
Chlorophyll derivatives as catalysts and comonomers for atom transfer radical polymerizations†
Bernadetta
Gajewska
 a,
Samuel
Raccio
a,
Kyle J.
Rodriguez
a and
Nico
Bruns
*ab
a,
Samuel
Raccio
a,
Kyle J.
Rodriguez
a and
Nico
Bruns
*ab
aAdolphe Merkle Institute, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: nico.bruns@unifr.ch
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
First published on 13th November 2018
Abstract
Copper trisodium chlorophyllin is obtained from natural chlorophyll, and is widely used as a major green food colorant in cosmetics and in medical devices. Copper chlorophyllin also proves to be an efficient catalyst and comonomer for atom transfer radical polymerizations (ATRP). Aqueous ATRP of poly(ethylene glycol) acrylate (PEGA) results in PEGA-chlorophyllin copolymers with narrow molecular weight distributions and a controlled content of chlorophyllin. The reactions proceed with first order kinetics, and the polymer's molecular weight increases with conversion. The resulting copolymers could find application in drug delivery and in biomedical materials, or as solar energy harvesting materials. In order to suppress the incorporation of the catalyst into the growing polymer chains, the vinyl bond of chlorin e6, one of the major porphyrin components of chlorophyllin, is deactivated by hydrobromination and hydration. Complexation of copper by the porphyrin leads to a bio-derived catalyst which mediates the polymerization of PEGA, yielding homopolymers with molecular weights ranging from 4000–5000 g mol−1 and dispersities ≤ 1.11. UV-Vis spectroscopy indicates that chlorophyllin is stable during the polymerization. Copper chlorophyllin is a plant-derived compound from a renewable feedstock that can be used for a more environmentally friendly route to ATRP.
Introduction
Atom transfer radical polymerizations (ATRP) are the most widely applied reversible deactivation radical polymerization (RDRP) technique, and allow for the synthesis of polymers with predefined molecular weights, narrow molecular weight distributions and complex molecular architectures.1–5 As such, ATRP is a key enabling technology for the preparation of advanced polymeric materials.6 However, toxicity and negative environmental aspects of the required transition metal complex catalysts limit the application of ATRP-derived polymers in the medical sector or in the food industry, and push researchers to look for alternative solutions, such as highly active catalytic systems, non-toxic catalysts, or catalysts obtained from renewable resources.2,4,7 Since the first reported use by Wallace and coworkers in 1989, enzymes have been used for the production of polymers8 and have since been established as catalysts to initiate free radical polymerizations.9–15 However, the first use of enzymes in RDRP has only been reported in recent years by our group and by di Lena and coworkers.7,16–18 We found that horseradish peroxidase (HRP) catalyzed the polymerization of N-isopropylacrylamide (NIPAAm) under activator regenerated by electron transfer (ARGET) ATRP conditions16 whereas di Lena and coworkers showed that laccase and catalase could initiate polymerizations of poly(ethylene glycol) methyl ether methacrylate (PEGMA).17,18 Since then, our group and others have demonstrated that metal-containing enzymes such as HRP,19–22 laccase,19,23 hemoglobin,24–27 and catalase19,28 can act as ATRP catalysts with distinct advantages, such as the ability to control the polymerizations of difficult monomers, which traditional ATRP conditions fail,23 to confine polymerizations into nanoreactors,20,21 to prepare biosensors,26,27 and to tune surface-initiated polymerizations by the protein affinity of surfaces.25 Metalloenzymes can be deduced to their cofactors, which also have been shown to catalyze ATRP.28–34 For example, Matyjaszewski and coworkers reported RDRP of methacrylates in aqueous solutions mediated by synthetic analogs of hemin and mesohemin, which were modified with 2-methoxy poly(ethylene glycol) (MPEG).28,30,34 Additionally, cobalt porphyrins35–40 and cobalamin41 have been used as catalysts for organometallic mediated radical polymerizations (OMRP) and catalytic chain transfer polymerizations (CCTP). Thus, biocatalytic ATRP (bioATRP) is a feasible approach to generate well-defined polymeric architectures.7 However, a drawback of many of the aforementioned biocatalysts is that they are available in limited quantities at relatively high costs.Chlorophylls are some of the most abundant redox-active biomolecules, as every green plant and many marine microorgansims produce them to catalyze photosynthesis.42–45 Chlorophylls can be easily extracted from plant leaves and other biological materials, and have been used to catalyze a variety of native and non-native reactions, e.g. in photosynthesis research,46 to initiate RAFT polymerizations,47,48 and to catalyze photoreduction reactions.49 While extracted chlorophyll itself is not stable to heat and light, its water-soluble derivative, copper chlorophyllin, is.50,51 Chemically, copper chlorophyllin is a mixture of chlorin compounds whose main components are Cu chlorin e6, Cu chlorin p6 and Cu isochlorin e4 (Scheme 1).52,53 Copper chlorophyllin is widely used as a common colorant (E141), which has been approved by the Food and Drug Administration (FDA) and European Union for use in human food, drugs, cosmetics and medical devices.54–59 Moreover, copper chlorophyllin may be used as a drug, e.g. in odor treatment,60 photodynamic therapy and cancer chemoprevention.45 It has also been explored as a catalyst for the photoinduced reduction of methylviologen,61 and as photosensitizer in dye-sensitized solar cells62–66 and in semiconductors.67,68 For many materials applications it is advantageous to incorporate copper chlorophyllin into polymeric materials, such as hydrogels, nanogels and linear polymers, as they can be easily processed into three dimensional objects or into thin films. Light sensitive copolymers containing chlorophyllin and other monomers, e.g. N-isopropylacrylamide, n-butyl methacrylate, n-butyl acrylate, acrylic acid, or 2-(dimethylamino)ethyl methacrylate, have been synthesized to prepare recyclable aqueous two-phase systems and cation exchangers for protein purification.69–72 The chlorophyllin copolymer poly(N-isopropyl acrylamide-co-chlorophyllin) has been explored to improve drug release from nanogels.73 Other porphyrin-containing polymers have been used in organic solar cell applications,74 as nanosensors,75 and in drug and gene delivery.76 Chlorophyllin has a vinyl bond that can be used in radical polymerizations, however, the double bond is not reactive enough to form homopolymers. To date copolymers containing chlorophyllin have only been synthesized by free radical polymerizations, therefore, their molecular weights were not controlled.69–73,77
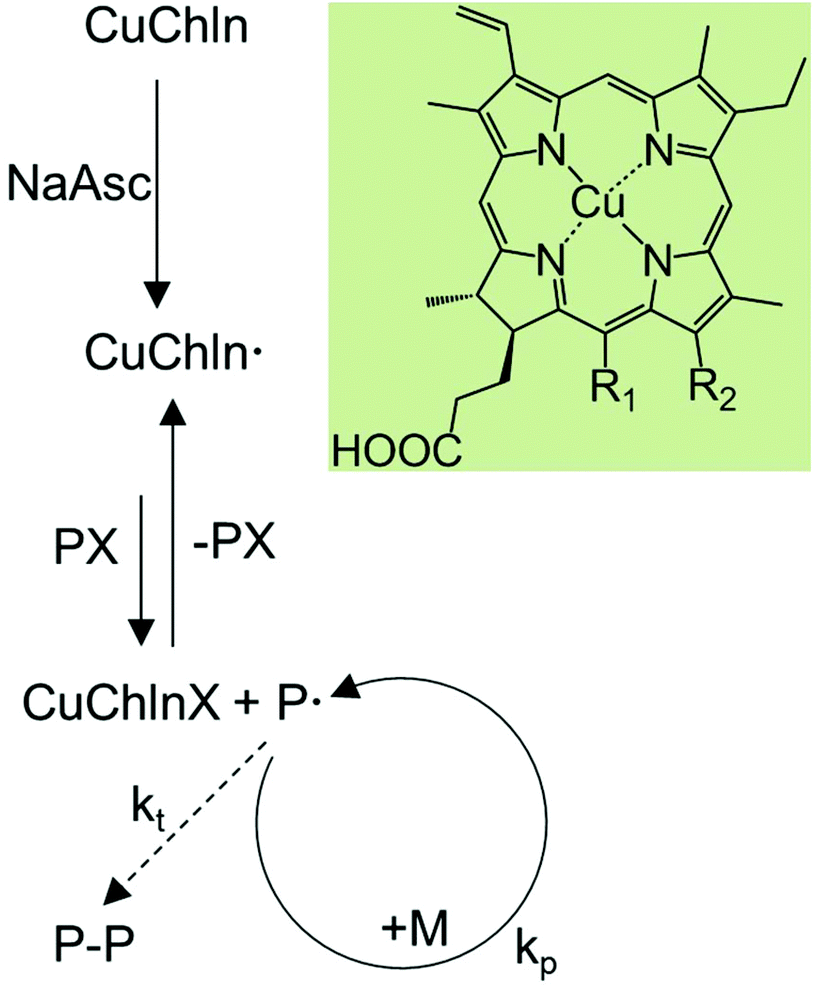 | ||
| Scheme 1 Proposed mechanism for activator generated by electron transfer ATRP (AGET ATRP) catalyzed by copper chlorophyllin and structure of the chlorins that copper chlorophyllin consists of (Cu chlorin e6: R1 = –CH2COOH, R2 = –COOH; Cu chlorin p6: R1 = R2 = –COOH; Cu isochlorin e4: R1 = –CH2COOH, R2 = –H). | ||
Here we show that copper chlorophyllin is not only a non-toxic, abundantly available, renewable and environmentally friendly catalyst for bioATRP, but also can be a useful functional monomer for ATRP-derived polymers. Copper chlorophyllin was used simultaneously as a catalyst and as a comonomer for the ATRP of poly(ethylene glycol) methyl ether acrylate (PEGA). The molecular weights of the polymers increased with conversion and the reactions resulted in copolymers with narrow molecular weight distributions and contained a controlled content of chlorophyllin. When homopolymers are desired, incorporation of chlorophyllin into the polymer chains could be suppressed by using a derivative of the main compound of copper chlorophyllin, chlorin e6, in which the vinyl bond was hydrated. The resulting bio-derived catalyst allowed for the controlled radical polymerizations of PEGA.
