DOI:
10.1039/C7RA12659J
(Paper)
RSC Adv., 2018,
8, 15725-15739
Identification of absorbed constituents and evaluation of the pharmacokinetics of main compounds after oral administration of yindanxinnaotong by UPLC-Q-TOF-MS and UPLC-QqQ-MS†
Received
21st November 2017
, Accepted 14th March 2018
First published on 26th April 2018
Abstract
Yindanxinnaotong capsule (YDXNT), a traditional Chinese formula, has been used to treat cardio-cerebrovascular diseases for several decades. Previous research has focused on evaluating the pharmacological properties and main compounds of YDXNT in vitro and in vivo. However, the multiple bioactive compounds in vivo remain poorly understood. In the present research, an integrative strategy using UPLC-Q-TOF-MS combined with UPLC-QqQ-MS was employed to detect the absorbed constituents and investigate the pharmacokinetics of main compounds in the plasma after oral administration of YDXNT. UPLC-Q-TOF-MS was developed to detect the absorbed constituents and their metabolites in the plasma after oral administration in rats. A total of 52 constituents, including 44 prototype compounds and 8 metabolites, were identified or tentatively characterized. Then, nine main compounds (quercetin, isorhamnetin, kaempferol, ginkgolide A, ginkgolide B, ginkgolide C, bilobalide, tanshinone IIA, and salvianolic acid B) were chosen to further investigate the pharmacokinetic behavior of YDXNT using UPLC-QqQ-MS. The concentration of nine main constituents were in the range of 27.85–76.54 ng mL−1. This research provides a systematic approach for rapid qualitative analysis of absorbed constituents and for evaluating the pharmacokinetics of the main ingredients of YDXNT following its oral administration. More importantly, this work provides key information on the identification of bioactive compounds and the clarification of their action mechanisms, as well as on the pharmacological actions of YDXNT.
1. Introduction
Traditional Chinese medicine (TCM), one of the oldest phytomedicine systems in health care, has been used in Asia, such as in China, Korea, and Japan, for thousands of years.1–4 The herbs used in TCM usually have complex formulas and function mainly via their multiple constituents, targets and modes of action (3M) in a network-based and holistic manner.5 Thus, the therapeutic effects of TCM formulae rely on the joint effect of multiple ingredients. Most TCM formulae are taken orally, and only ingredients absorbed into the blood can exert their bioactivities.6 Therefore, it is necessary to trace the compounds in a TCM prescription in vivo and evaluate their pharmacokinetics to provide more in-depth insight into the main active components and therapeutic mechanisms of TCM formulae. In recent years, an integrative strategy using ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF-MS) combined with ultra-high-performance liquid chromatography with triple quadrupole mass spectrometry (UPLC-QqQ-MS) has been widely used to identify the absorbed constituents and determine main compounds in complex matrices.7–11 Hence, they are valuable analytical techniques for identifying key compounds and evaluating the pharmacokinetics of TCM formulae in vivo.
Yindanxinnaotong capsule (YDXNT), a classic TCM formula, comprises eight herbs: Ginkgo biloba leaf (yinxingye, YXY), Salvia miltiorrhizae (danshen, DS1), Gynostemma gynostemmatis (jiaogulan, JGL), Erigerontis herba (dengzhanxixin, DZXX), Allii sativi bulbus (dasuan, DS2), Radix/rhizoma notoginseng (sanqi, SQ), Crataegi fructus (Shanzha, SZ), and Borneolum (tianranbingpian, TRBP). In China, YDXNT capsule is officially listed in the Chinese Pharmacopoeia 2015 as a prescription for patients suffering from angina pectoris in CHD, hyperlipidemia, stroke, and atherosclerosis.12 YDXNT, which has been used to treat cardio-cerebrovascular diseases for many years,13–15 is one of the stasis-eliminating formulated products listed in China's National Basic Medical Insurance Drugs Catalogue (http://www.nhfpc.gov.cn/, 2013 edition).
Our previous research focused on the chemical compounds, quality control, and in vitro and vivo pharmacological properties of YDXNT. In studying the chemical compounds of YDXNT, 70 chemical constituents, including 23 flavonoids, 4 ginkgolides, 15 phenolic compounds, 18 diterpenoid tanshinones, and 10 ginsenosides, were tentatively identified using various detection approaches. Furthermore, fingerprint analysis was established to evaluate the uniform quality of YDXNT soft capsules.16 In in vivo experiments, taking YDXNT and swimming may prevent atherosclerosis through a synergistic effect between YDXNT and swimming leading to improved blood circulation, hemorheological parameters, blood lipid levels and vascular endothelium in rats.17 In an isolated heart experiment, YDXNT and its main compounds elevated heart function, coronary flow and SOD levels and decreased levels of MDA and inflammatory factors.18 Ultimately, our research demonstrated that YDXNT and its major constituents relieve atherosclerosis by regulating lipids, reducing lipid particle deposition in the endothelial layer of arteries, enhancing antioxidant power, and repressing inflammatory activity by inhibiting the nuclear factor-kappa B signaling pathway both in vivo and vitro.19 Previous work focus on the pharmacokinetics of main constituents in YXY, DS1, SQ, SZ, and DZXX.20–24 However, until now, little has been known about how many constituents are absorbed into the blood after oral administration of YDXNT or about the final fate of the formula in vivo. This presents a huge obstacle to understanding the pharmacological mechanisms of YDXNT. Thus, the present research was designed to determine the absorbed compounds and metabolites in biological samples from rats and investigate the pharmacokinetics of the main abundant active components.
In this research, UPLC-Q-TOF-MS was used to analyze and identify the absorbed ingredients and possible metabolites in rat plasma after oral administration of YDXNT due to its high chromatographic resolution, high sensitivity, short analysis times, good separation, and highly accurate mass values.25–27 In addition, UPLC-QqQ-MS was used for simultaneous quantitation of the main components quercetin (QCT), kaempferol (KMF), isorhamnetin (ISR), ginkgolide A (GA), ginkgolide B (GB), ginkgolide C (GC), bilobalide (BB), tanshinone IIA (TSIIA), and salvianolic acid B (SAB) in rats, with baicalein used as the internal standard (IS).28,29 These compounds exert anti-atherosclerotic and endothelium-independent vasodilator effects, prevent inflammatory damage and have a protective effect on nitric oxide, antioxidant action, and anti-vascular inflammation.26–29 The pharmacokinetics of QCT, KMF, ISR, GA, GB, GC, BB, TSIIA, and SAB were investigated in rats after oral administration of YDXNT, and the pharmacokinetics properties of this formula were further speculated. This research represents the first detailed investigation into the absorbed constituents, metabolites, and pharmacokinetics of YDXNT. It may provide valuable information for better understanding the pharmacological mechanisms and clinical applications of YDXNT.
2. Experiments
2.1 Chemicals and reagents
YDXNT soft capsules were provided by Guizhou Bailing Group Pharmaceutical Co., Ltd. Standards of rutin and cryptotanshinone were purchased from Chengdu Chroma-Biotechnology Co., Ltd. (Sichuan, China). Kaempferol, quercetin, tanshinone IIA, salvianolic acid B, ginkgolide A, ginkgolide B, bilobalide, and baicalein were purchased from Chengdu Herb Purify Co., Ltd. (Sichuan, China). Isorhamnetin, ginkgolide C, ginsenoside Rg1, ginsenoside Re, ginsenoside Rd, and notoginsenoside R1 were purchased from the National Institutes for Food and Drug Control (Beijing, China), Ltd., and HPLC-grade acetonitrile and methanol were obtained from Thermo Fisher Scientific Inc. (Shanghai, China). Formic acid was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and the water used in the experiments was purified with a Milli-Q system (Sartorius Arium® Pro, Germany).
2.2 Preparation of YDXNT sample
The samples were first processed by completely removing the outer capsule layer. Then, a 48.0 g sample was precisely weighed and soaked with 100 mL de-ionized water. The sample was ultrasonically dissolved for 30 min and then stored at −4 °C until it was orally administered to rats.
2.3 Animal experiments
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Center for Laboratory Animal Care, China Academy of Chinese Medical Science and approved by the Animal Ethics Committee of Institute of Chinese Material Medica, China Academy of Chinese Medical Science.
Twenty-four male Sprague-Dawley rats (250 ± 30 g) were obtained from the Laboratory Animal Center of the Academy of Military Medical Sciences (Beijing, China) and divided into four groups (six rats each). The rats were housed and fed at a temperature of 25 ± 2 °C and a relative humidity of 50% for three days in the breeding room. Standard chow and deionized water were provided prior to the oral treatment. All rats were fasted for 12 h with free access to water before the experiment. YDXNT was administered orally to rats at a dose of 4.8 g kg−1 d−1, and blood was collected from the abdominal aorta after 20% urethane anesthesia and then centrifuged at 3000 rpm for 10 min at 4 °C to obtain the plasma. All samples were stored at −20 °C until analysis. Blank plasma samples were prepared in the same manner.
