
Selective synthesis of pyrrolidin-2-ones and 3-iodopyrroles via the ring contraction and deformylative functionalization of piperidine derivatives†
Fang
Wang
,
Xinying
Zhang
 ,
Yan
He
and
Xuesen
Fan
*
,
Yan
He
and
Xuesen
Fan
*
Henan Key Laboratory of Organic Functional Molecule and Drug Innovation, Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Environment, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China. E-mail: xuesen.fan@htu.cn
First published on 4th December 2018
Abstract
In this paper, a selective synthesis of pyrrolidin-2-ones and 3-iodopyrroles via the cascade reactions of N-substituted piperidines is presented. Mechanistically, the formation of pyrrolidin-2-ones involves a domino process including the in situ formation of pyrrolidine-2-carbaldehyde followed by carboxylic acid formation, decarboxylation and ipso-oxidation. On the other hand, 3-iodopyrroles are believed to be formed via the initial generation of pyrrolidine-2-carbaldehyde followed by carboxylic acid formation, decarboxylation, dehydrogenation, iodination and aromatization. Interestingly, either pyrrolidin-2-ones or 3-iodopyrroles could be obtained selectively from the same substrates, and the selectivity was easily tuned by using a specific oxidant and additive.
Introduction
Pyrrolidin-2-ones (γ-lactams) are common structural motifs widely encountered in natural products and synthetic compounds possessing potent biological activities and diverse functional properties (Fig. 1).1,2 In addition, pyrrolidin-2-ones are routinely used as versatile synthons in organic synthesis due to their inherently rich reactivity.1,3 Owing to their importance, a number of synthetic approaches toward pyrrolidin-2-ones have been established, which mainly included: (1) amination and cyclization of functionalized acyclic substrates,1,4 (2) oxidation of pyrrolidine derivatives,5 (3) ring expansion of β-lactams6 or cyclopropylamides,7etc.8 Meanwhile, iodo-substituted pyrroles are highly valuable intermediates in the synthesis of drugs, dyes, pigments and other fine chemicals.9 Traditionally, iodopyrroles are accessed either by electrophilic iodination of the existing pyrrole unit10 or through electrophilic iodocyclization of functionalized acyclic substrates.11 While these above-mentioned synthetic methods toward pyrrolidin-2-one and iodopyrrole derivatives are generally efficient and reliable, some of them suffer from poor regioselectivity, require multi-step preparation of the highly functionalized substrates, or employ expensive transition-metal catalysts. Therefore, the development of more selective and easy-to-run methods starting from simple and readily obtainable substrates without using precious metal catalysts is highly desirable.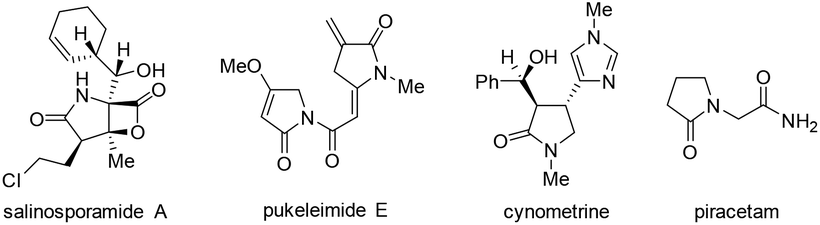 | ||
| Fig. 1 Some important pyrrolidin-2-one derivatives. | ||
Recently, direct functionalization of the inert C(sp3)–H bond has attracted much attention owing to its elimination of substrate pre-activation and minimization of by-product formation. In particular, C(sp3)–H bond functionalization of inactivated cyclic amines is a highly promising strategy in the preparation of diversely functionalized N-heterocyclic compounds.12,13 In this regard, we have recently disclosed a novel synthesis of 1-phenylpyrrolidine-2-carbaldehyde (A, Scheme 1, (1)) through the oxidative ring contraction of 1-phenylpiperidine (1a) under the promotion of a combination of Cu(OAc)2 with O2 and KI.13b During this study, we have been trying to use Oxone14 as a co-oxidant or DMAP as a co-additive in order to improve the yield of A. To our surprise, in the presence of Oxone or DMAP, the reaction of 1a mainly afforded 1-phenylpyrrolidin-2-one (2a, Scheme 1, (2)) or 3-iodo-1-phenyl-1H-pyrrole (3a, Scheme 1, (3)) instead of the expected A.
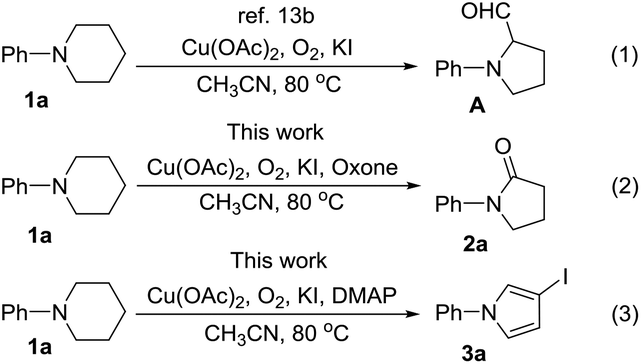 | ||
| Scheme 1 Transformations of 1-phenylpiperidine under different conditions. | ||
Notwithstanding that we did not obtain our initially expected results, it occurred to us that the facile formation of pyrrolidin-2-ones and 3-iodopyrroles directly from piperidine is synthetically significant. This is not only owing to the importance of pyrrolidin-2-one and 3-iodopyrrole derivatives, but also due to the shortage of selective and sustainable synthetic methods for their preparation. Under this circumstance, we decided to carry out a systematic study of these serendipitously found transformations with the aim to develop them into efficient and reliable synthetic methods toward pyrrolidin-2-ones and 3-iodopyrroles. Herein we report the detailed results we obtained on this aspect.