Experimental
Reagents
2-Hydroxyethyl-2-bromoisobutyrate (HEBIB), poly(ethylene glycol) methyl ether 2-bromoisobutyrate (PEG-Br; Mn = 600 g mol−1), sodium L-ascorbate (NaAsc), ammonium persulfate (APS, 98%), N,N,N′,N′-tetramethylethylenediamine (TEMED, 99%), neutral and basic aluminum oxide, anhydrous magnesium sulfate (MgSO4), copper(II) acetate, sodium bromide, sodium hydrogen carbonate, anhydrous sodium acetate, methanol (99.9%), N,N-dimethylformamide (DMF, 99.8%), tert-butyl methyl ether (TBME; 99.0%) and 33 wt% hydrobromic acid in acetic acid were purchased from Sigma-Aldrich and used as received. Methanol, dichloromethane and hexane of technical grade were purchased from Reactolab and used as received. Copper trisodium chlorophyllin was purchased from Alfa Aesar and used as received. Chlorin e6 (Ce6; 93–98%) was purchased from Frontier Scientific and used as received. Tetrahydrofuran (THF; GPC grade with 250 ppm of BHT) was purchased from Scharlau Chemicals and used as received. Deuterium oxide (D2O; 99.9%) and dimethyl sulfoxide-d6 (DMSO-d6; 99.9%) were purchased from Cambridge Isotope Laboratories. Poly(ethylene glycol) methyl ether acrylate (PEGA; Mn = 480 g mol−1) was purchased from Sigma Aldrich and purified prior to a reaction on basic aluminum oxide in a syringe equipped with a PTFE filter (0.20 μm CHROMAFIL O-20/15 MS) in order to remove polymerization inhibitors. Polymerizations were carried out in phosphate–citrate buffers pH: 2.0, 3.0, 4.0, 5.0, 6.0, 7.0 and 8.0, which were prepared by addition of 0.1 M citric acid to 0.2 M disodium phosphate according to McIlvaine et al.78Hydration of chlorin e6
Hydration of the vinyl bond of Ce6 was carried out according to Clezy et al. with the modification of a Markovnikov addition of HBr to the vinyl bond, followed by nucleophilic substitution of the bromide with water.79 50 mg of Ce6 (0.084 mmol) was treated with 50 mL of 33% HBr in acetic acid in a 1 L round bottom flask equipped with a magnetic stirring bar, a condenser outfitted with a balloon, a scrubber filled with a saturated aqueous solution of sodium bicarbonate, and a septum pierced with a glass Pasteur pipette. The solution was bubbled with argon through the Pasteur pipette for 45 minutes. The septum with a glass Pasteur pipette was replaced with a glass connector and the balloon on the condenser was filled with argon. Then, the glass connector was exchanged with a glass stopper under argon flow from the balloon. The reaction was heated to 55 °C for 3 h. After, the reaction continued to stir at room temperature overnight. The flask was purged with argon in order to lead acidic gasses through the scrubber. 500 mL of a 5% HClaq was then added to the reaction and the solution was bubbled with argon for 45 minutes. Following the addition, the reaction was stirred at room temperature overnight. The reaction mixture was then split into 100 mL aliquots. The first 100 mL volume was extracted into 200 mL TBME over 200 mL aqueous 10% sodium acetate solution. The colorless aqueous fraction was removed and 200 mL aqueous 10% sodium acetate solution was added to the TBME fraction. The mixture was shaken and the TBME fraction was collected. The aqueous fraction changed color to light violet due to residual porphyrin content. The next 100 mL aliquot of the reaction mixture was extracted into the next 200 mL TBME over the previously obtained violet aqueous fraction. The colorless aqueous fraction was removed and the extraction continued as described above. By repeating these steps, all aliquots of the reaction mixture were extracted. The organic phases were combined and the ether was evaporated to dryness by rotary evaporation under reduced pressure. The product was purified by flash column chromatography on Biotage SNAP Ultra 10 g HP-Sphere 25 μm cartridge using a Biotage ISOLERA. The dried product was loaded on a cartridge with maximum 10 mL of 10 vol% methanol in dichloromethane. The purification used a gradient mixture composed of methanol and dichloromethane. The gradient started with 10% methanol for 9 column volumes to elute all unreacted less polar compounds. Then, the gradient increased to 50% methanol within 1 column volume and continued with 50% methanol for 3 column volumes, which eluted more polar side products. At the end, the eluent composition was increased to 100% methanol and the pure product was collected as the last fraction, as monitored by the UV detector set to a wavelength of 380 nm. Methanol was evaporated by rotary evaporation under reduced pressure, and the product was dried in a desiccator under vacuum overnight. The overall yield of the reaction was 40%. The purified 2-(1-hydroxyethyl) chlorin e6 (Hydrated Ce6) was characterized by ESI-MS: m/z = 615.3+, UV-vis: Soret maximum at 395 nm, Q band maximum at 654 nm, and 1H NMR (300 MHz, DMSO-d6): δ = 12.01 (bs), 10.05 (d, 1H), 9.75 (s, 1H), 9.08 (d, 1H), 6.78–6.06 (m, 2H), 5.56–4.96 (m, 2H), 4.57–4.09 (m, 5H), 3.84 (q, 2H), 3.52–3.50 (m, 9H), 2.03–1.67 (m, 9H, 1OH), −2.06 to −2.60 (dd, 2NH).Insertion of copper to 2-(1-hydroxyethyl) chlorin e6
42.4 mg hydrated Ce6 (0.0691 mmol) and 9.1 mg copper(II) acetate (0.050 mmol) were dissolved in 10 mL methanol in a 25 mL round bottom flask equipped with the condenser and a magnetic stirring bar and refluxed for 2 h. The reaction was monitored by UV-vis spectroscopy using the shift of the Soret band of Hydrated Ce6 from 395 nm to 402 nm and the Q-band from 654 nm to 618 nm. Solvent was evaporated on a rotary evaporator and the product (Hydrated Ce6 copper(II) acetate) was dried in a desiccator under vacuum overnight. The product was characterized by UV-Vis spectroscopy: Soret maximum at 402 nm and Q band maximum at 618 nm.Copolymerizations of PEGA with copper trisodium chlorophyllin under AGET ATRP conditions
Unless otherwise stated, 0.38 mL PEG-Br (0.76 mmol) and 151.0 mg NaAsc (0.7622 mmol) were dissolved in 2.62 ml Milli-Q water in a 5 mL round bottom flask equipped with a septum and a magnetic stirring bar. 184.2 mg copper trisodium chlorophyllin (0.2543 mmol) and, unless otherwise stated, 78.5 mg sodium bromide (0.763 mmol) were dissolved in 7.0 mL phosphate–citrate buffer (pH 4 or 8) and 7.0 mL PEGA (16 mmol) in a 25 mL round bottom flask equipped with a septum and a stirring bar. Solutions were bubbled with argon for 1 h after which 1 mL of the PEG-Br/NaAsc solution was transferred via a syringe purged with argon to the reaction flask. The flask was equipped with a balloon filled with argon and the reaction was stirred at room temperature for 50 h. In regular time intervals, analytical samples (0.5 mL) were withdrawn from the reaction mixture and analyzed by 1H NMR spectroscopy and GPC as described below. After 50 h the reaction flask was opened to air by removal of the septum and the balloon. The reaction mixture was diluted in THF (stabilized with BHT) to the volume of 50 mL followed by addition of MgSO4. The mixture was filtered through a Büchner filter. The liquid phase was passed through basic aluminum oxide and poured into cold hexane. The solution was filtered on a Büchner filter, and the precipitated polymer was dried in a desiccator under vacuum. If polymerizations were carried out under different conditions (e.g. HEBIB as initiator, other pH, other ratios or concentrations of reagents) the same protocol was followed and the differences are specified in the results and discussion section.Polymerizations with the addition of NaAsc during the reaction were set-up following the procedure described above. However, NaAsc solutions were prepared prior to addition and were added after 24 h, 48 h and 72 h according to the following procedure. 201.6 mg NaAsc (1.018 mmol) was dissolved in 2 mL Milli-Q water in a 5 mL round bottom flask equipped with a septum and a magnetic stirring bar. The solution was bubbled with argon for 1 h. After, 0.5 mL of the NaAsc solution was transferred via a syringe purged with argon to the reaction flask. Analytical samples were withdrawn and analyzed as described above.