2.4 Serum sample preparation
Six milliliters of 75% methanol was added to two milliliters of prepared plasma and then vortexed for 60 s. The mixture was centrifuged at 4000 rpm for 10 min at 4 °C. The supernatant was transferred to another EP tube and dried using a pressure blowing concentrator. The residue was redissolved with 200 μL of 75% methanol, vortexed for 60 s, and then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. To an aliquot of 200 μL plasma, 120 μL of 0.2 mol L−1 acetic acid–sodium acetate buffer solution (pH = 4.6), 100 μL of the IS baicalein and 80 μL of 980 U mL−1 β-glucuronidase and sulfatase was added and then vortexed for 1 min. The mixture was hydrolyzed in a water bath at 37 °C for 30 min, and 600 μL acetone was added to stop the hydrolysis. The sample was cooled, followed by vortexing for 2 min and then centrifugation at 13000 rpm for 10 min. The supernatant fluid was evaporated to dryness under a flow of nitrogen gas.
000 rpm for 10 min. To an aliquot of 200 μL plasma, 120 μL of 0.2 mol L−1 acetic acid–sodium acetate buffer solution (pH = 4.6), 100 μL of the IS baicalein and 80 μL of 980 U mL−1 β-glucuronidase and sulfatase was added and then vortexed for 1 min. The mixture was hydrolyzed in a water bath at 37 °C for 30 min, and 600 μL acetone was added to stop the hydrolysis. The sample was cooled, followed by vortexing for 2 min and then centrifugation at 13000 rpm for 10 min. The supernatant fluid was evaporated to dryness under a flow of nitrogen gas.
2.5 Preparation of standard, calibration, and quality control solutions
Stock solutions of QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, SAB and IS were separately prepared by dissolving the compounds in methanol at a concentration of 200 ng mL−1; all solutions were stored at 4 °C. Working solutions for calibration and QC samples were prepared by adding the diluted stock solutions to blank rat plasma. Calibration standards of plasma-derived working solutions with QCT, ISR, KMF, GA, GB, GC, BB, and TSIIA were all prepared with final concentrations ranging from 1–100 ng mL−1. The SAB solution was prepared with final concentrations ranging from 5–500 ng mL−1. The IS solution was prepared at a final concentration of 100 ng mL−1 in methanol. For method validation, QC plasma samples of QCT, ISR, KMF, GA, GB, GC, BB, and TSIIA were all prepared separately at three concentrations (1, 10 and 100 ng mL−1). The QC plasma sample of SAB was prepared at three concentrations (5, 50, and 500 ng mL−1).
2.6 UPLC-Q-TOF-MS for qualitative analysis
UPLC data were produced using the Waters Synapt G2-S UPLC system (Waters, Milford, USA) equipped with a quaternary pump system, online degasser, autosampler, thermostatically controlled column compartment, and diode array detector. The system was controlled with Masslynx V4.1 software. The chromatographic column ACQUITY BEH C18 (100 × 2.1 mm, 1.7 μm) equipped with VanGuard™ BEHC18 1.7 μm was used. The column was eluted with a gradient of A (0.1% formic acid in deionized water) and B (CAN) at a flow rate of 0.3 mL min−1 and a temperature of 30 °C: 0–2.5 min, 10–16% B; 2.5–6.5 min, 16–16% B; 6.5–9.5 min, 16–25% B; 9.5–11.0 min, 25–25% B; 11.0–12.0 min, 25–45% B; 12.0–15.0 min, 45–55% B; 15.0–18.0, 55–98% B; 18.0–21.0 min, 98–98% B; 21.0–25.0 min, 98–10% B. An injection volume of 10 μL was used for analysis.
For mass detection, mass data were recorded using the Waters Synapt G2-S Q-TOF Mass Spectrometer system equipped with an atmospheric pressure chemical ionization (APCI) electrospray ionization (ESI) interface (Waters, Milford, USA). The conditions of the TOF/MS were set as follows: capillary, 2.5 kV; source temperature, 120 °C; cone gas flow, 50 L h−1; nebulizer, 6 bar; desolvation temperature, 450 °C; sheath gas flow, 12 L min−1; nozzle voltage, 10–1000 V; The mass spectrometer was operated from 100 to 1700 m/z. Accurate masses and compositions of the precursor and fragment ions were calculated using MassLynx 4.1 software. The UPLC and Q-TOF-MS methods have been optimised (ESI 1†).
2.7 UPLC-QqQ-MS for quantitative analysis
Chromatographic analysis was performed using an Agilent 1290 Series UPLC from Agilent Technologies (Palo Alto, CA, USA), equipped with a quaternary pump an online degasser, auto-sampler and column oven. Chromatographic separation was performed on the Agilent ZORBAX SB-Aq C18 column (100 mm × 2.1 mm, 1.8 μm, Agilent, USA). The analytical column temperature was maintained at 30 °C and eluted with a mobile phase comprising 0.1% formic acid in water (A) and acetonitrile (B) using the following gradient program: 25–90% B from 0 to 1 min, 90% B from 1 to 3.5 min, and 90–25% B from 3.5 to 5 min at a flow rate of 0.3 mL min−1. The equilibration time was 5 min, and the injection volume of sample and reference constituents was 5 μL.
Mass spectrometry was performed on the Agilent 6490 triple-quadrupole mass spectrometer (Palo Alto, CA, USA) equipped with an atmospheric pressure chemical ionization (APCI) interface. The mass spectrometer was set in negative ionization mode, with the capillary voltage set at 3500 V. The other parameters in the source were set as follows: desolvation gas flow, 15 L min−1; source temperature, 290 °C; N2 sheath gas temperature, 350 °C; nebulizer gas (N2) pressure, 50 psi; flow, 12 L min−1. The mass spectrometer scanned in multiple reaction monitoring (MRM) mode. The UPLC and QqQ-MS methods have been optimised (ESI 2†). Fig. 1 shows the MS/MS product ion spectra of the analytes and IS. The optimized extraction ion pairs and parameter values are shown in Table 1.
 |
| | Fig. 1 Product iron mass spectra of the analytes and internal standard (IS). QCT: quercetin; ISR: isorhamnetin; KMF: kaempferol; GA: ginkgolide A; GB: ginkgolide B; GC: ginkgolide C; BB: bilobalide; TSIIA: tanshinone IIA; SAB: salvianolic acid B; IS: baicalein. | |
Table 1 MRM transitions and parameters for the detection of QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, SAB and IS
| Analyte |
MRM transitions |
Fragementor (V) |
Collision energy (V) |
Dwell time (ms) |
| QCT |
301.1 → 151.0 |
350 |
26 |
20 |
| ISR |
315.0 → 300.1 |
350 |
22 |
20 |
| KMF |
285.0 → 142.9 |
350 |
35 |
20 |
| GA |
453.1 → 351.1 |
350 |
16 |
20 |
| GB |
423.1 → 367.1 |
350 |
15 |
20 |
| GC |
439.1 → 383.1 |
350 |
12 |
20 |
| BB |
325.1 → 163.1 |
350 |
16 |
20 |
| TSIIA |
295.0 → 249.0 |
350 |
26 |
20 |
| SAB |
717.1 → 519.0 |
350 |
38 |
20 |
| IS |
269.0 → 195.0 |
350 |
28 |
20 |
2.8 Method validation
Method validation was performed according to the guidelines for bioanalytical method validation published by the US FDA.30 The analytical method used in this research was validated to demonstrate its specificity, selectivity, linearity, lower limit of quantification (LLOQ), precision, accuracy, stability, extraction recovery rates and matrix effects of the samples (ESI 3†).
2.9 Pharmacokinetics study
Six male rats were used in the pharmacokinetics study. All the rats were orally administered 2.4 g kg−1 YXCNT suspended in an aqueous solution of 0.5% carboxymethyl cellulose sodium (QCT: 4.118 mg; KMF: 5.067 mg; ISR: 0.965 mg; GA: 1.416 mg; GB: 0.680 mg; GC: 0.796 mg; BB: 0.326 mg; TSIIA: 0.624 mg; SAB: 3.84 mg). After dosing for 0 min, 5 min, 15 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, and 24 h, blood samples (500 μL) were drawn from the retro-orbital plexus of each mouse and collected in heparinized tubes according to the specific schedule. All the collected blood samples were immediately centrifuged at 13000 rpm for 10 min. The plasma layer was transferred to clean tubes and stored at −20 °C until analysis. The plasma concentrations of QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, and SAB were expressed as the mean ± standard deviation (SD), and the mean concentration–time curve was plotted. Non-compartmental pharmacokinetic parameters were computed using DAS 2.1.1 software.
3. Results and discussion
3.1 Identification of prototype constituents and metabolites in drug-containing plasma
The optimized LC-MS protocol was used for the separation and identification of compounds in drug-containing plasma. Exogenous constituents of YDXNT in the plasma were divided into prototype components and metabolites. Ultimately, a total of 52 compounds, including 44 prototype constituents and 8 metabolites, were detected by comparison to the blank plasma sample (Fig. 2, Tables 2 and 3).