Results and discussion
At the outset of our investigation, the reaction conditions for the formation of pyrrolidin-2-ones 2 were optimized by using 1-phenylpiperidine (1a) as the model substrate. Initially, 1a was treated with Cu(OAc)2 (1 equiv.), KI (1 equiv.) and Oxone (1 equiv.) in CH3CN under O2 at 80 °C for 12 h. From this reaction, 1-phenyl pyrrolidin-2-one (2a) was obtained in 31% yield (Table 1, entry 1). Increasing the loading of Cu(OAc)2 from 1 equiv. to 2 equiv. could improve the yield of 2a slightly (entry 2). On the other hand, reducing it from 1 equiv. to 0.5 equiv. diminished the reaction efficiency dramatically (entry 3). Our study on the suitable loading of Oxone showed that the reaction using 2 equiv. of Oxone gave the best yield of 2a (entry 4 vs. 1 and 5). Next, CuSO4, CuBr2 and CuCl2 were tried to replace Cu(OAc)2. However, they were found to be much less effective compared to Cu(OAc)2 (entries 6–8 vs. 4). When K2S2O8, TEMPO or TBHP was used to replace Oxone as the co-oxidant, the yield of 2a decreased significantly (entries 9–11). Next, screening of different solvents including DMF, DCE and dioxane revealed that CH3CN is the most efficient in mediating this reaction (entries 12–14 vs. 4). Delightfully, increasing the loading of Cu(OAc)2 from 1 equiv. to 2 equiv. in the presence of 2 equiv. of Oxone could further improve the yield of 2a (entry 15). When the loading of KI was also increased to 2 equiv., 2a could be obtained in a yield of 58% (entry 16). Finally, control experiments showed that in the absence of either Cu(OAc)2 or Oxone the formation of 2a was not observed (entries 17 and 18).|
|
||||
|---|---|---|---|---|
| Entry | Cu(II) salt (eq.) | Co-oxidant (eq.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1 (0.5 mmol), KI (0.5 mmol), solvent (5 mL), 80 °C, O2 (1 atm, balloon), 12 h. b Isolated yield. c KI (1 mmol). | ||||
| 1 | Cu(OAc)2 (1) | Oxone (1) | CH3CN | 31 |
| 2 | Cu(OAc)2 (2) | Oxone (1) | CH3CN | 32 |
| 3 | Cu(OAc)2 (0.5) | Oxone (1) | CH3CN | 23 |
| 4 | Cu(OAc)2 (1) | Oxone (2) | CH3CN | 50 |
| 5 | Cu(OAc)2 (1) | Oxone (3) | CH3CN | 41 |
| 6 | CuSO4 (1) | Oxone (2) | CH3CN | Trace |
| 7 | CuBr2 (1) | Oxone (2) | CH3CN | 20 |
| 8 | CuCl2 (1) | Oxone (2) | CH3CN | 21 |
| 9 | Cu(OAc)2 (1) | K2S2O8 (2) | CH3CN | 8 |
| 10 | Cu(OAc)2 (1) | TEMPO (2) | CH3CN | Trace |
| 11 | Cu(OAc)2 (1) | TBHP (2) | CH3CN | Trace |
| 12 | Cu(OAc)2 (1) | Oxone (2) | DMF | 14 |
| 13 | Cu(OAc)2 (1) | Oxone (2) | DCE | 13 |
| 14 | Cu(OAc)2 (1) | Oxone (2) | Dioxane | 13 |
| 15 | Cu(OAc)2 (2) | Oxone (2) | CH3CN | 53 |
| 16c | Cu(OAc)2 (2) | Oxone (2) | CH3CN | 58 |
| 17c | — | Oxone (2) | CH3CN | — |
| 18c | Cu(OAc)2 (2) | — | CH3CN | — |
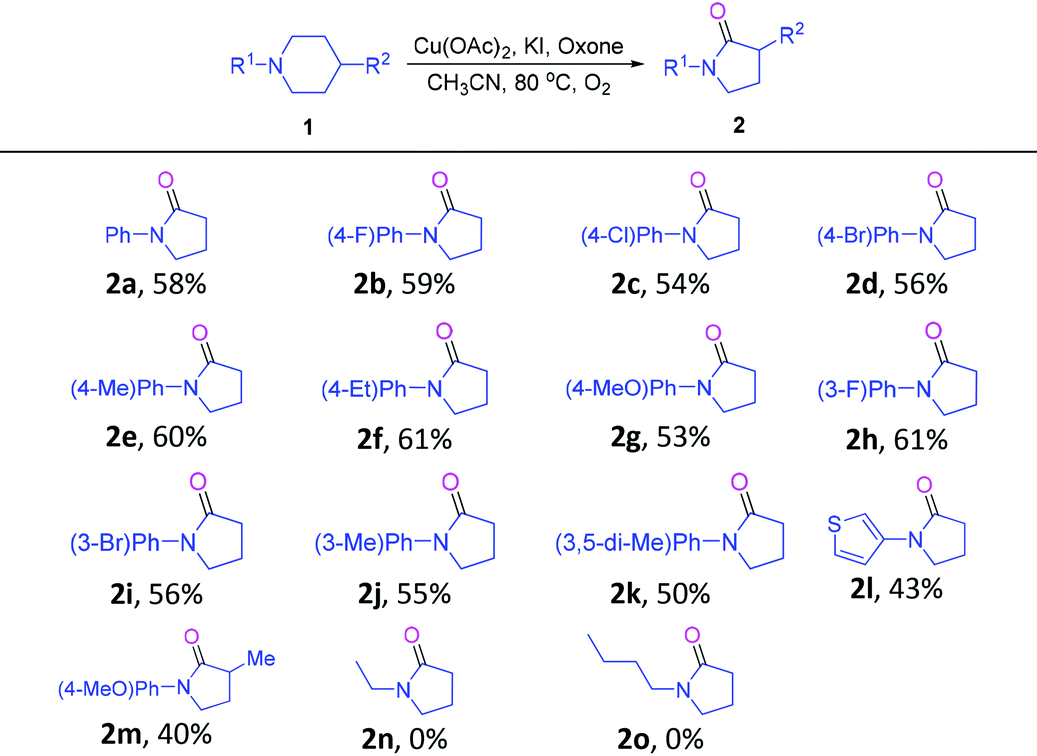
With the optimum conditions established, the scope and generality of this novel transformation was studied, and the results are included in Table 2. First, a range of 1-phenylpiperidines 1 bearing different substituents on the para-position of the phenyl ring underwent this cascade reaction smoothly to give products 2b–2g in yields ranging from 53–61%. Notably, a number of functional groups, from the electron-donating methyl, ethyl and methoxy to the electron-withdrawing fluoro and chloro, were well compatible with the reaction conditions. In addition, substrates with meta substitutions on the phenyl ring were also compatible with this transformation to give 2h–2k. Interestingly, 1-(thiophen-3-yl)-piperidine was a suitable substrate to give 2l. In addition, 1 bearing a methyl group on the para-position of the piperidine ring could also take part in this cascade reaction to give a 1,3-disubstituted pyrrolidin-2-one derivative 2m in moderate yield. Unfortunately, when 1-ethylpiperidine and 1-butylpiperidine were tried as substrates for this reaction, the formation of the desired products (2n, 2o) was not observed.