For polymerizations in the following solvents: phosphate–citrate buffers with varying pHs between 2 and 8, water, and a mixture of water and 20% DMF (v/v), the reactions were stopped after 24 h and a final time point was taken. The polymerizations were conducted in a total volume of 4 mL. 0.65 mL HEBIB (4.6 mmol) and 359.8 mg NaAsc (1.816 mmol) were dissolved in 4.35 mL and 4 mL Milli-Q water, respectively, in 10 mL round bottom flasks equipped with a septum and magnetic stirring bar. 2.5 mL PEGA (5.7 mmol) was dissolved in 1.2 mL phosphate–citrate buffer, or 0.9 mL water and 0.3 mL DMF in the case of the reaction in 20% v/v DMF. Solutions were sparged with argon for 1 h. After, 0.1 mL of the HEBIB solution and 0.2 mL of the NaAsc solution were transferred via an argon purged syringe to the reaction flask. After 24 h, a 0.5 mL analytical sample was withdrawn from the reaction mixture and analyzed by 1H NMR spectroscopy and GPC as described below.
The theoretical content of chlorophyllin, cCHLNth, in the precipitated polymer was calculated according to the equation below,

Polymerizations of PEGA with 2-(1-hydroxyethyl) chlorin e6 under AGET ATRP conditions
The polymerizations were carried out as described in the previous section, but chlorophyllin was replaced with the same molar amount of hydrated CuCe6.Free radical copolymerization of PEGA in the presence of copper trisodium chlorophyllin
290.2 mg APS (1.27 mmol) was dissolved in 2.5 mL Milli-Q water in a 5 mL round bottom flask equipped with a septum and a magnetic stirring bar. 190.5 μL TEMED (1.27 mmol) was dissolved in 2 mL and 309.5 μL Milli-Q water in a 5 mL round bottom flask equipped with a septum and a magnetic stirring bar. 184.2 mg copper trisodium chlorophyllin (0.254 mmol) was dissolved in 7.0 mL phosphate–citrate buffer pH 4, and 7.0 mL PEGA (16.0 mmol) was added to a 25 mL round bottom reaction flask equipped with a septum and a stirring bar. The solutions were sparged with argon for 1 h. After, 0.5 mL of the ammonium persulfate solution and 0.5 mL of the TEMED solution were transferred via an argon purged syringe to the reaction flask. The flask, equipped with a balloon filled with argon, was allowed to react with stirring for 50 h at room temperature. Analytical samples (0.5 mL) were taken from the reaction mixture in regular time intervals and analyzed by 1H NMR spectroscopy and GPC as described below.Copolymerization of PEGA with copper trisodium chlorophyllin carried out in a UV-Vis spectrophotometer
170 μL PEG-Br (0.344 μmol) and 49.58 mg NaAsc (0.250 mmol) were dissolved in 9.83 mL Milli-Q water after which 1.0 mL was withdrawn and diluted with 9.0 mL Milli-Q water. 100 μL of this solution was transferred to a 10 mL round bottom flask, diluted with 4.9 mL Milli-Q water and equipped with a septum and a magnetic stirring bar. 400 μL PEGA (0.908 μmol), 10.45 mg copper trisodium chlorophyllin (0.014 mmol) and 4.5 mg sodium bromide (0.044 mmol) were dissolved in 9.6 mL Milli-Q water after which 1.0 mL was withdrawn and diluted with 9.0 mL Milli-Q water. 100 μL of this solution was transferred to a 3.5 mL quartz cuvette (Hellma Analytics type 117-QS) equipped with a magnetic stirring bar. 1.52 mL Milli-Q water and 380 μL buffer pH 4 were added to the cuvette and the cuvette was sealed with a septum. After 40 minutes of sparging with argon, 0.29 mL of the PEG-Br/NaAsc solution was transferred via an argon purged syringe to the cuvette. The cuvette was equipped with an argon-filled balloon and the reaction was run in a UV-Vis spectrophotometer under stirring at room temperature. 24 UV-Vis spectra were recorded in cycle mode with intervals of 1 s, from λ = 200 nm to λ = 800 nm with a scanning speed of 0.5 nm s−1 and Δλ 1.0 nm.Reaction of copper trisodium chlorophyllin or HEBIB with sodium ascorbate in Milli-Q water carried out in a UV-Vis spectrophotometer
1.3 mg copper trisodium chlorophyllin (1.8 μmol) was dissolved in 13 mL Milli-Q water and 200 μL was transferred to a 3.5 mL quartz cuvette (Hellma Analytics type 117-QS) equipped with a magnetic stirring bar. 2.4 mL Milli-Q water was added to the cuvette and it was sealed with a septum. 54.7 mg NaAsc was dissolved in 10 mL Milli-Q water. A 100 μL stock solution of NaAsc was transferred to a 25 mL round bottom flask, diluted with 9.9 mL Milli-Q water and equipped with a septum and a magnetic stirring bar. The solution was sparged with argon for 40 min. After, 100 μL of the NaAsc solution was transferred via an argon purged syringe to the cuvette. The cuvette was equipped with a balloon filled with argon and the reaction was run in a UV-Vis spectrophotometer under stirring at room temperature. 96 UV-Vis spectra were recorded in cycle mode with intervals of 5 s, from λ = 200 nm to λ = 800 nm with a scanning speed of 2.0 nm s−1 and Δλ 1.0 nm.The reaction of HEBIB with NaAsc was carried out as described above, but chlorophyllin was replaced with the same molar amount of HEBIB.
Instrumentation
Proton Nuclear Magnetic Resonance (1H NMR) spectroscopy was recorded at room temperature on a Bruker Avance III 300 MHz spectrometer using deuterated solvents.For kinetic investigations by NMR spectroscopy, 0.2 mL samples from a polymerization reaction were passed through neutral aluminum oxide columns in Pasteur pipettes plugged with cotton. The columns were additionally rinsed with 0.5 mL D2O.