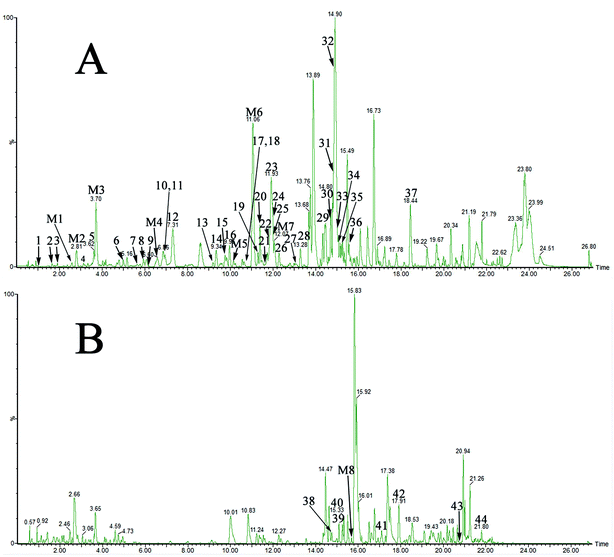 |
| | Fig. 2 The UPLC-Q-TOF-MS results for drug-containing plasma. (A) negative ionisation mode-analysis of drug-containing plasma. (B) positive ionisation-mode analysis of drug-containing plasma. | |
Table 2 (1) Chromatographic and MS data of absorbed constituents analysed by UPLC-Q-TOF-MS in negative mode. (2) Chromatographic and MS data of absorbed constituents analysed by UPLC-Q-TOF-MS in positive mode
| Peak no. |
RT (min) |
[M − H]− |
Tolerance ppm |
MS2 |
Molecular formula |
Tentatively identity |
Source |
| 1 |
1.01 |
217.0338 |
|
|
|
Unknown |
16 |
| 2 |
2.03 |
197.0449 |
2.284 |
135.0470 [M − H–C2H2O]− |
C9H10O5 |
Tanshinol |
16 |
| 3 |
2.37 |
153.0188 |
3.691 |
109.0326 [M − H–CO2]− |
C7H6O4 |
Protocatechuic acid |
16 |
| 4 |
3.18 |
137.0233 |
−0.150 |
108.0453 |
C7H6O3 |
Protocatechualdehyde |
16 |
| 5 |
3.62 |
179.0349 |
5.668 |
|
C9H8O4 |
Caffeic acid |
16 |
| 6 |
4.99 |
755.2035 |
6.4 |
301.0343 [M − H–2C6H10O4–C6H10O5]− |
C33H40O20 |
Quercetin 3-O-[2-O,6-O-bis(α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 7 |
5.82 |
193.0501 |
0.0 |
178.9783, 134.9963 |
C10H10O4 |
Ferulic acid |
16 |
| 8 |
6.00 |
739.2123 |
5.810 |
593.1466 [M − H–C6H10O4]− |
C33H40O19 |
Kaempferol 3-O-[2-O,6-O-bis(α-L-rhamnosyl)-β-D-glucoside] (c) |
16 |
| 447.0882 [M − H–2C6H10O4]− |
| 285.0379 [M − H–2C6H10O4–C6H10O5]− |
| 9 |
6.19 |
769.2180 |
0.741 |
315.0522 [M − H–2C6H10O4–C6H10O5]− |
C34H42O20 |
Isorhamnetin 3-O-[2-O,6-O-bis (α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 10 |
6.73 |
609.1464 |
2.280 |
463.0923 [M − H–C6H10O4]− |
C27H30O16 |
Quercetin 3-O-[6-O-(α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 301.0345 [M − H–C6H10O4–C6H10O5]− |
| 11 |
6.83 |
325.0914 |
−0.9 |
163.1106 |
C15H18O8 |
Bilobalide |
16 |
| 12 |
7.31 |
439.1245 |
2.305 |
411.1272 [M − H–CO]− |
C20H24O11 |
Ginkgolide C |
16 |
| 383.1348 [M − H–2CO]− |
| 13 |
9.20 |
609.1459 |
2.280 |
477.0924 [M − H–C6H10O5]− |
C27H30O16 |
Quercetin 3-O-[6-O-(α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 301.0343 [M − H–C6H10O4–C6H10O5]− |
| 14 |
9.34 |
593.1501 |
0.006 |
285.0379 [M − H–C6H10O4–C6H10O5]− |
C27H30O15 |
Kaempferol 3-O-[6-O-(α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 15 |
9.76 |
623.1625 |
2.951 |
315.0522 [M − H–C6H10O4–C6H10O5]− |
C28H32O16 |
Isorhamnetin 3-O-[6-O-(α-L-rhamnosyl)-β-D-glucoside] |
16 |
| 16 |
9.96 |
577.1567 |
2.630 |
269.0482 [M − H–C6H10O4–C6H10O5]− |
C27H30O14 |
Apigenin 7-O-rutinoside |
16 |
| 17 |
10.52 |
359.0775 |
3.777 |
161.0258 [M − H–C9H10O5]− |
C18H16O8 |
Rosmarinic acid |
16 |
| 18 |
10.57 |
593.1501 |
0.006 |
285.0382 [M − H–C6H10O5–C6H10O4]− |
C27H30O15 |
Kaempferol 3-O-[2-O-(β-D-glucosyl)-α-L-rhamnoside] (a) |
16 |
| 19 |
11.30 |
755.1843 |
3.323 |
609.1464 [M − H–C6H10O4]− |
C36H36O18 |
Quercetin 3-O-[2-O-(6-O-β-hydroxy-trans-cinnamoyl)-β-D-glucosyl)-α-L-rhamnoside] |
16 |
| 301.0343 [M − H–2C6H10O4–C6H10O5]− |
| 20 |
11.39 |
977.5346 [M + HCOO−]− |
3.099 |
931.5281 [M − H]− |
C47H81O18 |
Panaxnotoginosides R1 |
16 |
| 799.4871 [M − H–C5H8O4]− |
C48H82O20 |
| 637.4261 [M − H–C5H8O4–C6H10O5]− |
|
| 475.3802 [M − H–C5H8O4–2C6H10O5]− |
|
| 21 |
11.67 |
717.1407 |
−0.6011 |
519.0934 [M − H–C9H10O5]− |
C36H30O16 |
Salvianolic acid B |
16 |
| 321.0412 [M − H–2C9H10O5]− |
| 22 |
11.91 |
407.1346 |
2.312 |
379.1448 [M − H–CO]− |
C20H24O9 |
Ginkgolide A |
16 |
| 351.1444 [M − H–2CO]− |
| 23 |
11.93 |
423.1321 |
8.335 |
395.1347 [M − H–CO]− |
C20H24O10 |
Ginkgolide B |
16 |
| 367.1410 [M − H–2CO]− |
| 24 |
11.98 |
845.4844 [M + HCOO−]− |
−5.810 |
799.4748 [M − H]− |
C42H73O14 |
Ginsenoside Rg1 |
16 |
| 637.4261 [M − H–C6H10O5]− |
C43H74O16 |
| 475.3802 [M − H–2C6H10O5]− |
|
| 25 |
12.01 |
991.5478 [M + HCOO−]− |
0.584 |
945.5412 [M − H]− |
C48H83O18 |
Ginsenoside Re |
16 |
| 783.4870 [M − H–C6H10O5]− |
C49H84O20 |
| 621.4357 [M − H–2C6H10O5]− |
|
| 459.3839 [M − H–3C6H10O5]− |
|
| 26 |
12.29 |
739.1883 |
2.332 |
593.1501 [M − H–C6H10O4]− |
C36H36O17 |
Kaempferol 3-O-[2-O-(6-O-β-hydroxy-trans-cinnamoyl)-β-D-glucosyl)-α-L-rhamnoside] (b) |
16 |
| 285.0379 [M − H–2C6H10O4–C6H10O5]− |
| 27 |
13.10 |
301.0343 |
0.07 |
273.0384 [M − H–CO]− |
C15H10O7 |
Quercetin |
16 |
| 151.0067 [M − H–C8H8O3]− |
| 28 |
13.28 |
637.1726 |
|
|
C29H34O16 |
Unknown |
16 |
| 29 |
14.47 |
269.0482 |
3.939 |
225.1175 [M − H–CO2]− |
C15H10O5 |
Apigenin |
16 |
| 30 |
14.66 |
285.0453 |
−5.138 |
211.0411 [M − H–2CO–H2O]− |
C15H10O6 |
Kaempferol |
16 |
| 31 |
14.83 |
1153.5948 [M + HCOO−]− |
−4.546 |
1107.5925 [M − H]− |
C54H93O23 |
Ginsenoside Rb1 |
16 |
| 945.5376 [M − H–C6H10O5]− |
C55H94O25 |
| 783.4909 [M − H–2C6H10O5]− |
|
| 621.4412 [M − H–3C6H10O5]− |
|
| 459.3687 [M − H–4C6H10O5]− |
|
| 32 |
14.88 |
315.0522 |
7.208 |
271.0235 [M − H–CO2]− |
C16H12O7 |
Isorhamnetin |
16 |
| 33 |
14.97 |
829.4974 [M + HCOO−]− |
3.619 |
783.4909 [M − H]− |
C41H71O13 |
Ginsenoside F2/20(s)-ginsenoside Rg3 |
16 |
| 637.4261 [M − H–C6H10O4]− |
C42H72O15 |
| 475.3802 [M − H–C6H10O4–C6H10O5]− |
|
| 34 |
15.05 |
683.4370 [M + HCOO−]− |
0.748 |