After having established a convenient synthesis of pyrrolidin-2-ones 2via the cascade reactions of 1 under the promotion of Cu(OAc)2/KI/Oxone/O2, we then moved forward to search for the suitable reaction conditions for the efficient formation of 3-iodopyrroles 3. Initially, 1a was treated with Cu(OAc)2 (1 equiv.), KI (1 equiv.) and DMAP (0.5 equiv.) in CH3CN under O2 at 80 °C for 10 h. From this reaction, 3-iodo-1-phenyl-1H-pyrrole (3a) was obtained in 30% yield (Table 3, entry 1). To our pleasure, increasing the loading of DMAP resulted in an obvious increase in the yield of 3a (entries 2 and 3). When DMAP was replaced by triethylamine or pyridine, the formation of 3a was not observed (entries 4 and 5). On the other hand, when KI was replaced by I2, the yield of 3a improved (entries 6–8). Next, CuBr2, CuCl2 and CuSO4 were found to be much less effective compared to Cu(OAc)2 (entries 9–11). When DCE, dioxane or DMSO was used as the solvent, the yield of 3a decreased (entries 12–14). Our further optimization showed that increasing the loading of Cu(OAc)2 from 1 equiv. to 2 equiv. could improve the yield of 3a (entry 15). More promisingly, when a combination of 2 equiv. of Cu(OAc)2 and 1 equiv. of I2 was used, 3a could be obtained in a yield of 65% (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cu(II) salt (eq.) | Additive 1 (eq.) | Additive 2 (eq.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), solvent (5 mL), 80 °C, O2 (1 atm, balloon), 10 h. b Isolated yield. | |||||
| 1 | Cu(OAc)2 (1) | KI (1) | DMAP (0.5) | CH3CN | 30 |
| 2 | Cu(OAc)2 (1) | KI (1) | DMAP (1) | CH3CN | 40 |
| 3 | Cu(OAc)2 (1) | KI (1) | DMAP (2) | CH3CN | 39 |
| 4 | Cu(OAc)2 (1) | KI (1) | Et3N (1) | CH3CN | — |
| 5 | Cu(OAc)2 (1) | KI (1) | Pyridine (1) | CH3CN | — |
| 6 | Cu(OAc)2 (1) | I2 (0.5) | DMAP (1) | CH3CN | 51 |
| 7 | Cu(OAc)2 (1) | I2 (0.25) | DMAP (1) | CH3CN | 25 |
| 8 | Cu(OAc)2 (1) | I2 (1) | DMAP (1) | CH3CN | 45 |
| 9 | CuBr2 (1) | I2 (0.5) | DMAP (1) | CH3CN | Trace |
| 10 | CuCl2 (1) | I2 (0.5) | DMAP (1) | CH3CN | Trace |
| 11 | CuSO4 (1) | I2 (0.5) | DMAP (1) | CH3CN | 10 |
| 12 | Cu(OAc)2 (1) | I2 (0.5) | DMAP (1) | DCE | 13 |
| 13 | Cu(OAc)2 (1) | I2 (0.5) | DMAP (1) | Dioxane | 15 |
| 14 | Cu(OAc)2 (1) | I2 (0.5) | DMAP (1) | DMSO | 17 |
| 15 | Cu(OAc)2 (2) | I2 (0.5) | DMAP (1) | CH3CN | 57 |
| 16 | Cu(OAc)2 (2) | I2 (1) | DMAP (1) | CH3CN | 65 |
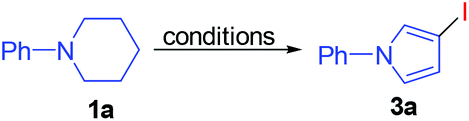
With the optimum reaction conditions in hand, the substrate scope for the preparation of 3-iodopyrroles 3 was explored. The results included in Table 4 demonstrated that 1-phenylpiperidines bearing either the electron-withdrawing or the electron-donating groups on the para-, meta- or ortho-position of the phenyl ring were well suitable for this reaction, affording 3-iodopyrrole derivatives 3b–3l in moderate to good yields. In addition, 1-(3,5-dimethylphenyl)piperidine could also take part in this reaction to give 3m. In another aspect, this reaction proceeded smoothly when the substrate bearing a phenyl group on the para-position of the piperidine ring gave 3-iodo-1,4-diphenyl-1H-pyrrole (3n). It is worth noting that the structure of 3d was unambiguously confirmed by its X-ray single crystal diffraction analysis.15