Gel Permeation Chromatography (GPC) experiments were performed on an Agilent 1200 series HPLC system equipped with an Agilent PLgel mixed guard column (particle size = 5 μm) and two Agilent PLgel mixed-D columns (ID = 7.5 mm, L = 300 mm, particle size = 5 μm). Signals were recorded by a UV detector (Agilent 1200 series), an Optilab REX interferometric refractometer, and a miniDawn TREOS light scattering detector (Wyatt Technology Corp.). Samples were run using THF with 250 ppm of BHT as the eluent at 30 °C and a flow rate of 1.0 mL min−1. Data were analyzed on Astra software (Wyatt Technology Corp.) and molecular weights were determined based on narrow molecular weight polystyrene calibration (from 2340 to 275![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1). 0.3 mL samples from a polymerization reaction were passed through neutral aluminum oxide columns in Pasteur pipettes. The columns were additionally rinsed with 3 mL THF (stabilized by 250 ppm BHT). Then, the combined filtrates were dried by the addition of MgSO4 and filtered through Macherey-Nagel Chromafil PTFE disposable syringe filters of 0.2 μm pore diameter O-20/15 MS.
300 g mol−1). 0.3 mL samples from a polymerization reaction were passed through neutral aluminum oxide columns in Pasteur pipettes. The columns were additionally rinsed with 3 mL THF (stabilized by 250 ppm BHT). Then, the combined filtrates were dried by the addition of MgSO4 and filtered through Macherey-Nagel Chromafil PTFE disposable syringe filters of 0.2 μm pore diameter O-20/15 MS.
Electrospray Ionization Mass Spectrometry (ESI-MS) was recorded on a Bruker – Ion Trap MS Esquire HCT in positive mode from 300 to 1000 m/z. Data were analyzed with DataAnalysis software (Bruker Esquire HCT). Samples were prepared by dissolution of pure product in methanol (HPLC grade) in order to reach a concentration between 0.1 to 1 mg mL−1, followed by filtration through a Macherey-Nagel Chromafil PTFE disposable syringe filter of 0.2 μm pore diameter O-20/15 MS.
Ultraviolet–visible (UV-Vis) spectra were recorded on a Specord 50 Plus spectrophotometer (Analytik Jena). Extinction coefficient of chlorophyllin was calculated from Beer–Lambert law by measurements of absorbance at 405 nm of chlorophyllin at the following concentrations: 0.025 mg mL−1, 0.020 mg mL−1, 0.015 mg mL−1, 0.010 mg mL−1, 0.005 mg mL−1, 0.001 mg mL−1.
Results and discussion
Influence of the solvent, pH and initiator on the copolymerization of PEGA with chlorophyllin
To determine a good reaction medium for the polymerizations, activators generated by electron transfer (AGET) ATRP of PEGA with chlorophyllin were conducted in phosphate–citrate buffers with pH values ranging between 2 and 8, as well as in water and in a mixture of water and 20% DMF (v/v). These initial polymerizations were performed with 16.5 mg mL−1 of copper chlorophyllin and a 62.5:1:1:1 ratio of PEGA to the initiator HEBIB, copper chlorophyllin, and the reducing agent NaAsc. No additional salts were added to these polymerizations. Conversions of PEGA in the buffers were above 70% after 24 h, and were higher at acidic pH (Fig. S1†). Number average molecular weights (Mn) of the polymers were between 16000 and 21000 g mol−1. Dispersities (Đ = Mw/Mn) varied from 1.37 to 1.65 and were lower at acidic pH. Polymerizations in water and the mixture of water and 20% DMF (v/v) resulted in similar conversions (64% and 87%, respectively), lower number average molecular weights (8600 g mol−1 and 12600 g mol−1, respectively), and higher dispersities (1.75 and 1.89, respectively) in comparison to the ones obtained in buffer. Based off the results of these initial polymerizations, buffer at pH 4 was chosen for further experiments, unless otherwise noted.
An optimal ratio of NaAsc to chlorophyllin was found by varying the concentration of NaAsc (Table S1†). Reactions without NaAsc did not yield polymer. In contrast, reactions carried out with one or more equivalents of the reducing agent (with respect to the catalyst) yielded polymers. Their dispersities varied from 1.45 to 1.92 and was the lowest at a 1:1 ratio of NaAsc to chlorophyllin. An increase of the ratio to 2.5:1 and 5:1 did not influence conversion significantly, but resulted in higher dispersities of 1.92 and 1.74. Thus, the ratio of 1:1 was chosen for further experiments.
The results of the reaction without NaAsc are a first indication that the polymerization occurs via an AGET ATRP-like mechanism, and that chlorophyllin needs to be reduced to its active form at the beginning of the reaction (Scheme 1). This agrees with the fact that chlorophyllin is a chlorin type of metalloporphyrin with copper in the +2 oxidation state.
A kinetic experiment at a concentration of 12.3 mg mL−1 of chlorophyllin and 62.5:1:1:1 ratio of PEGA to the initiator, HEBIB, chlorophyllin, and NaAsc revealed the character of the polymerization. Monomer conversion reached 68% within 50 h (Fig. 1). The semi-logarithmic plot, ln([M]0/[M]) vs. time, is linear, which indicates that the polymerization followed first order kinetics, and that the radical concentration was constant. The GPC elugrams show monomodal polymer peaks with slight tailing towards lower molecular weights. The peaks shift to higher molecular weights with increased conversion. Polystyrene-apparent Mn increased with conversion during the whole reaction. The values were higher than the theoretical molecular weights (Mth) during the initial 25 h of the polymerization. After 50 h of the reaction, Mn values were lower than Mth. Dispersities broadened from 1.09 at the beginning of the reaction to 1.40 within the next 6 h and reached 1.77 after 25 h and 1.67 after 50 h. This data illustrates that the polymerization follows an ATRP mechanism and is controlled, even though the degree of control is not perfect.
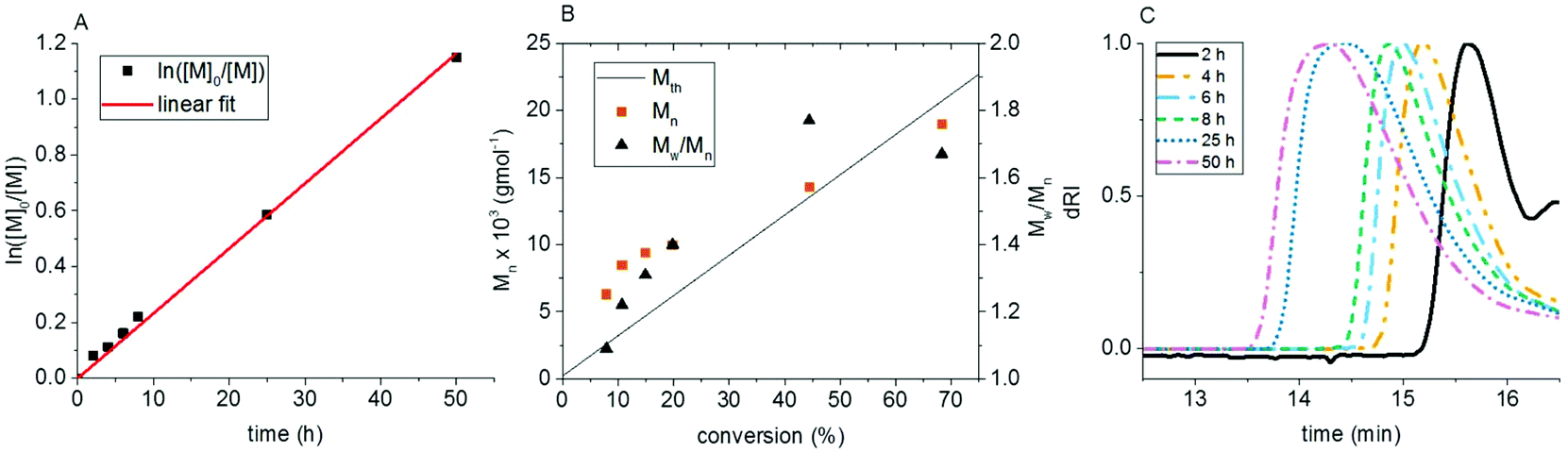 | ||
| Fig. 1 Kinetic data for the CuChln-catalyzed copolymerization of PEGA and CuChln with HEBIB as the initiator in the absence of NaBr. (A) ln([M]0/[M]) vs. time, (B) molecular weight and dispersity vs. conversion, (C) GPC elugrams. Reaction conditions: Ratio PEGA:HEBIB:CuChln:NaAsc: 62:1:1:1; solvent: phosphate–citrate buffer (pH 4). | ||
In order to increase the control over the polymerization and to obtain polymers with narrower dispersities, sodium bromide was added to the reaction. Monomer conversion reached 54% within 50 h (Fig. 2). The semi-logarithmic plot, ln([M]0/[M]) vs. time, is linear for the first 25 h of the polymerization. At longer reaction times, the polymerization slowed down and the log value stayed below the linear fit, indicating that termination reactions occurred. The GPC elugrams show monomodal distributions of the polymer peaks. The peaks shift to higher molecular weights with increasing conversion for 25 h, after which the polymer chain growth stops. The polymer peaks also show slight tailing towards lower molecular weights. Polystyrene-apparent Mn increased linearly with conversion during the whole reaction and was higher than the theoretical molecular weight during the first 8 h of the polymerization. After 25 h and 50 h of the reaction, Mn was lower than Mth. Dispersities broadened from 1.07 at the beginning of the reaction to 1.38 after 25 h. The reaction proceeds in a more controlled fashion, and a bit slower than the polymerization in the absence of sodium bromide, which is typical for the addition of halide salts to aqueous ATRP reactions.80 A Mn that is higher than Mth in the beginning of the polymerization and the tailing of the polymer peaks at lower molecular weights could suggest slow initiation. However, the logarithmic kinetic plot is linear from the start of the reaction, thus slow initiation is unlikeley.81 This could mean, however, that transfer of the active propagating species took place. Since the average molecular weight increased linearly until the end of the reaction, termination by recombination is not the main cause for the irreversible stop of chain growth after the 25 h of the polymerization.81 These data suggest that the termination occurred through chain transfer.
