRef.
- M. D. T. Nguyen, H. S. Dang and D. Kim, J. Membr. Sci., 2015, 496, 13–20 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- X. He, J. Liu, H. Zhu, Y. Zheng and D. Chen, RSC Adv., 2015, 5, 63215–63225 RSC.
- A. K. Mishra, S. Bose, T. Kuila, N. H. Kim and J. H. Lee, Prog. Polym. Sci., 2010, 37, 842–869 CrossRef.
- D. Marani, A. D'Epifanio, E. Traversa, M. Miyayama and S. Licoccia, Chem. Mater., 2010, 22, 1126–1133 CrossRef CAS.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Int. J. Hydrogen Energy, 2010, 35, 9349–9384 CrossRef CAS.
- Y. Kim, Y. Choi, H. K. Kim and J. S. Lee, J. Power Sources, 2010, 195, 4653–4659 CrossRef CAS.
- H. Zhang, C. Ma, J. Wang, X. Wang, H. Bai and J. Liu, Int. J. Hydrogen Energy, 2014, 39, 974–986 CrossRef CAS.
- C. Zhao, H. Lin, K. Shao, X. Li, H. Ni, Z. Wang and H. Na, J. Power Sources, 2006, 162, 1003–1009 CrossRef CAS.
- C. Zhao, H. Lin and H. Na, Int. J. Hydrogen Energy, 2010, 35, 2176–2182 CrossRef CAS.
- K. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- Y. Wei, X. Li, Q. Hu, C. Ni, B. Liu, M. Zhang, H. Zhang and W. Hu, RSC Adv., 2016, 6, 65072–65080 RSC.
- M. A. Hickner, C. H. Fujimoto and C. J. Cornelius, Polymer, 2006, 47, 4238–4244 CrossRef CAS.
- W. Gao, G. Wu, M. T. Janicke, D. A. Cullen, R. Mukundan, J. K. Baldwin, E. L. Brosha, C. Galande, P. M. Ajayan, K. L. More, A. M. Dattelbaum and P. Zelenay, Angew. Chem., Int. Ed., 2014, 53, 3588–3593 CrossRef CAS PubMed.
- J. L. Yan and M. A. Hickner, Macromolecules, 2010, 43, 2349–2356 CrossRef CAS.
- H. J. Lee, J. H. Kim, J. H. Won, J. M. Lim, Y. T. Hong and S. Y. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 5034–5043 CAS.
- Y. L. Liu, Y. H. Su, C. M. Chang, D. M. Wang and J. Y. Lai, J. Mater. Chem., 2010, 20, 4409–4416 RSC.
- C. M. Chang, Y. L. Liu and Y. M. Lee, J. Mater. Chem., 2011, 21, 7480–7486 RSC.
- B. Smitha, S. Sridhar and A. A. Khan, Macromolecules, 2004, 37, 2233–2239 CrossRef CAS.
- Y. S. Kim, M. A. Hickner, L. Dong, B. S. Pivovara and J. E. McGrath, J. Membr. Sci., 2004, 243, 317–326 CrossRef CAS.
- H. Bai, H. Zhang, Y. He, J. Liu, B. Zhang and J. Wang, J. Membr. Sci., 2014, 454, 220–232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01731j |
| This journal is © The Royal Society of Chemistry 2018 |