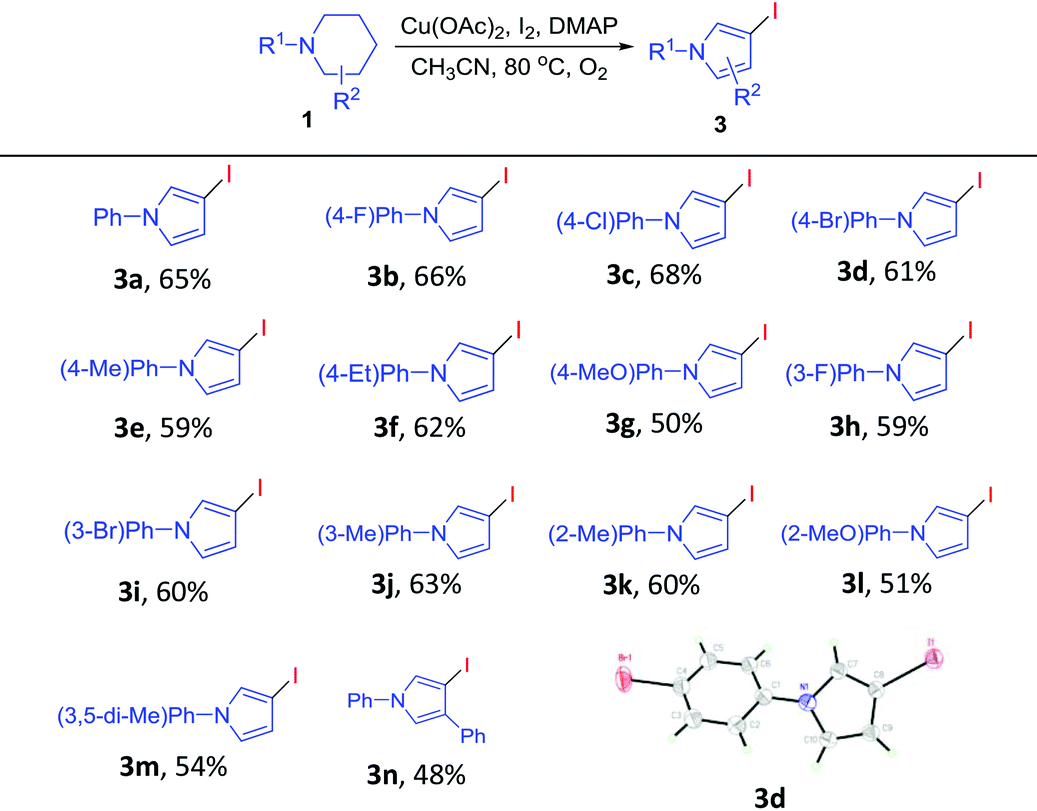
To gain some insight into the reaction mechanism accounting for the formation of 2a, the following control experiments were conducted. First, 1a was subjected to the optimum reaction conditions (Table 1, entry 16) used for the preparation of 2 for 2 h. From the resulting mixture, A and 2a were isolated in 35% and 15% yields (Scheme 2, (1)). Second, A was subjected to the same reaction conditions for 6 h to give 2a in a yield of 82% (Scheme 2, (2)). These results indicated that A should be a key intermediate in the formation of 2a from 1a. Third, A was treated with KI and Oxone in the absence of Cu(OAc)2. Under this circumstance, the formation of 2a was not observed (Scheme 2, (3)). Fourth, A was treated with Cu(OAc)2, KI and Oxone under nitrogen instead of oxygen. From this reaction, 2a was formed only in a trace amount (Scheme 2, (4)). These results showed that in addition to Oxone and Cu(OAc)2, O2 is also indispensable for the efficient formation of 2a from A.
 | ||
| Scheme 2 Control experiments (I). | ||
Based on our experimental results and literature reports, a plausible pathway accounting for the formation of 2a is proposed in Scheme 3. Initially, an oxidative ring contraction occurs with 1a to give A,13b which is then oxidized by Oxone/O2 to give intermediate B.14 Next, B undergoes a decarboxylation and ipso-oxidation under the reaction conditions to give 2a, presumably via the formation of intermediates C, D, E and F.16 As for the role played by KI, it is postulated that it might be firstly converted into I2, which can activate the C–N bond of 1a, and thus facilitate the C–N bond cleavage to give A.13b
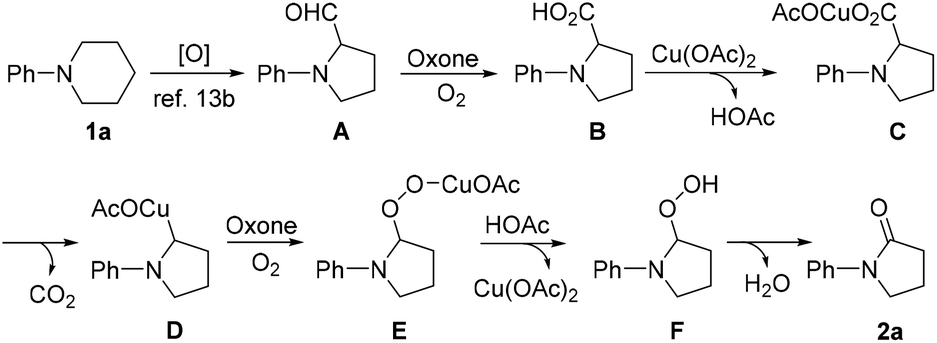 | ||
| Scheme 3 Plausible mechanism accounting for the formation of 2a. | ||
To elucidate the reaction mechanism accounting for the formation of 3a, another set of control experiments were carried out. First, 1a was subjected to the optimum reaction conditions (Table 3, entry 16) used for the preparation of 3 for 2 h. From the resulting mixture, A and 3a were isolated in 36% and 19% yields (Scheme 4, (1)). Second, A was subjected to the same reaction conditions for 6 h to give 3a in a yield of 78% (Scheme 4, (2)). These results indicated that A should be a key intermediate in the formation of 3a from 1a. Third, A was treated with Cu(OAc)2 and I2 in the absence of DMAP. Under this circumstance, the formation of 3a was not observed (Scheme 4, (3)). Fourth, A was treated with I2 and DMAP but in the absence of Cu(OAc)2. From this reaction, the formation of 3a was not observed (Scheme 4, (4)). These results showed that the presence of DMAP and Cu(OAc)2 is indispensable for the formation of 3a from intermediate A.