 | ||
| Fig. 2 Kinetic data for the CuChln-catalyzed copolymerization of PEGA and CuChln with HEBIB as the initiator in the presence of NaBr. (A) ln([M]0/[M]) vs. time, (B) molecular weight and dispersity vs. conversion, (C) GPC elugrams. Reaction conditions: Ratio PEGA:HEBIB:CuChln:NaAsc: 62:1:1:1; solvent: phosphate–citrate buffer (pH 4) + NaBr. | ||
HEBIB is not an ideal initiator for aqueous polymerizations even though it is often used in aqueous ATRP. Although it contains a hydroxyl group in its structure, HEBIB forms dispersions in water at high concentrations in the absence of co-solvents. To circumvent this potential problem, an initiator bearing a short PEG chain was used in further experiments. Poly(ethylene glycol) methyl ether 2-bromoisobutyrate (PEG-Br; Mn = 600 g mol−1) is not only well soluble in the reaction mixture, but it is also miscible with the monomer due to its structural similarity. Polymerizations at pH 4 and 8 were performed. The polymerization at pH 8 resulted in 64% conversion after 4 h, giving a copolymer with chlorophyllin of Mn = 5470 g mol−1 and Đ = 1.18. Unfortunately, after 8 h reaction time, the copolymer gelled, and it was not possible to characterize it further. At pH 4 the polymerization followed first order kinetics and did not gel. The logarithmic plot, ln([M]0/[M]) vs. time, is linear for 50 h (Fig. 3). GPC elugrams show monomodal distributions of molecular weights of the polymers that shift towards higher molecular weight values with time. The peaks slightly tail towards lower molecular weights. Mn grew constantly with conversion of PEGA and dispersities remained below 1.3. The molecular weight is higher than Mth at the beginning of the reaction and falls below it after 25 h of polymerization. The use of PEG-Br as initiator resulted in slower polymerizations and polymers with lower dispersities than the polymerizations initiated by HEBIB.
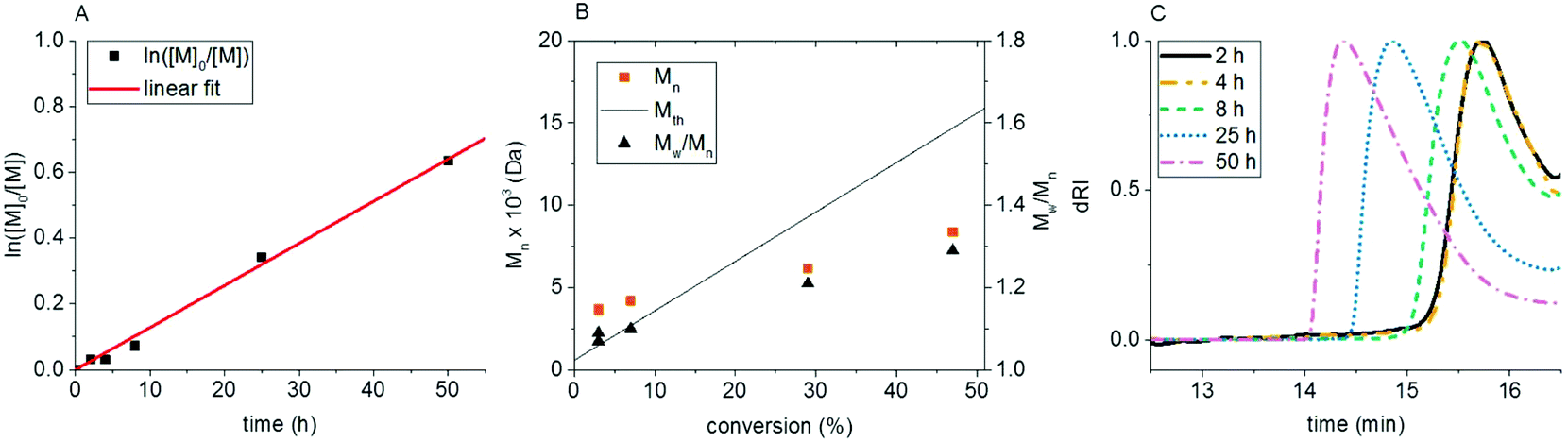 | ||
| Fig. 3 Kinetic data for the CuChln-catalyzed copolymerization of PEGA and CuChln with PEG-Br as the initiator in the presence of NaBr. (A) ln([M]0/[M]) vs. time, (B) molecular weight and dispersity vs. conversion, (C) GPC elugrams. Reaction conditions: Ratio PEGA:PEG-Br:CuChln:NaAsc: 62:1:1:1; solvent: phosphate–citrate buffer (pH 4) + NaBr. | ||
As mentioned before, the role of NaAsc is to reduce chlorophyllin. The fact that polymerizations do not proceed without NaAsc suggests that it has to activate the catalyst i.e. to reduce chlorophyllin, before the catalyst can take part in the ATRP equilibrium. Moreover, reducing agents can regenerate activating species during a polymerization, e.g. in an ARGET ATRP. To asses if the reactions proceed with a higher degree of control when the reducing agent is added throughout the reaction, a polymerization was carried out for 80 h with the addition of NaAsc every 24 h (Fig. S5†). The polymerization proceeded with first order kinetics during the entire experiment. GPC elugrams showed monomodal distributions of molecular weights of the polymers and a shift towards higher molecular weights during the first 24 h. Polymer peaks also showed slight tailing towards lower molecular weights. At longer reaction times, i.e. after addition of an additional equivalent of NaAsc (in ratio to the catalyst), the polymers did not proceed to grow. Instead, smaller chains formed that increased the intensity of the shouldering in the elugram. As a result, the average molecular weight of the polymer sample eventually decreased. Thus, we conclude that chain transfer led to the initiation of new chains and terminated existing ones at longer reaction times.