637.4310 [M − H]− |
C36H62O9 |
Ginsenoside F1/ginsenoside Rh1 |
16 |
| 475.3784 [M − H–C6H10O5]− |
C37H63O11 |
| 35 |
15.22 |
683.4365 [M + HCOO−]− |
0.016 |
637.4312 [M − H]− |
C36H62O9 |
Ginsenoside F1/ginsenoside Rh1 |
16 |
| 475.3791 [M − H–C6H10O5]− |
C37H63O11 |
| 36 |
15.54 |
991.5486 [M + HCOO−]− |
1.391 |
945.5411 [M − H]− |
C48H83O18 |
Ginsenoside Rd |
16 |
| 783.4902 [M − H–C6H10O5]− |
C49H84O20 |
| 621.4377 [M − H–2C6H10O5]− |
|
| 459.3886 [M − H–3C6H10O5]− |
|
| 37 |
18.44 |
313.1482 |
4.3 |
269.1556 [M − H–CO2]− |
C17H14O6 |
Salvianolic acid F |
16 |
| 213.1271 [M − H–CO2–2CO]− |
| Peak no. |
RT (min) |
[M + H]+ |
Tolerance ppm |
MS2 |
Molecular formula |
Tentatively identity |
|
| 38 |
14.72 |
311.1254 |
−9.3 |
293.0783 [M + H–H2O]+ |
C19H18O4 |
Tanshinone II B |
16 |
| 265.1241 [M + H–H2O–CO]+ |
| 39 |
15.10 |
311.1256 |
−7.3 |
293.1178 [M + H–H2O]+ |
C19H18O4 |
3α-Hydroxytanshinone II A |
16 |
| 275.1057 [M + H–2H2O]+ |
| 40 |
15.33 |
309.1118 |
−1.085 |
291.1050 [M + H–H2O]+ |
C19H16O4 |
Tanshinaldehyde |
16 |
| 263.1079 [M + H–H2O–CO]+ |
| 41 |
17.07 |
311.1254 |
−9.3 |
293.1173 [M + H–H2O]+ |
C19H18O4 |
Przewaquinone A |
16 |
| 275.1066 [M + H–2H2O]+ |
| 247.1115 [M + H–2H2O–CO]+ |
| 42 |
17.91 |
279.1017 |
1.179 |
261.0872 [M + H–H2O]+ |
C18H14O3 |
Dihydrotanshinone I |
16 |
| 233.0954 [M + H–H2O–CO]+ |
| 43 |
20.77 |
297.1495 |
3.295 |
279.1382 [M + H–H2O]+ |
C19H20O3 |
Cryptotanshinone |
16 |
| 251.1421 [M + H–H2O–CO]+ |
| 44 |
21.80 |
295.1323 |
−1.935 |
277.1293 [M + H–H2O]+ |
C19H18O3 |
Tanshinone IIA |
16 |
| 249.1198 [M + H–H2O–CO]+ |
Table 3 (1) Chromatographic and MS data of metabolites analysed by UPLC-Q-TOF-MS in negative mode. (2) Chromatographic and MS data of metabolites analysed by UPLC-Q-TOF-MS in positive mode
| Peak no. |
RT (min) |
[M − H]− |
Tolerance ppm |
MS2 |
Molecular formula |
Tentatively identity |
Source |
| M1 |
2.59 |
313.0555 |
−1.6 |
137.0233 [M − H–Glu]− |
C13H14O9 |
Protocatechuicaldehyde glucuronide |
33–35 |
| M2 |
2.81 |
441.1423 |
7.168 |
423.1321 [M − H–H2O]− |
C20H26O11 |
Hydrolysis ginkgolides B |
36,37,39,40 |
| 367.1410 [M–H2O–2CO]− |
| M3 |
3.70 |
425.1468 |
6.061 |
407.1346 [M − H–H2O]− |
C20H26O10 |
Hydrolysis ginkgolides A |
36,37,39,40 |
| 351.1444 [M − H–H2O–2CO]− |
| M4 |
6.60 |
457.1345 |
0.979 |
439.1267 [M − H–H2O]− |
C20H26O12 |
Hydrolysis ginkgolides C |
36,37,39,40 |
| 383.1348 [M − H–H2O–2CO]− |
| M5 |
10.16 |
445.0768 |
−0.7 |
269.0482 [M − H–Glu]− |
C21H18O11 |
Apigenin glucuronide |
41–45 |
| 225.1175 [M − H–CO2]− |
| M6 |
11.06 |
443.1534 |
−1.9 |
369.1520 [M − H–H2O–2CO]− |
C20H28O11 |
Bi-ionized ginkgolides A |
36,37,39,40 |
| 351.1444 [M − H–2H2O–2CO]− |
| M7 |
12.07 |
461.1082 |
2.3 |
285.0754 [M − H–Glu] |
C21H18O12 |
Kaempferol-O-glucuronide |
41–45 |
| 211.0458 [M − H–Glu–2CO–H2O] |
| Peak no. |
RT (min) |
[M + H]+ |
Tolerance ppm |
MS2 |
Molecular formula |
Tentatively identity |
Source |
| M8 |
15.76 |
293.0816 |
0.7 |
278.1023 [M + H–CH3] |
C18H12O4 |
1,2; 1,3; 2,3-dehydrotanshinone IIA |
35,46–48 |
| 275.1273 [M + H–H2O]− |
| 247.1256 [M + H–H2O–2CO]− |
3.2 Analysis of prototype constituents of YDXNT in rat plasma
Owning to the high sensitivity of UPLC-Q-TOF-MS and extracted ion chromatograms, 44 prototype constituents were tentatively identified by comparison with standards, mass data, fragment information, and our previous compound research.16 Of the 44 peaks, 36 were detected in negative-ionization mode, and 7 peaks were detected in positive-ionization mode. The peaks included 15 flavonoids, 3 ginkgolide, 8 phenolic, 8 ginsenosides, and 7 diterpenoid tanshinones. Among them, 14 peaks were further confirmed with standards by comparing retention time and MS data, including peaks 10, 11, 12, 20, 21, 22, 23, 24, 26, 27, 30, 32, 36, 43, and 44.
3.3 Analysis of the metabolites of YDXNT in rat plasma
The full-scan mass spectrum was achieved from the plasma sample after YDXNT was orally administered to rats and compared to the blank plasma sample to discover possible metabolites in rat plasma. Eight metabolites, including 1 monophenol, 4 ginkgolides, 2 flavonoids, and 1 diterpenoid tanshinone, were tentatively identified from the plasma by comparing their retention times, deprotonated molecules, and characteristic fragments to those reported in the literature.31–47 The precursors of these metabolites were deduced to be protocatechuic aldehyde, ginkgolide A, ginkgolide B, ginkgolide C, apigenin, kaempferol, and tanshinone IIA. Characterization of the structures of these metabolites is described below.
In our research, one monophenolic-related metabolite (M1) was detected. M1 (tR = 2.59 min) showed a deprotonated molecular ion at m/z 313.0555, and product ions of m/z 137.0233 were obtained. The neutral loss of 176 Da (C6H8O6) indicated that M1 was the glucuronide conjugate32,33 (Fig. 3). Based on retention time, accurate quasi-molecular ion, and MS/MS data, as well as the related literature,32–34 M1 was tentatively identified as the characteristic ion of protocatechuic aldehyde glucuronide.