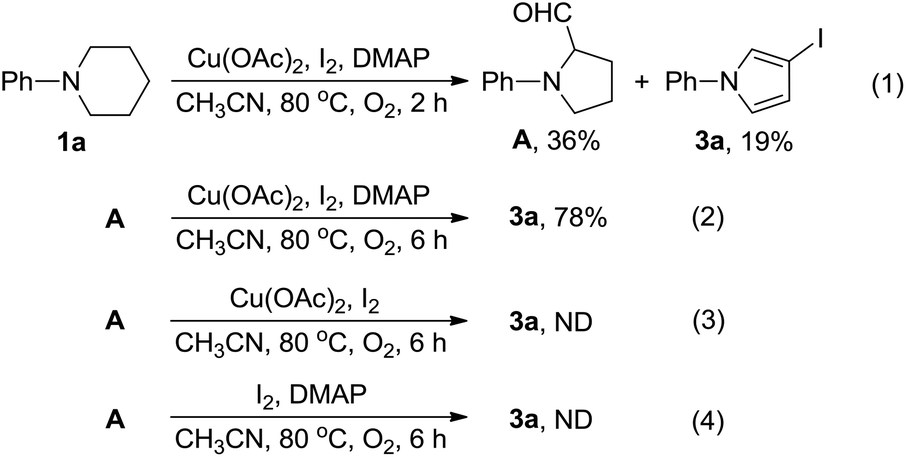 | ||
| Scheme 4 Control experiments (II). | ||
Based on the above-described control experiments and literature reports, a plausible pathway accounting for the formation of 3a from 1a is proposed in Scheme 5. Initially, A is formed through an oxidative ring contraction of 1a,13b and then oxidized by Cu(II)/O2/DMAP to give B. The following decarboxylation and dehydrogenation of B under the reaction conditions afforded dihydropyrrole G. Since the in situ formed enamine moiety in intermediate G is nucleophilic on the β-position, it then undergoes a regioselective iodination with I2 to give H. Finally, an oxidative aromatization occurs with H to give 3a. As for the role played by DMAP, it is postulated that it may help to facilitate this cascade reaction by activating the Cu(II) catalyst through coordination17 and promoting the dehydrogenation of D to form Gvia hydrogen abstraction.18,19 Alternatively, G may be firstly oxidized to give I, which is then iodinated to give 3a.
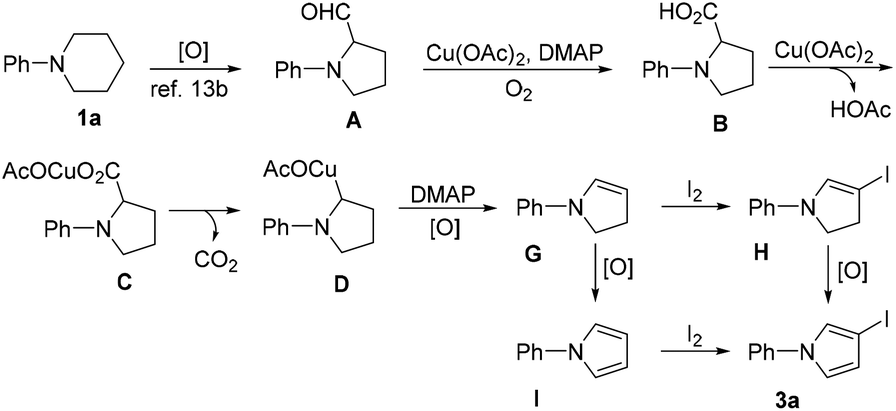 | ||
| Scheme 5 Plausible mechanism accounting for the formation of 3a. | ||
To showcase the utility of the 3-iodopyrroles obtained above, 3e was reacted with piperidine, phenylacetylene and phenylboronic acid under different reaction conditions to give diversely functionalized pyrrole derivatives 4, 5, and 6 in moderate yields (Scheme 6). Interestingly, a Pd-catalyzed carbonylation and esterification of 3e with CO and ethanol proceeded smoothly to give pyrrole-3-carboxylate 7 in 72% yield (Scheme 6).