In order to further characterize the polymers that formed during chlorophyllin-catalyzed polymerizations, PEGA was polymerized at pH 4 (ratio 62.5:1:1:1 PEGA:PEG-Br:chlorophyllin:NaAsc; c(chlorophyllin) = 12.3 mg mL−1). After the reaction had proceeded to 47% conversion, the polymer was purified by precipitation. A green product with Mn = 3890 g mol−1 and Đ = 1.15 was obtained. The UV-Vis spectrum of the product showed absorption bands at 405 nm and 627 nm that are typical for chlorophyllin (Fig. 4). Thus, the purified polymer contained chlorophyllin. The concentration of chlorophyllin in the copolymer was 1.40 ± 0.06 wt% according to UV-Vis spectroscopy and 2.37 ± 0.04 wt% according to ICP-OES measurements (Table S2†). Most likely, the out-of-plane vinyl bond located on the porphyrin macrocycle reacted with the polymer radicals, so that chlorophyllin not only performed as a catalyst, but also acted as a comonomer during the polymerization. Provided that the conversion of chlorophyllin was as high as the monomer conversion, i.e. 47%, the ICP-OES result corresponds to the theoretical content of chlorophyllin in the polymer (2.21 wt%).
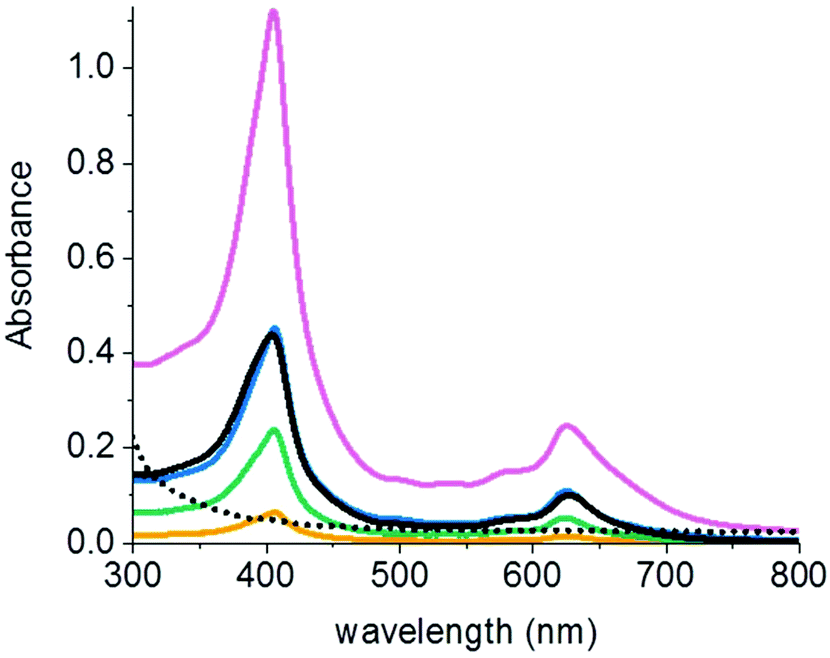 | ||
| Fig. 4 UV-vis spectra of polymers synthesized by CuChln and hydrated CuCe6 catalyzed ATRP of PEGA. Products of CuChln-catalyzed copolymerization of PEGA with (—) 1 equiv. of chlorophyllin (c(polymer) = 0.1 mg mL−1), (—) 2 equiv. of chlorophyllin (c(polymer) = 0.1 mg mL−1), (—) 5 equiv. of chlorophyllin (c(polymer) = 0.1 mg mL−1), (—) 10 equiv. of chlorophyllin (c(polymer) = 0.151 mg mL−1). (…) product of polymerization of PEGA catalyzed by 1 equiv. of hydrated CuCe6 (c(polymer) = 0.057 mg mL−1). For comparison, the spectrum of a 0.010 mg mL−1 solution of CuChln is also shown (—). Reaction conditions of the polymerizations: Ratio PEGA:PEG-Br:CuChln:NaAsc: 62:1:1:1; solvent: phosphate–citrate buffer (pH 4) + NaBr; reaction time 50 h. The polymers were purified from the reaction mixtures by precipitation and redissolved in water for the spectroscopic measurements. | ||
Control over the content of chlorophyllin in poly(PEGA-co-chlorophyllin) copolymers
As outlined in the introduction, polymers that contain chlorophyll derivatives are useful for a variety of applications. We therefore tested if the content of chlorophyllin in the copolymer could be controlled by changing the ratio of chlorophyllin to initiator from 1: 2, 5 and 10 equivalents. The polymerizations were conducted as described above. The polymers were purified from unreacted monomers and chlorophyllin by precipitation and characterized by GPC, UV-vis spectroscopy, and ICP-OES (Table S2† and Fig. 4). The polymerization with 2 equivalents of chlorophyllin resulted in a copolymer with a chlorophyllin content of 4.4 wt% (UV-Vis spectroscopy) and 7.8 wt% (ICP-OES). These values are in agreement with the theoretical content of chlorophyllin (4.47 wt%), assuming the same conversion for monomer and chlorophyllin. 5 equivalents of the catalyst yielded a copolymer that contained 9.6 wt% chlorophyllin according to UV-Vis spectroscopy and 16.5 wt% according to ICP-OES. The theoretical content of chlorophyllin was 10.5 wt%. When the concentration of chlorophyllin in the reaction mixture was increased to 10 equivalents, the precipitated copolymer contained 15.9 wt% chlorophyllin according to UV-Vis spectroscopy, whereas ICP-OES measured 43.1 wt%. It was not possible to calculate the theoretical content of chlorophyllin as the high concentration of Cu resulted in broadened 1H NMR spectra, due to the paramagnetic nature of metal, and therefore made it impossible to accurately determine conversion. The data revealed a clear correlation between the concentration of the catalyst in the reaction mixture and the content of chlorophyllin in the purified copolymers. Thus, polymers with a defined content of chlorophyllin can be prepared by tailoring the chlorophyllin to initiator ratio.A higher chlorophyllin concentration in the reaction mixture should also manifest itself in the rate of the reaction. Indeed, when 2 equivalents of chlorophyllin were used, the polymerization was significantly faster than with 1 equivalent of chlorophyllin (Fig. S2†). The logarithmic plot, ln([M]0/[M]) vs. time, was linear during the first 25 h of the polymerization and the reaction stopped at approx. 50% conversion. The polymerization carried out with 5 equivalents of chlorophyllin was even faster (Fig. S3†). 60% monomer conversion was observed within only 8 h, after which the polymerization slowed down and stopped. In both reactions, the polymer chains grew with conversion, and the polymer peaks in the GPC elugrams shifted to shorter retention times. Increase of the chlorophyllin concentration did not only increase the speed of the polymerization, but faster polymerizations also resulted in slightly broader dispersities of the obtained copolymers. Nevertheless, the molecular weight distributions are still relatively narrow, especially when compared to the molecular weight distribution of a polymer obtained by the free radical copolymerization of PEGA in the presence of chlorophyllin (Fig. S6†).
It was not possible to measure conversions of reactions that were carried out with 10 equivalents of chlorophyllin because the copper content in the sample was too high for quantitative 1H NMR measurements. The GPC elugrams showed monomodal, but relatively broad distributions of molecular weights that only shift slightly towards higher molecular weights with increased time (Fig. S4†).
These experiments show that the content of chlorophyllin in the copolymers scales with the concentration of chlorophyllin in the reaction mixture and that the copolymers can be synthesized via a reversible-deactivation radical polymerization using chlorophyllin simultaneously as a catalyst and comonomer.