 |
| | Fig. 3 The mass spectra of protocatechuic aldehyde glucuronide in negative mode and the proposed fragmentation pathway. | |
Ginkgolide-related metabolites were the main possible xenobiotic metabolites in plasma.35,36 In the present study, 3 ginkgolides and 4 ginkgolide-related metabolites were detected. The 3 ginkgolides (ginkgolide A, ginkgolide C and ginkgolide B) were identified by comparison to standards. The characteristic ions at m/z 351, m/z 383 and m/z 367 were observed in ginkgolide A, ginkgolide C, and ginkgolide B, respectively. The common fragmentation pattern of ginkgolide was the successive loss of 2CO.37 M2, M3 and M4 have [M − H]− ions at m/z 441.1423, m/z 425.1468, and m/z 457.1345, which were 18 Da heavier than ginkgolide B, ginkgolide A, and ginkgolide C, respectively. The main fragment ions of M2, M3, and M4 in the MS/MS spectra were 367.1410, 351.1444, and 383.1348 by the loss of 74 Da. Three metabolites both exhibited fragment ions of [M − H–H2O–2CO], which have a similar mass schizolysis rule as seen for ginkgolide. According to the above analysis, we tentatively presumed that M2 is a hydrolyzed metabolite of ginkgolide B, M3 is a hydrolyzed metabolite of ginkgolide A, and M4 is a hydrolyzed metabolite of ginkgolide C (Fig. 4).35,36,38,39 Furthermore, M6 (m/z 443.1534) was tentatively identified as another hydrolyzed metabolite of ginkgolide A (bi-ionized ginkgolide A) by comparing deprotonated molecular [M − H]− and characteristic fragments [M − H-74] to those reported in the literature.36
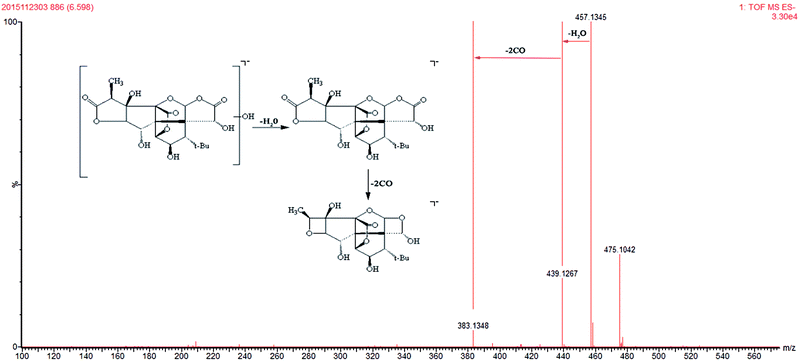 |
| | Fig. 4 The mass spectra of hydrolysis ginkgolides C in negative mode and the proposed fragmentation pathway. | |
Two flavonoid-related metabolites were detected. M5 (tR = 10.16) and M7 (tR = 12.05) showed quasi-molecular ions at m/z 445.0768 and m/z 461.1082, respectively, in the MS spectra. Two metabolites showed product ions at m/z 269.0482 and m/z 285.0754 in the MS/MS spectra. The loss of 176 Da indicated that M5 and M7 were glucuronide conjugates. Alternatively, loss of CO2 from the product ion (m/z 269.0482) [M − H-176-44]− and loss of 2CO and H2O from the product ion (m/z 285.0754) [M − H-176-74]− occurred. The pathway of further fragmentation was similar to that of apigenin and kaempferol;16 therefore, M5 and M7 were tentatively identified as apigenin-O-glucuronide (Fig. 5) and kaempferol-O-glucuronide by comparing retention times, accurate quasi-molecular ions, and MS/MS fragmentation data as well as by referring to the related literature.40–44
 |
| | Fig. 5 The mass spectra of apigenin glucuronide in negative mode and the proposed fragmentation pathway. | |
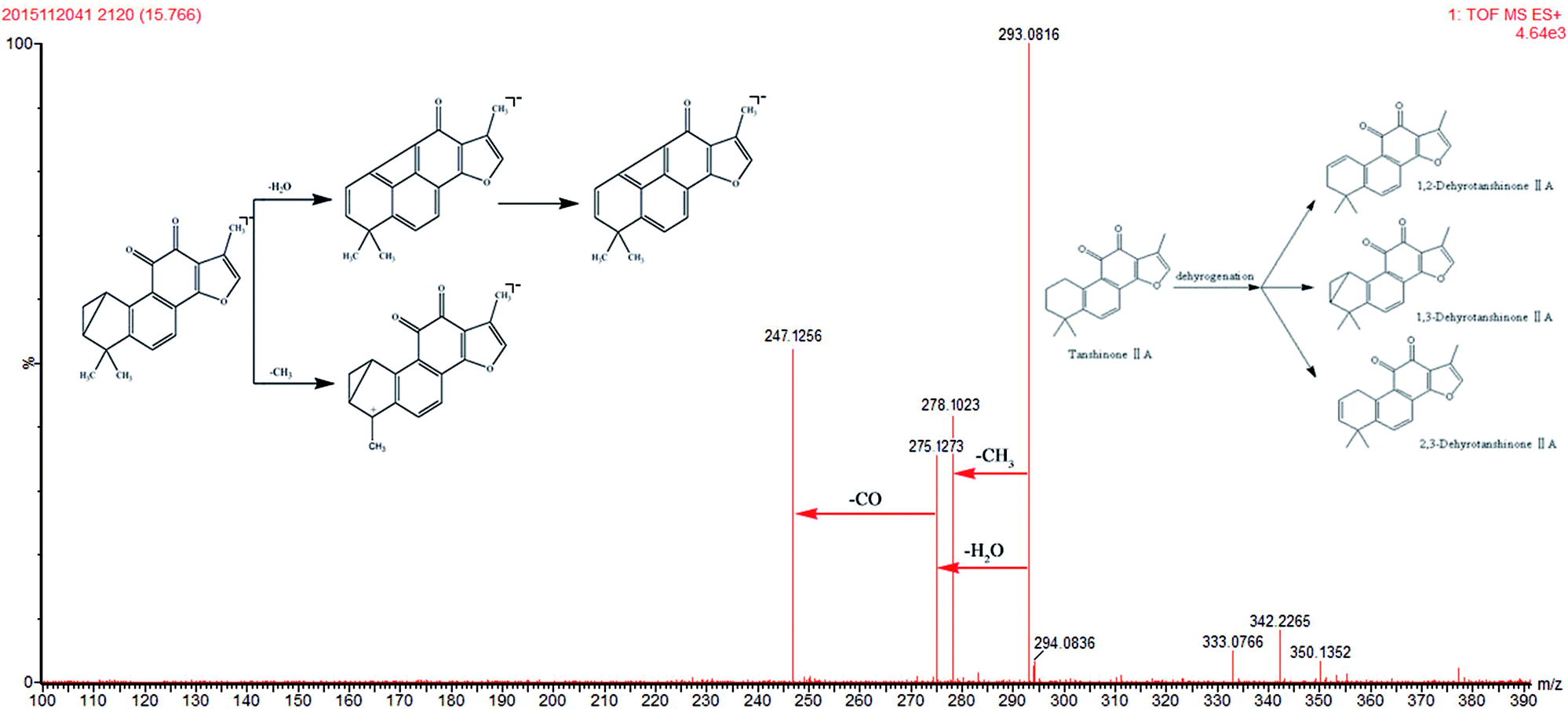
One tanshinone-related metabolite was tentatively identified from rat plasma on the basis of accurate deprotonated molecular data, MS/MS fragmentation data, and previous reports.45–47 M7 gives rise to a protonated molecule [M + H]+ at m/z 293.0816. This molecule was 2 Da smaller than TSIIA. The neutral loss of m/z 15, m/z 18, and m/z 28 indicated the loss of CH3, H2O, and CO, respectively (Fig. 6). Corresponding to the deprotonated molecular ion and its fragments, M7 was tentatively identified as dehydrogenated TSIIA, which has been reported in the literature.34,45–47
 |
| | Fig. 6 The mass spectra of 1,2; 1,3; 2,3-dehydrotanshinone IIA in positive mode and the proposed fragmentation pathway. | |
3.4 Assay validation
3.4.1 Specificity. Under the UPLC-MS/MS conditions, a specific voltage at the optimal value was set to obtain the best sensitivity and specificity for each analyte. According to the results, there was no obvious interference from endogenous components, indicating good method selectivity. The chromatographic peaks were at retention times of 2.435, 2.580, 2.557, 2.499, 2.494, 2.324, 2.454, 3.384, 2.285 and 2.612 min for QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, SAB and IS, respectively.
3.4.2 Linearity. The calibration curves showed good linearity (R2 > 0.9931) over the concentration ranges of the nine constituents in rat plasma. The correlation coefficients of the standard curves and the linear ranges of the plasma are listed in Table 4.
Table 4 Regression data and LLOQs of BB, GA, GB, GC, QCT, KMF, and ISR
| Biosamples |
Analyte |
Linear range (ng mL−1) |
Linear equation |
R2 |
LLOQ (ng mL−1) |
| Plasma |
QCT |
1–100 |
Y = 0.0443X + 0.0621 |
0.9993 |
1 |
| ISR |
1–100 |
Y = 1.8431X − 16.561 |
0.9982 |
1 |
| KMF |
1–100 |
Y = 0.0031X + 0.0222 |
0.9971 |
1 |
| GA |
1–100 |
Y = 0.00741X + 0.3012 |
0.9987 |
1 |
| GB |
1–100 |
Y = 0.1355X + 1.3265 |
0.9987 |
1 |
| GC |
1–100 |
Y = 0.0541X + 0.4030 |
0.9995 |
1 |
| BB |
1–100 |
Y = 0.0204X + 0.1212 |
0.9978 |
1 |
| TSIIA |
1–100 |
Y = 0.3066X + 0.3920 |
0.9992 |
1 |
| SAB |
5–500 |
Y = 0.0063X + 0.0542 |
0.9985 |
5 |
3.4.3 Precision and accuracy. The precision and accuracy data for six replicates of the QC samples at three concentrations are shown in Table 5. The results demonstrated that the precision (RSD%) of the method was 10.26%.