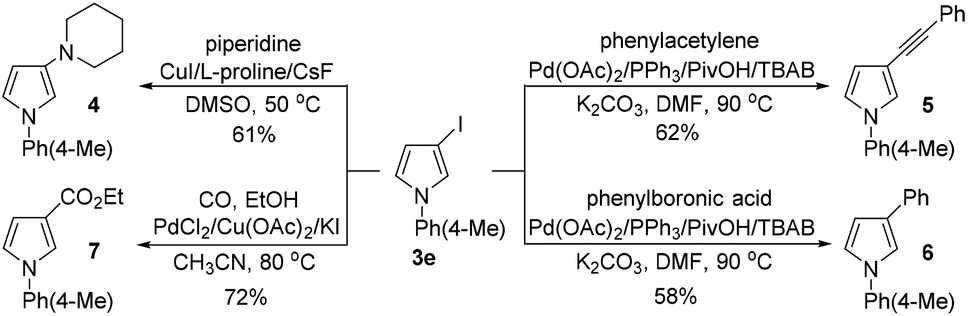 | ||
| Scheme 6 Structural elaboration of 3e. | ||
Conclusions
In summary, we have developed a novel and selective synthesis of pyrrolidin-2-one and 3-iodopyrrole derivatives via the cascade reactions of piperidine derivatives under different conditions. To the best of our knowledge, this is the first example in which pyrrolidin-2-ones and 3-iodopyrroles were directly and selectively synthesized from inactivated cyclic amines featuring with an oxidative ring contraction and deformylative functionalization. The use of simple and inactivated cyclic amines as the substrates, air-stable and low-cost copper salt as the promoter, non-poisonous and cost-effective Oxone as the oxidant and DMAP as the additive makes this approach an attractive and practical method for the preparation of the title compounds. A further study on the detailed reaction mechanism and application of these methods is in progress.Experimental section
Typical procedure for the synthesis of 2a
A tube containing 1-phenylpiperidine (1a, 81 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol), KI (166 mg, 1 mmol) and Oxone (615 mg, 1 mmol) was evacuated and back-filled with O2 three times. Then, CH3CN (5 mL) was added, and the mixture was stirred at 80 °C under O2 (balloon) for 12 h. Upon completion, it was quenched with saturated brine, and extracted with EtOAc (10 mL × 3). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with petroleum ether/ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent to give 2a in 58% yield. 2b–2m were obtained in a similar manner.
:1) as the eluent to give 2a in 58% yield. 2b–2m were obtained in a similar manner.
Typical procedure for the synthesis of 3a
A tube containing 1-phenylpiperidine (1a, 81 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol), I2 (127 mg, 0.5 mmol) and DMAP (61 mg, 0.5 mmol) was evacuated and back-filled with O2 three times. Then, CH3CN (5 mL) was added, and the mixture was stirred at 80 °C under O2 (balloon) for 10 h. Upon completion, it was quenched with saturated ammonium chloride (5 mL), and extracted with EtOAc (10 mL × 3). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with petroleum ether/ethyl acetate (100:1) as the eluent to give 3a in 65% yield. 3b–3n were obtained in a similar manner.
349 [M]+. HRMS (APCI) calcd for C10H8BrIN: 347.8879 [M + H]+, found: 347.8877.
Synthetic applications of 3e
:1) as the eluent to give 4 in 61% yield.
1-(1-(p-Tolyl)-1H-pyrrol-3-yl)piperidine (4). Yellow oil (73 mg, 61%). 1H NMR (600 MHz, CDCl3) δ 1.45–1.49 (m, 2H), 1.63–1.67 (m, 4H), 2.27 (s, 3H), 2.88 (t, J = 5.4 Hz, 4H), 6.02 (t, J = 2.4 Hz, 1H), 6.46 (t, J = 1.8 Hz, 1H), 6.85 (t, J = 2.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 20.8, 24.2, 25.7, 52.1, 101.9, 103.4, 118.1, 119.4, 130.0, 134.3, 138.7, 142.5. HRMS calcd for C16H20N2Na: 263.1519 [M + Na]+, found: 263.1521.
:1) as the eluent to give 5 in 62% yield.
3-(Phenylethynyl)-1-(p-tolyl)-1H-pyrrole (5). Brown solid (80 mg, 62%). 1H NMR (600 MHz, CDCl3) δ 2.31 (s, 3H), 6.41 (dd, J1 = 3.0 Hz, J2 = 1.2 Hz, 1H), 6.90 (t, J = 2.4 Hz, 1H), 7.15–7.26 (m, 8H), 7.42–7.43 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 20.9, 84.6, 88.5, 106.2, 113.6, 119.8, 120.6, 123.2, 124.1, 127.6, 128.3, 130.2, 131.3, 136.1, 137.8. HRMS calcd for C19H15NNa: 280.1097 [M + Na]+, found: 280.1097.
:1) as the eluent to give 6 in 58% yield.
3-Phenyl-1-(p-tolyl)-1H-pyrrole (6). Yellow oil (68 mg, 58%). 1H NMR (600 MHz, CDCl3) δ 2.28 (s, 3H), 6.27 (t, J = 3.6 Hz, 1H), 6.35 (dd, J1 = 3.6 Hz, J2 = 1.8 Hz, 1H), 6.84 (dd, J1 = 2.4 Hz, J2 = 1.8 Hz, 1H), 6.97–6.99 (m, 2H), 7.03–7.05 (m, 2H), 7.06–7.09 (m, 3H), 7.12–7.14 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 21.0, 109.0, 110.4, 124.5, 125.6, 126.2, 128.0, 128.3, 129.6, 133.1, 133.8, 136.4, 138.1. HRMS calcd for C17H16N: 234.1277 [M + H]+, found: 234.1270.
:1) as the eluent to give 7 in 72% yield.
Ethyl 1-(p-tolyl)-1H-pyrrole-3-carboxylate (7)13a. Yellow oil (82 mg, 72%). 1H NMR (600 MHz, CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 4.23 (q, J = 7.2 Hz, 2H), 6.67 (s, 1H), 6.90 (s, 1H), 7.17 (d, J = 7.8 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.58 (s, 1H). 13C NMR (150 MHz, CDCl3) δ 14.5, 20.9, 59.8, 111.4, 118.0, 120.6, 120.9, 124.4, 130.2, 136.8, 137.6, 164.8. HRMS calcd for C14H15NNaO2: 252.0995 [M + Na]+, found: 252.0998.