Kinetic study of polymerizations catalyzed by hydrated chlorin e6 copper(II)
If the aim of a chlorophyllin-catalyzed polymerization is not to obtain copper chlorophyllin-containing copolymers, but to synthesize homopolymers, the out-of-plane vinyl bond of chlorophyllin has to be modified so that it cannot undergo radical attack. To test this, chlorine e6 (Ce6), the main porphyrin of chlorophyllin, was hydrated by an addition reaction of HBr to the double bond, followed by nucleophilic substitution of the bromine with water (Scheme S1†). The material was characterized and confirmed by 1H NMR spectroscopy and ESI-MS (Fig. S7–S9†). Then, Cu(II) was inserted into the modified chlorine, yielding hydrated CuCe6. The resulting copper chlorophyllin derivative was used to catalyze the ATRP of PEGA. When carried out at pH 4, the reaction yielded only a 7% monomer conversion within 24 h (Mn = 4350 g mol−1, Mw/Mn = 1.07) and the molecular weight of the polymer did not increase with reaction time (Fig. S10†). Thus, polymerizations were performed at pH 8, where the reaction was faster and resulted in a 16% monomer conversion after 24 h (Mn = 4290 g mol−1, Mw/Mn = 1.11). Within the first 8 h, the reaction followed first order kinetics as indicated by a linear ln([M]0/[M]) vs. time plot (Fig. 5). GPC elugrams showed monomodal and symmetric distributions of molecular weights of the polymers. The molecular weight increased with conversion. Tailing of the peaks towards lower molecular weights were less pronounced than in copolymerization reactions. Another difference to the polymerizations with unmodified chlorophyllin is that the polymerization is drastically slower with the hydrated chlorophyllin. | ||
| Fig. 5 Kinetic data for the polymerization of PEGA catalyzed by hydrated CuCe6 at pH 8 with PEG-Br as the initiator. (A) ln([M]0/[M]) vs. time, (B) molecular weight and dispersity versus conversion, (C) GPC elugrams. Reaction conditions: Ratio PEGA:PEG-Br:hydrated CuCe6:NaAsc: 62:1:1:1; solvent: phosphate–citrate buffer (pH 8) + NaBr. | ||
In order to show that the hydrated chlorophyllin did not copolymerize, a polymer was purified after 50 h by precipitation in hexane. The UV-Vis spectrum of the precipitated polymer does not contain absorbance bands at the characteristic wavelengths of copper or porphyrin (Fig. 4). Precipitated poly(PEGA) was also digested in 10% nitric acid and analyzed by ICP-OES for the presence of copper. The signal intensity of copper in the precipitated polymer was lower than the standard error of the copper calibration. Thus, it can be assumed that the product is copper free. As the precipitated poly(PEGA) was free of porphyrin and copper, the modification of the vinyl bond prevented the copolymerization of chlorophyllin into the product.
Stability of chlorophyllin during copolymerization
Some porphyrins, like hemes, are sensitive to oxidative cleavage of the macrocycle in the presence of reductants such as ascorbate.82 Thus it is essential to assess the stability of chlorophyllin in order to understand its fate during these polymerization conditions. The first indication for the stability of the catalysts is that the reaction solutions remained the same green color during the entirety of the polymerization, as observed by naked eye. Moreover, the UV-Vis spectra of copper chlorophyllin and of hydrated CuCe6 were recorded during polymerizations (Fig. 6 and S11†). There were no hypso- or bathochromic shifts of the Q-bands for both catalysts. The maximum of the Q-band stayed at 627 nm in the case of chlorophyllin and 623 nm in the case of its modified analog. This absorption band is closely related to the oxidation state of copper in the porphyrin and the complexation of the metal center. A shift of that peak to longer wavelengths would indicate demetalation of the complex or a change of the oxidation state of copper. The Soret band remained at 405 nm, which indicates that the porphyrin ring of the catalyst is stable during the polymerization. The intensity of the entire spectra decreases slightly within the time of the experiment. This could be explained by partial photodegradation of chlorophyllin.50,51 In the UV region, absorbance intensity at 265 nm decreased. As the monomer and citrate, which is present in the buffer, absorb in this spectral range,83 the reaction of chlorophyllin with NaAsc was investigated in pure water, i.e. in buffer free and monomer free conditions (Fig. 6 and S11†). Again, the Q- and the Soret bands did not change over time. A peak with a maximum at 265 nm decreased. This absorption band arises from NaAsc. Its decrease shows that NaAsc oxidizes in the presence of copper chlorophyllin. In contrast, in the absence of the catalyst the peak of NaAsc is stable (Fig. S12 and S13†). Thus, sodium ascorbate reduces copper chlorophyllin. As the absorption band that arises from the metal center does not change, the reduction happens on the porphyrin ring. This reduction is speculated to be necessary for the catalysis of the polymerization, i.e. it produces the activating species of the catalyst. | ||
| Fig. 6 Development of UV-vis spectra (A) during the CuChln-catalyzed copolymerization of PEGA with chlorophyllin at pH 4, (B) during the polymerization of PEGA catalyzed by hydrated CuCe6 at pH 8, and (C) during the reaction of NaAsc with chlorophyllin in water (every 4 spectra from the original 96 depicted). The arrows indicate the direction of the evolution of the spectra. | ||
Conclusions
Although a lot of emphasis is placed on screening various metalloenzymes to catalyze ATRP, there remains the opportunity to explore other naturally occurring ATRP catalysts. Here we report that copper chlorophyllin, a stable derivative of chlorophyll, can catalyze ATRP. Chlorophyllin acts as both a catalyst and a comonomer through its out-of-plane vinyl bond, enabling access to chlorophyllin-containing copolymers with well-defined molecular weights, narrow molecular weight distributions, and a tunable chlorophyllin content. Such copolymers could find application in biomedical and drug delivery applications, or as light-harvesting materials. Modification of the vinyl bond of chlorin e6, one of the major components of chlorophyllin, followed by the insertion of copper into its porphyrin ring lead to a bio-derived catalyst that can catalyze ATRP without copolymerizing with the monomer. Plant-derived metalloporphyrin complexes are exceptional “green” catalysts and functional co-monomers, because they are abundantly produced in nature, predominantly in green plants. In contrast to ATRP-active enzymes like hemoglobin, chlorophyllin is not extracted from animals and therefore meets requirements for the vegan production of polymers that might be used in food and cosmetics applications. Nevertheless, in order to judge the environmental impact of such reactions, the whole process, including the reagents used for the modification of chlorophyll, the monomers, and the solvents used will have to be holistically assessed.84Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support by the Swiss National Science Foundation (projects PP00P2_144697 and PP00P2_172927). The authors thank Prof. Dr Stephan Hörtensteiner (University of Zürich) for good advice and help in modification of the vinyl bond in chlorin e6, Dr Diederik Balkenende and Mark Karman for perfectly operating the GPC, Dr Laetitia Haeni for ICP-OES measurements, and Wojciech Gajewski for the TOC images.Notes and references
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 2603–2614 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- K. J. Rodriguez, B. Gajewska, J. Pollard, M. M. Pellizzoni, C. Fodor and N. Bruns, ACS Macro Lett., 2018, 7, 1111–1119 CrossRef CAS.
- J. S. Wallace and C. J. Morrow, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2553–2567 CrossRef CAS.
- O. Emery, T. Lalot, M. Brigodiot and E. Marechal, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3331–3333 CrossRef CAS.
- D. Teixeira, T. Lalot, M. Brigodiot and E. Marechal, Macromolecules, 1999, 32, 70–72 CrossRef CAS.
- A. Singh, S. Roy, L. Samuelson, F. Bruno, R. Nagarajan, J. Kumar, V. John and D. L. Kaplan, J. Macromol. Sci., Pure Appl. Chem., 2001, A38, 1219–1230 CrossRef CAS.
- A. Singh, D. Ma and D. L. Kaplan, Biomacromolecules, 2000, 1, 592–596 CrossRef CAS.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS.
- F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793 CrossRef.
- S.-i. Shoda, H. Uyama, J.-i. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS.
- Y.-H. Ng, F. di Lena and C. L. L. Chai, Polym. Chem., 2011, 2, 589–594 RSC.
- Y.-H. Ng, F. di Lena and C. L. L. Chai, Chem. Commun., 2011, 47, 6464–6466 RSC.
- G. Gao, M. A. Karaaslan, J. F. Kadla and F. Ko, Green Chem., 2014, 16, 3890–3898 RSC.
- M. V. Dinu, M. Spulber, K. Renggli, D. Wu, C. A. Monnier, A. Petri-Fink and N. Bruns, Macromol. Rapid Commun., 2015, 36, 507–514 CrossRef CAS.
- K. Renggli, N. Sauter, M. Rother, M. G. Nussbaumer, R. Urbani, T. Pfohl and N. Bruns, Polym. Chem., 2017, 8, 2133–2136 RSC.
- L. P. Kreuzer, M. J. Männel, J. Schubert, R. P. M. Höller and M. Chanana, ACS Omega, 2017, 2, 7305–7312 CrossRef CAS.
- C. Fodor, B. Gajewska, O. Rifaie-Graham, E. A. Apebende, J. Pollard and N. Bruns, Polym. Chem., 2016, 7, 6617–6625 RSC.
- T. B. Silva, M. Spulber, M. K. Kocik, F. Seidi, H. Charan, M. Rother, S. J. Sigg, K. Renggli, G. Kali and N. Bruns, Biomacromolecules, 2013, 14, 2703–2712 CrossRef CAS.