Table 5 Precision and accuracy for bioactive compounds in rat plasma and brain homogenates (n = 18, 6 replicates per day for 3 days)
| Biosamples |
Analytes |
Analyte concentration (ng mL−1) |
Inter-day |
Intra-day |
| Accuracy (%) |
RSD (%) |
Accuracy (%) |
RSD (%) |
| Plasma |
QCT |
1 |
94.16 ± 2.36 |
2.51 |
94.56 ± 4.05 |
4.28 |
| 10 |
107.52 ± 4.82 |
4.48 |
101.12 ± 5.32 |
5.26 |
| 100 |
98.73 ± 6.29 |
6.37 |
96.18 ± 7.43 |
7.73 |
| ISR |
1 |
97.75 ± 7.39 |
7.56 |
98.33 ± 6.71 |
6.82 |
| 10 |
93.12 ± 5.48 |
5.88 |
95.26 ± 7.93 |
8.32 |
| 100 |
102.48 ± 3.32 |
3.24 |
100.11 ± 5.19 |
5.18 |
| KMF |
1 |
96.49 ± 5.19 |
6.71 |
94.20 ± 6.90 |
7.32 |
| 10 |
103.28 ± 8.48 |
8.21 |
99.89 ± 10.15 |
10.16 |
| 100 |
92.27 ± 4.31 |
4.67 |
96.37 ± 6.54 |
6.79 |
| GA |
1 |
100.35 ± 6.40 |
6.38 |
101.18 ± 9.64 |
9.53 |
| 10 |
97.57 ± 2.45 |
2.51 |
95.47 ± 3.19 |
3.34 |
| 100 |
103.63 ± 8.81 |
8.5 |
101.54 ± 9.30 |
9.16 |
| GB |
1 |
95.53 ± 4.17 |
4.37 |
94.15 ± 6.99 |
7.42 |
| 10 |
101.13 ± 3.23 |
3.19 |
98.24 ± 5.59 |
5.69 |
| 100 |
99.28 ± 7.45 |
9.87 |
97.11 ± 8.97 |
9.24 |
| GC |
1 |
94.25 ± 8.31 |
7.5 |
95.35 ± 7.80 |
8.18 |
| 10 |
101.37 ± 9.22 |
9.1 |
99.40 ± 10.20 |
10.26 |
| 100 |
97.46 ± 5.34 |
5.48 |
95.27 ± 6.24 |
6.55 |
| BB |
1 |
105.38 ± 7.24 |
6.87 |
101.33 ± 7.20 |
7.11 |
| 10 |
94.21 ± 4.22 |
4.48 |
93.62 ± 5.27 |
5.63 |
| 100 |
96.48 ± 5.29 |
5.48 |
94.71 ± 6.03 |
6.37 |
| TSIIA |
1 |
97.36 ± 6.53 |
6.71 |
94.41 ± 4.23 |
3.85 |
| 10 |
102.42 ± 4.73 |
4.62 |
98.56 ± 6.73 |
2.73 |
| 100 |
106.26 ± 10.49 |
9.87 |
99.18 ± 5.59 |
8.93 |
| SAB |
5 |
92.65 ± 6.78 |
7.32 |
103.11 ± 6.94 |
4.97 |
| 50 |
94.57 ± 4.69 |
4.96 |
95.44 ± 7.26 |
6.20 |
| 500 |
101.77 ± 7.82 |
7.68 |
98.84 ± 5.38 |
9.44 |
3.4.4 Extraction recovery rates and matrix effects. The extraction recovery and matrix effects of each QC concentration are listed in Table 6. The extraction recoveries ranged from 76.86% to 95.77% at different concentrations in plasma. All ratios for matrix effects were in the range of 94.56–103.25% in plasma samples. Thus, no significant matrix effects were observed for the analytes.
Table 6 Extraction recovery and matrix effect for the analytes or bioactive compounds in rat plasma and brain homogenates (n = 6)
| Biosamples |
Analytes |
Analyte concentration (ng mL−1) |
Matrix effect |
Extraction recovery |
| Accuracy (%) |
RSD (%) |
Accuracy (%) |
RSD (%) |
| Plasma |
QCT |
1 |
100.69 ± 9.02 |
8.96 |
84.42 ± 8.51 |
10.08 |
| 10 |
97.51 ± 3.91 |
4.51 |
79.87 ± 6.59 |
9.26 |
| 100 |
96.51 ± 5.94 |
6.16 |
93.89 ± 3.40 |
3.62 |
| ISR |
1 |
97.56 ± 4.78 |
4.9 |
83.03 ± 4.57 |
5.5 |
| 10 |
96.33 ± 8.08 |
8.38 |
79.84 ± 2.27 |
2.85 |
| 100 |
102.33 ± 10.89 |
10.64 |
90.31 ± 6.74 |
7.46 |
| KMF |
1 |
96.13 ± 5.84 |
6.08 |
79.73 ± 5.88 |
7.37 |
| 10 |
100.04 ± 8.39 |
8.38 |
89.14 ± 3.13 |
3.52 |
| 100 |
98.81 ± 5.84 |
5.91 |
95.77 ± 5.63 |
5.88 |
| GA |
1 |
103.25 ± 7.22 |
6.99 |
83.17 ± 3.24 |
3.89 |
| 10 |
102.75 ± 8.69 |
8.45 |
87.72 ± 3.75 |
4.27 |
| 100 |
98.48 ± 6.99 |
7.09 |
93.89 ± 3.40 |
3.62 |
| GB |
1 |
99.29 ± 5.99 |
6.04 |
87.26 ± 3.27 |
5.26 |
| 10 |
100.39 ± 5.82 |
5.8 |
85.72 ± 3.28 |
3.83 |
| 100 |
94.71 ± 3.26 |
3.44 |
91.12 ± 5.14 |
5.64 |
| GC |
1 |
102.91 ± 11.93 |
11.59 |
80.17 ± 2.12 |
2.64 |
| 10 |
101.18 ± 9.23 |
9.13 |
87.72 ± 3.75 |
4.27 |
| 100 |
98.50 ± 4.62 |
4.69 |
83.47 ± 6.25 |
7.49 |
| BB |
1 |
94.25 ± 7.36 |
7.8 |
76.86 ± 5.23 |
6.8 |
| 10 |
97.57 ± 5.82 |
5.97 |
81.33 ± 4.89 |
4.27 |
| 100 |
94.56 ± 3.50 |
3.7 |
91.22 ± 3.18 |
6.01 |
| TSIIA |
1 |
98.63 ± 5.99 |
6.07 |
82.84 ± 4.63 |
5.59 |
| 10 |
94.63 ± 3.91 |
4.13 |
87.72 ± 3.75 |
4.27 |
| 100 |
93.28 ± 5.21 |
5.59 |
93.92 ± 8.18 |
8.71 |
| SAB |
5 |
99.11 ± 12.27 |
12.38 |
72.43 ± 6.26 |
8.64 |
| 50 |
96.92 ± 5.83 |
6.02 |
77.65 ± 5.72 |
7.37 |
| 500 |
103.64 ± 11.20 |
10.80 |
83.97 ± 7.24 |
8.62 |
3.4.5 Stability. The data for freeze–thaw stability, short-term temperature stability, long-term stability, and autosampler stability under different storage conditions are summarized in Table 7. The results were well within the acceptable limits, therefore validating the established method for sample extraction, storage and intermittent analysis and indicating its suitability for pharmacokinetic study.
Table 7 Stability of the analytes or bioactive compounds in rat plasma and brain homogenates (n = 6)
| Biosamples |
Analytes |
Analyte concentration (ng mL−1) |
Short-term stability |
Autosampler stability |
Freeze–thaw strability (three cycles) |
Long-term stability |
| Accuracy (%) |
RSD (%) |
Accuracy (%) |
RSD (%) |
Accuracy (%) |
RSD (%) |
Accuracy (%) |
RSD (%) |
| Plasma |
QCT |
1 |
100.23 ± 1.69 |
1.6 |
103.73 ± 5.52 |
5.32 |
98.27 ± 6.33 |
6.44 |
97.57 ± 3.42 |
3.51 |
| 10 |
97.45 ± 4.23 |
4.34 |
99.34 ± 6.73 |
6.76 |
100.31 ± 4.27 |
4.26 |
96.41 ± 5.79 |
6.01 |
| 100 |
95.97 ± 2.36 |
2.46 |
94.22 ± 5.14 |
5.46 |
98.35 ± 7.84 |
7.97 |
93.17 ± 7.35 |
7.89 |
| ISR |
1 |
99.24 ± 6.68 |
6.73 |
98.06 ± 8.29 |
8.45 |
103.15 ± 5.43 |
5.26 |
98.35 ± 6.27 |
6.38 |
| 10 |
96.28 ± 5.89 |
6.12 |
97.72 ± 3.83 |
3.92 |
94.52 ± 3.68 |
3.89 |
96.44 ± 6.33 |
6.56 |
| 100 |
102.18 ± 4.95 |
4.84 |
98.34 ± 7.61 |
7.74 |
99.63 ± 3.38 |
3.52 |
95.76 ± 8.18 |
8.54 |
| KMF |
1 |
96.50 ± 6.47 |
6.71 |
99.17 ± 4.39 |
4.43 |
101.23 ± 8.76 |
8.65 |
98.70 ± 9.35 |
9.47 |
| 10 |
101.22 ± 2.09 |
2.06 |
102.65 ± 5.28 |
5.14 |
103.21 ± 4.14 |
4.01 |
100.48 ± 7.44 |
7.4 |
| 100 |
97.10 ± 3.69 |
3.8 |
95.96 ± 3.11 |
3.24 |
94.24 ± 6.63 |
7.04 |
95.21 ± 5.96 |
6.26 |
| GA |
1 |
100.59 ± 6.08 |
6.04 |
97.28 ± 2.19 |
2.25 |
102.76 ± 5.39 |
5.25 |
98.92 ± 5.81 |
5.87 |
| 10 |
102.54 ± 7.33 |
7.15 |
98.33 ± 3.46 |
3.52 |
98.42 ± 2.84 |
2.89 |
95.48 ± 10.28 |
10.77 |
| 100 |
98.31 ± 5.29 |
5.39 |
96.83 ± 8.30 |
7.09 |
95.44 ± 7.51 |
7.87 |
92.19 ± 8.81 |
9.56 |
| GB |
1 |
96.95 ± 2.87 |
2.96 |
101.66 ± 4.73 |
8.57 |
96.53 ± 5.48 |
5.68 |
98.45 ± 5.75 |
5.84 |
| 10 |
98.25 ± 5.91 |
6.01 |
95.45 ± 5.97 |
6.25 |
97.82 ± 4.31 |
4.41 |
95.72 ± 6.59 |
3.83 |
| 100 |
98.74 ± 4.06 |
4.11 |
99.11 ± 7.23 |
7.3 |
96.55 ± 5.94 |
4.64 |
97.38 ± 4.39 |
6.88 |
| GC |
1 |
100.24 ± 4.70 |
3.1 |
98.49 ± 6.22 |
6.32 |
104.74 ± 8.47 |
8.09 |
100.07 ± 10.14 |
10.13 |
| 10 |
104.11 ± 3.38 |
3.25 |
102.24 ± 5.11 |
5 |
100.15 ± 4.52 |
4.52 |
100.48 ± 8.62 |
8.58 |
| 100 |
94.58 ± 5.63 |
5.96 |
91.17 ± 5.33 |
5.85 |
96.22 ± 5.73 |
5.96 |
93.97 ± 4.38 |
4.66 |
| BB |
1 |
98.10 ± 7.83 |
7.98 |
103.47 ± 6.75 |
6.52 |
99.85 ± 6.27 |
6.28 |
99.15 ± 5.73 |
5.78 |
| 10 |
99.78 ± 5.25 |
5.26 |
95.29 ± 2.77 |
2.91 |
98.88 ± 3.59 |
3.63 |