Control experiments
:1 to 3:1) as the eluent to give A and 2a in 35% and 15% yields, respectively.
1-Phenylpyrrolidine-2-carbaldehyde (A)13b. Oil (31 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 1.96–2.01 (m, 2H), 2.09–2.14 (m, 2H), 3.26–3.32 (m, 1H), 3.56–3.62 (m, 1H), 3.95–3.99 (m, 1H), 6.52 (d, J = 8.0 Hz, 2H), 6.69 (t, J = 7.2 Hz, 1H), 7.14–7.18 (m, 2H), 9.47 (d, J = 3.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 23.4, 27.2, 48.1, 65.9, 111.1, 116.3, 128.4, 146.1, 202.6. HRMS calcd for C11H14NO: 176.1070 [M + H]+, found: 176.1070.
A tube containing A (88 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol), KI (166 mg, 1 mmol) and Oxone (615 mg, 1 mmol) was evacuated and back-filled with O2 three times. Then CH3CN (5 mL) was added, and the mixture was stirred at 80 °C under O2 (balloon) for 6 h. Upon completion, it was quenched with saturated brine, and extracted with EtOAc (10 mL × 3). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with petroleum ether/ethyl acetate (3:1) as the eluent to give 2a in 82% yield.
A tube containing A (88 mg, 0.5 mmol), KI (166 mg, 1 mmol) and Oxone (615 mg, 1 mmol) was evacuated and back-filled with O2 three times. Then CH3CN (5 mL) was added, and the mixture was stirred at 80 °C under O2 (balloon) for 6 h, from which the formation of 2a was not observed (TLC).
A tube containing A (88 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol), KI (166 mg, 1 mmol) and Oxone (615 mg, 1 mmol) was evacuated and back-filled with N2 three times. Then CH3CN (5 mL) was added, and the mixture was stirred at 80 °C under N2 (balloon) for 6 h, from which 2a was formed in a trace amount (TLC).
:1 to 20:1) to afford A and 3a in 36% and 19% yields, respectively.
A tube containing A (88 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol), I2 (127 mg, 0.5 mmol) and DMAP (61 mg, 0.5 mmol) was evacuated and back-filled with O2. Then, CH3CN (5 mL) was added, and the resulting mixture was stirred at 80 °C under O2 (balloon) for 6 h. Upon completion, it was quenched with saturated ammonium chloride (5 mL), and extracted with EtOAc (10 mL × 3). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with petroleum ether/ethyl acetate (100:1) to afford 3a in 78% yield.
A tube containing A (88 mg, 0.5 mmol), Cu(OAc)2 (181 mg, 1 mmol) and I2 (127 mg, 0.5 mmol) was evacuated and back-filled with O2. Then CH3CN (5 mL) was added, and the resulting mixture was stirred at 80 °C under O2 (balloon) for 6 h, from which the formation of 3a was not observed (TLC).
A tube containing A (88 mg, 0.5 mmol), I2 (127 mg, 0.5 mmol) and DMAP (61 mg, 0.5 mmol) was evacuated and back-filled with O2. Then CH3CN (5 mL) was added, and the resulting mixture was stirred at 80 °C under O2 (balloon) for 6 h, from which the formation of 3a was not observed (TLC).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21572047, 21702050), the Plan for Scientific Innovation Talents of Henan Province (184200510012), and the 111 Project (D17007) for financial support.Notes and references
- (a) L.-W. Ye, C. Shu and F. Gagosz, Org. Biomol. Chem., 2014, 12, 1833 RSC; (b) J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134 RSC.
- X. Lin, W. Chen, Z. Qiu, L. Guo, W. Zhu, W. Li, Z. Wang, W. Zhang, Z. Zhang, Y. Rong, M. Zhang, L. Yu, S. Zhong, R. Zhao, X. Wu, J. C. Wong and G. Tang, J. Med. Chem., 2015, 58, 2809 CrossRef CAS PubMed.
- (a) S. Goudedranche, C. Besnard, L. Egger and J. Lacour, Angew. Chem., Int. Ed., 2016, 55, 13775 CrossRef CAS PubMed; (b) S. Verhasselt, B. I. Roman, O. De Wever, K. Van Hecke, R. Van Deun, M. E. Bracke and C. V. Stevens, Org. Biomol. Chem., 2017, 15, 2104 RSC; (c) S. Verhasselt, C. V. Stevens, T. Van den Broecke, M. E. Bracke and B. I. Roman, Bioorg. Med. Chem. Lett., 2017, 27, 2986 CrossRef CAS PubMed.
- (a) S. Comesse, M. Sanselme and A. Daïch, J. Org. Chem., 2008, 73, 5566 CrossRef CAS PubMed and references cited therein; (b) C. Shu, M.-Q. Liu, S.-S. Wang, L. Li and L.-W. Ye, J. Org. Chem., 2013, 78, 3292 CrossRef CAS PubMed; (c) C. Wu, H. Zhang, B. Yu, Y. Chen, Z. Ke, S. Guo and Z. Liu, ACS Catal., 2017, 7, 7772 CrossRef CAS.
- (a) Y. Zhao, J. Q. L. Ang, A. W. T. Ng and Y.-Y. Yeung, RSC Adv., 2013, 3, 19765 RSC; (b) J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998 CrossRef CAS PubMed; (c) S. Ghosh and C. K. Jana, J. Org. Chem., 2018, 83, 260 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, G. Cabrero and M. P. Ruiz, Chem. Commun., 2008, 615 RSC.