- M. Divandari, J. Pollard, E. Dehghani, N. Bruns and E. M. Benetti, Biomacromolecules, 2017, 18, 4261–4270 CrossRef CAS PubMed.
- Y. Sun, H. Du, Y. Lan, W. Wang, Y. Liang, C. Feng and M. Yang, Biosens. Bioelectron., 2016, 77, 894–900 CrossRef CAS.
- Y. Sun, J. Zhang, J. Li, M. Zhao and Y. Liu, RSC Adv., 2017, 7, 28461–28468 RSC.
- A. Simakova, M. Mackenzie, S. E. Averick, S. Park and K. Matyjaszewski, Angew. Chem., Int. Ed., 2013, 52, 12148–12151 CrossRef CAS.
- K. Yamashita, K. Yamamoto and J.-i. Kadokawa, Polymer, 2013, 54, 1775–1778 CrossRef CAS.
- S. Smolne, M. Buback, S. Demeshko, K. Matyjaszewski, F. Meyer, H. Schroeder and A. Simakova, Macromolecules, 2016, 49, 8088–8097 CrossRef CAS.
- I. Proietti Silvestri and F. Cellesi, Macromol. Chem. Phys., 2015, 216, 2032–2039 CrossRef CAS.
- H. Zhou, W. Jiang, N. An, Q. Zhang, S. Xiang, L. Wang and J. Tang, RSC Adv., 2015, 5, 42728–42735 RSC.
- H. Zhou, X. Wang, J. Tang and Y.-W. Yang, Polymers, 2016, 8, 277 CrossRef.
- L. Fu, A. Simakova, M. Fantin, Y. Wang and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 26–30 CrossRef CAS PubMed.
- B. B. Wayland, G. Poszmik and M. Fryd, Organometallics, 1992, 11, 3534–3542 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943–7944 CrossRef CAS.
- R. M. Islamova and Y. B. Monakov, Polym. Sci., Ser. C, 2011, 53, 27–34 CrossRef CAS.
- R. Islamova, S. Nazarova and O. Koifman, J. Charact. Dev. Novel Mater., 2014, 6, 105–122 CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3659 CrossRef CAS PubMed.
- L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127–156 CrossRef CAS.
- Y.-H. Ng, F. di Lena and C. L. L. Chai, Macromol. Res., 2012, 20, 473–476 CrossRef CAS.
- H. Scheer, Chlorophylls, CRC Press, Inc., 1991 Search PubMed.
- P. Evans, The colours of life: an introduction to the chemistry of porphyrins and related compounds, ed. R. M. Lionel, Harwood Academic Publishers, 1999 Search PubMed.
- A. Hosikian, S. Lim, R. Halim and M. K. Danquah, Int. J. Chem. Eng., 2010, 2010, 11 Search PubMed.
- K. Solymosi and B. Mysliwa-Kurdziel, Mini-Rev. Med. Chem., 2017, 17, 1194–1222 CAS.
- H. K. Lichtenthaler, Applications of chlorophyll fluorescence in photosynthesis research, stress physiology, hydrobiology and remote sensing, Kluwer Academic Publishers, 1988 Search PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS.
- T. Itoh, K. Yano, Y. Inada and Y. Fukushima, J. Am. Chem. Soc., 2002, 124, 13437–13441 CrossRef CAS.
- M. L. Salin, L. M. Alvarez, B. C. Lynn, B. Habulihaz and A. W. Fountain, Free Radical Res., 1999, 31(Suppl), 97–105 CrossRef.
- M. G. Ferruzzi and S. J. Schwartz, J. Agric. Food Chem., 2005, 53, 7098–7102 CrossRef CAS PubMed.
- M. Sato, I. Fujimoto, T. Sakai, T. Aimoto, R. Kimura and T. Murata, Chem. Pharm. Bull., 1986, 34, 2428–2434 CrossRef CAS.
- A. Mortensen and A. Geppel, Innovative Food Sci. Emerging Technol., 2007, 8, 419–425 CrossRef CAS.
- F. Delgado-Vargas and O. Paredes-López, Natural colorants for food and nutraceutical uses, CRC press, 2002 Search PubMed.
- B. D. Berezin, S. V. Rumyantseva, A. P. Moryganov and M. B. Berezin, Russ. Chem. Rev., 2004, 73, 185–194 CrossRef CAS.
- C. Socaciu, Food colorants: chemical and functional properties, CRC Press. ph, Boca Raton, Fla., 2008, pp. 583–601 Search PubMed.
- R. E. Wrolstad and C. A. Culver, Annu. Rev. Food Sci. Technol., 2012, 3, 59–77 CrossRef CAS PubMed.
- M. Shahid and F. Mohammad, J. Cleaner Prod., 2013, 53, 310–331 CrossRef CAS.
- P. Esquivel, Handbook on natural pigments in food and beverages: industrial applications for improving food color, 2016 Search PubMed.
- R. W. Young and J. S. Beregi, J. Am. Geriatr. Soc., 1980, 28, 46–47 CrossRef CAS.
- S. Hidaka, E. Matsumoto and F. Toda, Bull. Chem. Soc. Jpn., 1985, 58, 207–210 CrossRef CAS.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- X.-F. Wang and O. Kitao, Molecules, 2012, 17, 4484–4497 CrossRef CAS PubMed.
- H. Hug, M. Bader, P. Mair and T. Glatzel, Appl. Energy, 2014, 115, 216–225 CrossRef CAS.
- N. A. Ludin, A. A.-A. Mahmoud, A. B. Mohamad, A. A. H. Kadhum, K. Sopian and N. S. A. Karim, Renewable Sustainable Energy Rev., 2014, 31, 386–396 CrossRef CAS.
- Q. Guan, R. Peng, Z. Liu, W. Song, R. Yang, L. Hong, T. Lei, X. Fan, Q. Wei and Z. Ge, J. Mater. Chem. A, 2018, 6, 464–468 RSC.
- P. V. Kamat, J. P. Chauvet and R. W. Fessenden, J. Phys. Chem., 1986, 90, 1389–1394 CrossRef CAS.
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Graetzel and S. Zhang, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- F. Kong, X. Cao, J. Xia and B.-K. Hur, J. Ind. Eng. Chem., 2007, 13, 424–428 CAS.
- W. Wen, J. Wan, X. Cao and J. Xia, Biotechnol. Prog., 2007, 23, 1124–1129 CAS.
- J. Chen, S. Miao, J. Wan, J. Xia and X. Cao, Process Biochem. (Amsterdam, Neth.), 2010, 45, 1928–1936 CAS.
- W. Wang, J. Wan, B. Ning, J. Xia and X. Cao, J. Chromatogr. A, 2008, 1205, 171–176 CrossRef CAS PubMed.
- W. Li, Q. Guo, H. Zhao, L. Zhang, J. Li, J. Gao, W. Qian, B. Li, H. Chen, H. Wang, J. Dai and Y. Guo, Nanomedicine, 2012, 7, 383–392 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- Q. Yan, J. Yuan, Y. Kang, Z. Cai, L. Zhou and Y. Yin, Chem. Commun., 2010, 46, 2781–2783 RSC.
- D. Ma, Q.-M. Lin, L.-M. Zhang, Y.-Y. Liang and W. Xue, Biomaterials, 2014, 35, 4357–4367 CrossRef CAS.
- N. Uri, J. Am. Chem. Soc., 1952, 74, 5808–5809 CrossRef CAS.
- T. McIlvaine, J. Biol. Chem., 1921, 49, 183–186 CAS.
- P. S. Clezy and J. Barrett, Biochem. J., 1961, 78, 798–806 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- P. Krys and K. Matyjaszewski, Eur. Polym. J., 2017, 89, 482–523 CrossRef CAS.
- K. M. Smith, Porphyrins and Metalloporphyrins: A New Edition Based on the Original Volume by J. E. Falk, Elsevier, 1975 Search PubMed.
- S. Krukowski, M. Karasiewicz and W. Kolodziejski, J. Food Drug Anal., 2017, 25, 717–722 CrossRef CAS.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/c8py01492b |
| This journal is © The Royal Society of Chemistry 2019 |