96.38 ± 8.63 |
8.95 |
| 100 |
94.05 ± 2.61 |
2.78 |
97.44 ± 3.13 |
3.21 |
95.47 ± 4.92 |
5.15 |
95.54 ± 7.76 |
8.12 |
| TSIIA |
1 |
96.50 ± 6.47 |
2.93 |
102.65 ± 5.28 |
4.33 |
100.31 ± 4.27 |
9.53 |
101.23 ± 8.76 |
3.47 |
| 10 |
101.22 ± 2.09 |
3.41 |
95.96 ± 3.11 |
5.70 |
101.22 ± 2.09 |
7.26 |
103.21 ± 4.14 |
3.11 |
| 100 |
95.96 ± 3.11 |
2.19 |
99.15 ± 5.73 |
4.92 |
95.97 ± 2.36 |
4.19 |
94.24 ± 6.63 |
3.58 |
| SAB |
5 |
97.28 ± 2.19 |
3.74 |
96.38 ± 8.63 |
6.51 |
99.24 ± 6.68 |
3.24 |
102.76 ± 5.39 |
4.25 |
| 50 |
96.44 ± 6.33 |
4.33 |
96.41 ± 5.79 |
9.18 |
98.88 ± 3.59 |
7.45 |
95.72 ± 6.59 |
5.67 |
| 500 |
95.76 ± 8.18 |
8.21 |
93.17 ± 7.35 |
6.88 |
95.47 ± 4.92 |
6.17 |
97.38 ± 4.39 |
7.69 |
3.5 Pharmacokinetics studies
The developed and validated UPLC-QqQ-MS/MS method was successfully applied in the pharmacokinetics study of QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, and SAB in rat plasma after an oral dose of 4.8 g kg−1 d−1 YDXNT. Fig. 7 shows the mean plasma concentration–time profiles of nine constituents in rat plasma. Table 8 shows the main pharmacokinetic parameters of QCT, ISR, KMF, GA, GB, GC, BB, TSIIA, and SAB in rat plasma.
 |
| | Fig. 7 Plasma concentration–time profiles of the analytes following oral administration of the YDXNT to rats (mean ± SD, n = 6). | |
Table 8 PK parameters of the seven analytes following oral administration of YXC to rats (n = 6)
| Analytes |
Cmax/(ng mL−1) |
Tmax (h) |
AUC0–t (ng mL−1 h−1) |
AUC0–INF (ng mL−1 h−1) |
T1/2 (h) |
MRT (h) |
| QCT |
45.02 ± 11.28 |
0.33 ± 0.11 |
410.34 ± 73.15 |
412.46 ± 82.67 |
2.69 ± 0.65 |
8.44 ± 1.24 |
| ISR |
27.85 ± 8.38 |
0.33 ± 0.14 |
319.17 ± 49.27 |
384.49 ± 55.13 |
8.19 ± 2.42 |
13.91 ± 3.71 |
| KMF |
49.90 ± 13.82 |
0.50 ± 0.23 |
401.33 ± 84.37 |
539.04 ± 82.29 |
8.19 ± 2.42 |
17.11 ± 4.42 |
| GA |
76.31 ± 18.19 |
0.75 ± 0.29 |
490.92 ± 72.63 |
545.10 ± 78.91 |
7.52 ± 2.08 |
9.42 ± 1.77 |
| GB |
76.54 ± 15.43 |
1.00 ± 0.35 |
610.18 ± 91.45 |
691.00 ± 96.83 |
7.84 ± 3.53 |
10.58 ± 2.63 |
| GC |
35.35 ± 10.28 |
1.50 ± 0.23 |
281.80 ± 41.33 |
297.49 ± 46.59 |
5.45 ± 1.48 |
7.77 ± 2.62 |
| BB |
48.70 ± 12.34 |
0.75 ± 0.50 |
365.97 ± 48.37 |
383.98 ± 51.49 |
5.55 ± 2.67 |
9.62 ± 3.18 |
| TSIIA |
38.34 ± 17.35 |
0.25 ± 0.23 |
167.20 ± 69.49 |
182.83 ± 77.41 |
7.04 ± 2.18 |
9.89 ± 1.50 |
| SAB |
32.00 ± 15.43 |
0.75 ± 0.18 |
118.17 ± 58.46 |
135.72 ± 64.24 |
12.17 ± 3.33 |
9.46 ± 2.37 |
GA and GB have similar pharmacokinetic parameters and may be connected to similar polar constituents, similar to the absorption and elimination processes.48 The Cmax of GC was the lowest among the four ginkgolides, indicating that GC may be partially converted to hydrolyzed ginkgolide C, as we detected. The time required to reach the maximum plasma concentration (Tmax) was 0.75 h for GA, 1.00 h for GB, 1.50 h for GC, and 0.75 h for BB. The T1/2 of four ginkgolides in YDXNT were 7.52 h, 7.84 h, 5.45 h, and 5.55 h. Absorption of the four ginkgolides was slower than that of the flavonols, TSIIA and SAB, while their distribution occurred relatively faster than that of the flavonols, TSIIA and SAB. Additionally, the flavonols showed double peaks in the mean plasma concentration curves. This phenomenon has been reported previously, indicating that these components might have enterohepatic recirculation.49,50 The double peaks suggested that enterohepatic recirculation, as well as inter-transformation among the compounds, might have occurred. One possible explanation for this phenomenon is that some of the other compounds with similar structures might have transformed into these compounds. Since drug absorption is a complex process, more detailed adsorption studies are needed to ascertain the mechanism of the double-peak phenomenon. The Cmax of TSIIA (38.34 ng mL−1) is higher than that of SAB (32.00 ng mL−1), and the Tmax and T1/2 of TSIIA (0.25 h, 7.04 h) are lower than those of SAB (0.75 h, 12.17 h), indicating the absorption and distribution rates of TSIIA are faster than those of SAB in rats.
4. Conclusion
In this research, we developed the UPLC-Q-TOF-MS method for analyzing and identifying the main absorbed constituents of YDXNT and their possible metabolites in rat plasma. A total of 52 constituents, including 44 prototype compounds and 8 metabolites, were identified or tentatively characterized. Among the prototype constituents, 15 flavonoids, 4 ginkgolides, 8 phenolic, 8 ginsenoside, and 7 diterpenoid tanshinones were identified. The metabolic routes of 8 metabolites in rat plasma after oral administration were hypothesized. The result indicated that the metabolites of YDXNT undergo the phase I metabolite pathway of hydrolysis (M2, M3, M4, and M6), dehydrogenation (M8) and the phase II metabolic route of glucuronide (M1, M5 and M7). Furthermore, a sensitive and reliable method based on UPLC-QqQ-MS was established for simultaneous quantification of nine abundant main compounds in rat plasma. The developed assay was successfully applied to assess the pharmacokinetics of ginkgetin aglycone, terpene lactones, salvianolic acid, and tanshinone from YDXNT in rat plasma. Our research clarifies for the first time the absorbed constituents, possible metabolites, and pharmacokinetics of YDXNT in vivo. This work provides more in-depth insight into the main active constituents of YDXNT in vivo as well as helpful information for clinical application. More importantly, our findings are helpful for better understanding the material foundation and action targets underlying the efficacy of YDXNT.
Author contributions
All the authors have approved the manuscript and agree with submission to your esteemed journal.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Financial support was provided by the Chinese drug discovery initiative project (No. 2012ZX09201201).
References
- L. Y. Kong and R. L. Tan, Nat. Prod. Rep., 2015, 32, 1617–1621 RSC.
- X. J. Wang, A. H. Zhang, H. Sun, Y. Han and G. L. Yan, TrAC, Trends Anal. Chem., 2016, 76, 86–94 CrossRef CAS.
- L. Ma, H. Liu, L. Meng, P. Qin, B. T. Zhang, Y. Y. Li, S. L. Man, Z. Liu, Z. X. Liu and A. P. Diao, RSC Adv., 2017, 7, 3408–3412 RSC.
- X. J. Wang, A. H. Zhang, H. Sun and P. Wang, Am. J. Chin. Med., 2012, 40, 1109 CrossRef PubMed.
- E. L. Leung, E. Clementi and W. S. Wong, Pharmacol. Res., 2017, 117, 65–66 CrossRef PubMed.
- Y. Tao, H. Y. Xu, S. S. Wang, B. Wang, Y. C. Zhang, W. H. Wang, B. Huang, H. W. Wu, D. F. Li, Y. Zhang, X. F. Xiao, Y. B. Li, H. J. Yang and L. Q. Huang, J. Chromatogr. B, 2013, 935, 1–9 CrossRef CAS PubMed.