- Y.-H. Yang and M. Shi, J. Org. Chem., 2005, 70, 8645 CrossRef CAS PubMed.
- (a) X. Lv and W. Bao, J. Org. Chem., 2007, 72, 3863 CrossRef CAS PubMed; (b) D. Kundu, S. Bhadra, N. Mukherjee, B. Sreedhar and B. C. Ranu, Chem. – Eur. J., 2013, 19, 15759 CrossRef CAS PubMed; (c) B. Y.-H. Tan and Y.-C. Teo, Synlett, 2015, 26, 1697 CrossRef CAS; (d) S. De, J. Yin and D. Ma, Org. Lett., 2017, 19, 4864 CrossRef CAS PubMed.
- (a) R. W. Bates and S. Sridhar, J. Org. Chem., 2011, 76, 5026 CrossRef CAS PubMed; (b) X. Ma, Y. Vo, M. G. Banwell and A. C. Willis, Asian J. Org. Chem., 2012, 1, 160 CrossRef CAS; (c) C. Dialer, D. Imbri, S. P. Hansen and T. Opatz, J. Org. Chem., 2015, 80, 11605 CrossRef CAS PubMed.
- (a) M. D'Auria, E. De Luca, G. Mauriello, R. Racioppi and G. Sleiter, J. Chem. Soc., Perkin Trans. 1, 1997, 2369 RSC; (b) J. A. Smith, S. Ng and J. White, Org. Biomol. Chem., 2006, 4, 2477 RSC.
- (a) D. W. Knight, H. C. Rost, C. M. Sharland and J. A. Singkhonrat, Tetrahedron Lett., 2007, 48, 7906 CrossRef CAS; (b) T. Okitsu, S. Yumitate, K. Sato, Y. In and A. Wada, Chem. – Eur. J., 2013, 19, 4992 CrossRef CAS; (c) C. Huang, Y. Zeng, H. Cheng, A. Hu, L. Liu, Y. Xiao and J. Zhang, Org. Lett., 2017, 19, 4968 CrossRef CAS.
- (a) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092 CrossRef CAS; (b) D. Seidel, Acc. Chem. Res., 2015, 48, 317 CrossRef CAS PubMed; (c) R. J. Griffiths, W. C. Kong, S. A. Richards, G. A. Burley, M. C. Willis and E. P. A. Talbot, Chem. Sci., 2018, 9, 2295 RSC and references cited therein; (d) W.-C. Gao, F. Hu, Y.-M. Huo, H.-H. Chang, X. Li and W.-L. Wei, Org. Lett., 2015, 17, 3914 CrossRef CAS PubMed; (e) W.-C. Gao, F. Hu, J. Tian, X. Li, W.-L. Wei and H.-H. Chang, Chem. Commun., 2016, 52, 13097 RSC; (f) S. Guha, I. Kazi, P. Mukherjee and G. Sekar, Chem. Commun., 2017, 53, 10942 RSC.
- (a) Y. He, F. Wang, X. Zhang and X. Fan, Chem. Commun., 2017, 53, 4002 RSC; (b) F. Wang, Y. He, M. Tian, X. Zhang and X. Fan, Org. Lett., 2018, 20, 864 CrossRef CAS PubMed; (c) X. Shi, Y. He, X. Zhang and X. Fan, Adv. Synth. Catal., 2018, 360, 261 CrossRef CAS; (d) X. Shi, X. Chen, M. Wang, X. Zhang and X. Fan, J. Org. Chem., 2018, 83, 6524 CrossRef CAS PubMed.
- For a review, see: H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329 CrossRef CAS PubMed . For recent examples, see: (a) B. R. Travis, M. Sivakumar, G. O. Hollist and B. Borhan, Org. Lett., 2003, 5, 1031 CrossRef CAS PubMed; (b) M.-Z. Zhang, P.-Y. Ji, Y.-F. Liu and C.-C. Guo, J. Org. Chem., 2015, 80, 10777 CrossRef CAS; (c) K. N. Parida and J. N. Moorthy, J. Org. Chem., 2015, 80, 8354 CrossRef CAS PubMed.
- CCDC 1859829 (3d)† contains the crystallographic data for this paper.
- S. Huang, N. Thirupathi, C.-H. Tung and Z. Xu, J. Org. Chem., 2018, 83, 9449 CrossRef CAS.
- (a) Y. Maegawa, T. Ohshima, Y. Hayashi, K. Agura, T. Iwasaki and K. Mashima, ACS Catal., 2011, 1, 1178 CrossRef CAS; (b) X. Zhang, A. Xia, H. Chen and Y. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS PubMed.
- (a) J. Peng, G. Huang, H.-J. Wang, F.-B. Li, C. Huang, J.-J. Xiang, Y. Huang, L. Liu, C.-Y. Liu, A. M. Asiri and K. A. Alamry, J. Org. Chem., 2018, 83, 85 CrossRef CAS PubMed; (b) M. H. Kim and J. Kim, J. Org. Chem., 2018, 83, 1673 CrossRef CAS PubMed.
- K. Sen and J. C. Hackett, J. Am. Chem. Soc., 2010, 132, 10293 CrossRef CAS PubMed.
- C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331 RSC.
- A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2007, 46, 8862 CrossRef CAS.
- E. Haldón, E. Álvarez, M. C. Nicasio and P. J. Pérez, Organometallics, 2009, 28, 3815 CrossRef.
- M. Hayashi, S. Bachman, S. Hashimoto, C. C. Eichman and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 8997 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data and NMR spectra of all products, and the X-ray crystal structures and data of 3d. CCDC 1859829. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob02640h |
| This journal is © The Royal Society of Chemistry 2019 |