- Z. Y. Tang, Y. Y. Hou, X. Y. Xu, A. N. Liu, L. F. Yau, T. T. Tong, Z. H. Jiang and G. Bai, RSC Adv., 2017, 7, 37459–37466 RSC.
- M. Q. Jia, Y. J. Xiong, Y. Xue, Y. Wang and C. Yan, Molecules, 2017, 22, 1–17 Search PubMed.
- S. P. Wang, P. Fu, L. Liu, L. L. Wang, C. C. Peng, W. D. Zhang and R. H. Liu, RSC Adv., 2016, 6, 32811–32822 RSC.
- H. Wu and F. Fang, RSC Adv., 2016, 6, 28279–28288 RSC.
- D. X. Chen, S. Lin, W. Xu, M. Q. Huang, J. F. Chu, F. Xiao, J. M. Lin and J. Peng, Molecules, 2015, 20, 18597–18619 CrossRef CAS PubMed.
- The State Pharmacopoeia Commission of PR China, The Pharmacopoeia of PR China, China Medical Science and Technology Press, PR China, 2015 Search PubMed.
- J. L. Wang, L. Wang, L. Chen, X. J. Yin, H. Y. Xu, W. D. Wang and R. X. Liang, China J. Chin. Mater. Med., 2014, 9, 2547–2552 Search PubMed.
- L. Cheng, Y. Liu and X. Sun, J. Evidence-Based Complementary Altern. Med., 2017, 2017, 1–14 Search PubMed.
- J. Huang, The Second China-Japan-Korea Congress on the Blood Stasis Syndrome, 2003, pp. 61–64 Search PubMed.
- L. L. Gong, H. Y. Xu, L. Wang, X. J. Yin, H. J. Yuan, S. S. Wang, L. Cheng, X. J. Ma, S. R. Gao, R. X. Liang and H. J. Yang, J. Sep. Sci., 2016, 39, 611–622 CrossRef CAS PubMed.
- J. L. Wang, L. Wang, H. J. Yang, Y. You, H. Y. Xu, L. L. Gong, X. J. Yin, W. D. Wang, S. R. Gao, L. Cheng, R. X. Liang and F. L. Liao, BMC Complementary Altern. Med., 2015, 15, 109–122 CrossRef PubMed.
- W. D. Wang, L. Wang, H. J. Yang, J. L. Wang, X. J. Yin, H. Y. Xu, L. Cheng and R. X. Liang, J. Tradit. Chin. Med., 2014, 34, 699–709 CrossRef PubMed.
- L. Cheng, G. F. Pan, X. D. Zhang, J. L. Wang, W. D. Wang, J. Y. Zhang, H. Wang, R. X. Liang and X. B. Sun, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- J. B. Fourtillan, A. M. Brisson, J. Girault, I. Ingrand, J. P. Decourt, K. Drieu, P. Jouenne and A. Biber, Thérapie, 1995, 50, 137–144 CAS.
- X. Li, C. Yu, Y. Lu, Y. Gu, J. Lu, W. Xu, L. Xuan and Y. Wang, Drug Metab. Dispos., 2007, 35, 234–239 CrossRef CAS PubMed.
- Z. Ouyang, M. Zhao, J. Tang and L. Pan, Pharmacogn. Mag., 2012, 8, 256 CrossRef PubMed.
- S. Li, X. Jiang, Q. Yang and Z. Jin, J. Biomed. Eng., 2003, 20, 692–694 CAS.
- L. Q. Sun, Asian Journal of Drug Metabolism & Pharmacokinetics, 2002, 2, 291–298 Search PubMed.
- Q. Chen, Z. Liang, E. Brand, H. Chen and Z. Zhao, Planta Med., 2017, 82, 263–272 Search PubMed.
- W. J. Chen, Y. Y. Liu, M. Y. Wei, L. Q. Shi, Y. Wu, Z. Y. Liu, S. Liu, F. R. Song and Z. Q. Liu, RSC Adv., 2017, 7, 42964–42972 RSC.
- G. Y. Ding, Y. S. Wang, A. N. Liu, Y. Y. Hong, T. J. Zhang, G. Bai and C. X. Liu, RSC Adv., 2017, 7, 22034–22044 RSC.
- J. W. Lee, H. J. Mok, D. Y. Lee, S. C. Park, G. S. Kim, S. E. Lee, Y. S. Lee, K. P. Kin and H. D. Kim, J. Proteome Res., 2017, 16, 1460–1469 CrossRef CAS PubMed.
- J. H. Lee, H. Shinohara, J. H. Jung, H. J. Mok, Y. Akao and K. P. Kim, RSC Adv., 2016, 6, 29512–29518 RSC.
- US Department of Health and Human Services, Guidance for Industry Analytical Procedures and Methods Validation for Drugs and Biologics, 2015, http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm386366.pdf Search PubMed.
- H. Wang, X. Bai, J. H. Sun, Y. Kano, T. Makino and D. Yuan, Planta Med., 2013, 79, 1552–1557 CrossRef CAS PubMed.
- Y. F. Zhang, P. Y. Shi, H. Yao, Q. Shao and X. H. Fan, Curr. Drug Metab., 2012, 13, 510–523 CrossRef CAS PubMed.
- M. Xu, Z. C. Zhang, G. Fu, S. F. Sun, J. H. Sun, M. Yang, A. H. Liu, J. Han and D. A. Guo, J. Chromatogr. B, 2007, 856, 100–107 CrossRef CAS PubMed.
- X. Zhang, D. H. Yang, F. Xu, S. Huang, L. Zhang, G. X. Liu and S. Q. Cai, Biomed. Chromatogr., 2015, 29, 285–304 CrossRef PubMed.
- D. L. Wang, Y. Liang, W. D. Chen, L. Xue, G. J. Wang and X. D. Liu, Acta Pharmacol. Sin., 2008, 29, 376–384 CrossRef CAS PubMed.
- X. G. Liu, L. D. Qi, Z. Y. Fan, X. Dong, R. Z. Guo, F. C. Lou, S. Fanali, P. Li and H. Yang, J. Chromatogr. A, 2015, 1388, 251–258 CrossRef CAS PubMed.
- S. Ding, E. Dudley, S. Plummer, J. Tang, R. P. Newton and A. G. Brenton, Phytochemistry, 2008, 69, 1555–1564 CrossRef CAS PubMed.
- X. J. Liu, K. Yang, G. Du, L. Xu and K. Lan, Anal. Bioanal. Chem., 2015, 407, 7945–7956 CrossRef PubMed.
- Y. Liu, M. L. Bandu and H. Desaire, Anal. Chem., 2005, 77, 6655–6663 CrossRef PubMed.
- H. Cai, D. J. Boocock, W. P. Steward and A. J. Gescher, Cancer Chemother. Pharmacol., 2007, 60, 257–266 CrossRef CAS PubMed.
- H. L. Ma, Y. Liu, X. Mai, Y. J. Liao, K. Zhang, B. Liu, X. Xie and Q. L. Du, J. Pharm. Biomed. Anal., 2016, 125, 194–204 CrossRef CAS PubMed.
- Y. J. Shi, Y. Wang, H. Z. Zhu, H. X. Zheng, W. D. Zhu, J. Xu and M. Y. Li, Chin. J. Pharm. Anal., 2015, 35, 1417–1423 CAS.
- Y. Z. Zhang, T. Wang, S. L. Zhou, H. L. Liu and C. Yan, China Pharmacy, 2016, 27, 479–482 CAS.
- J. H. Ding, X. X. Wang, H. Zhang, S. S. Pan, M. B. Luo, J. Q. Li and H. W. Chen, Chem. J. Chin. Univ., 2011, 32, 1714–1719 CAS.
- J. H. Sun, M. Yang, X. M. Wang, M. Xu, A. H. Liu and D. A. Guo, J. Pharmaceut. Biomed., 2007, 44, 564–574 CrossRef CAS PubMed.
- P. Li, G. J. Wang, J. Li, H. P. Hao and C. N. Zheng, J. Mass Spectrom., 2006, 41, 670–684 CrossRef CAS PubMed.
- P. Li, G. J. Wang, J. Li, H. P. Hao and C. N. Zheng, Rapid Commun. Mass Spectrom., 2006, 20, 815–822 CrossRef CAS PubMed.
- Z. Yan-Yan, G. Li-Li, S. Guo-Ming, R. Rong and T. Jing-Zhen, Drug Res., 2016, 66, 520–526 CrossRef CAS PubMed.
- Z. P. Chen, J. Sun, H. X. Chen, Y. Y. Xiao, D. Liu, J. Chen, H. Cai and B. C. Cai, Fitoterapia, 2010, 81, 1045–1052 CrossRef CAS PubMed.
- W. P. Wang, N. Liu, Q. Kang, P. P. Du, Y. Lan, B. C. Zhao, Y. Y. Chen, Q. Zhang, H. Li, Y. W. Zhang and Q. Wu, J. Zhejiang Univ., Sci., B, 2014, 15, 929–939 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12659j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Haiyu Xua,
Huijun Yuanb,
Lan Wanga,
Xiaojie Yina,
Moqi Fanc,
Long Cheng
a,
Haiyu Xua,
Huijun Yuanb,
Lan Wanga,
Xiaojie Yina,
Moqi Fanc,
Long Cheng