
Stereoselective synthesis of chromane derivatives via a domino reaction catalyzed by modularly designed organocatalysts†
Satish
Jakkampudi
,
Ramarao
Parella
and
John C.-G.
Zhao
 *
*
Department of Chemistry, University of Texas at San Antonio One UTSA Circle, San Antonio, TX 78249-0698, USA. E-mail: cong.zhao@utsa.edu
First published on 6th December 2018
Abstract
A highly enantio- and diastereoselective method for the synthesis of functionalized chroman-2-ones and chromanes was achieved by using an organocatalytic domino Michael/hemiacetalization reaction of aliphatic aldehydes and (E)-2-(2-nitrovinyl)phenols followed by a PCC oxidation and dehydroxylation, respectively. Using the modularly designed organocatalysts (MDOs) self-assembled from cinchona alkaloid derivatives and amino acids in the reaction media, the title products were obtained in good to high yields (up to 97%) and excellent diastereoselectivities (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and enantioselectivities (up to 99% ee).
:1 dr) and enantioselectivities (up to 99% ee).
Introduction
Michael addition to nitroalkenes is a powerful tool in organic synthesis that enables the synthesis of complex organic molecules bearing the synthetically useful nitro group. Not surprisingly, organocatalytic nitro-Michael reactions have been extensively investigated in the past few decades.1Chroman-2-one and chromane are important classes of benzopyran derivatives.1 The dihydrocoumarin and chromane scaffolds are found in many natural products and synthetic molecules that frequently exhibit unique biological and pharmacological activities,2 such as antineoplastic activity,3 antiherpetic activity,4 and inhibitive activities against protein kinases,5 aldose reductase,6 and HIV-1 reverse transcriptase.7 Owing to the importance of the chromane scaffold, its stereoselective synthesis has attracted considerable attention.8 Indeed, several organocatalytic methods have been developed to access this core structure in an asymmetric manner.9–13 For examples, Ramachary,9 Enders,10 Hong,11 and Gong12 have independently developed organocatalytic domino14 Michael/hemiacetalization reactions followed by an oxidation reaction for the efficient synthesis of chroman-2-one derivatives in a highly stereoselective manner.
Our group is interested in developing novel catalytic methods15 using the modularly designed organocatalysts (MDOs),16,17 which are self-assembled in the reaction media from cinchona alkaloid derivatives and amino acids. Herein, we wish to report that, using MDOs as the catalysts, the reaction between aliphatic aldehydes and (E)-2-(2-nitrovinyl)phenols gives the expected domino Michael/hemiacetalization products, which may be converted to functionalized chroman-2-ones and chromanes by PCC oxidation and dehydroxylation, respectively (Scheme 1). The desired chroman-2-ones and chromanes were both obtained in good yields and high stereoselectivities.
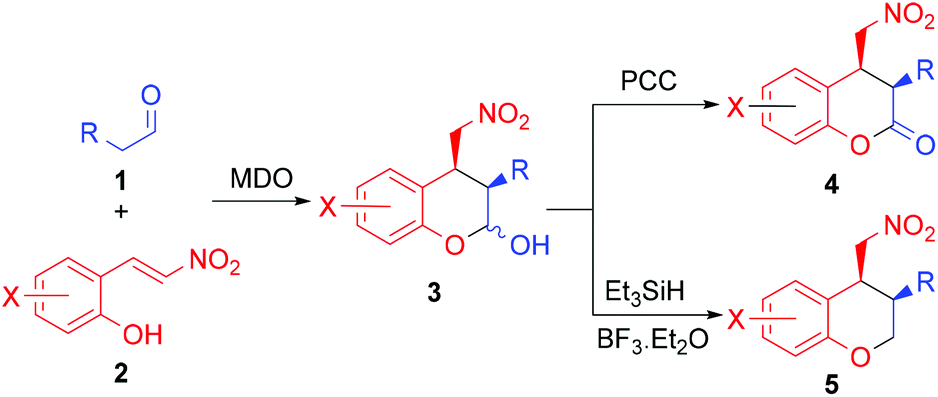 | ||
| Scheme 1 Synthesis of chroman-2-ones and chromanes using MDOs as the catalysts. | ||
Results and discussion
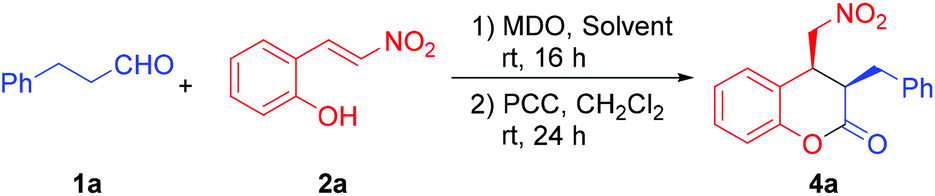
Hydrocinnamaldehyde (1a) and (E)-2-(2-nitrovinyl)phenol (2a) were adopted as the model substrates. Several cinchona alkaloid derivatives and amino acids (Fig. 1) were adopted as the precatalyst modules. These two modules have complementary basic and acidic functional groups that can help them self-assemble in situ in the reaction media. The most interesting results of the catalyst screening are collected in Table 1. As the results in Table 1 show, when quinidine thiourea 6a and L-proline (7a) were adopted as the stereocontrolling module and the reaction-center module, respectively, the reaction of 1a and 2a gave product 4a (after oxidation with PCC) in a high yield (94%) and excellent diastereoselectivity (96:4 dr) and ee value (99%, entry 1). Control experiments conducted with either 6a or 7a alone as the catalyst did not yield any product under otherwise identical conditions (entries 2 and 3). These results confirm that the observed catalytic activity is indeed due to the in situ generated MDO.
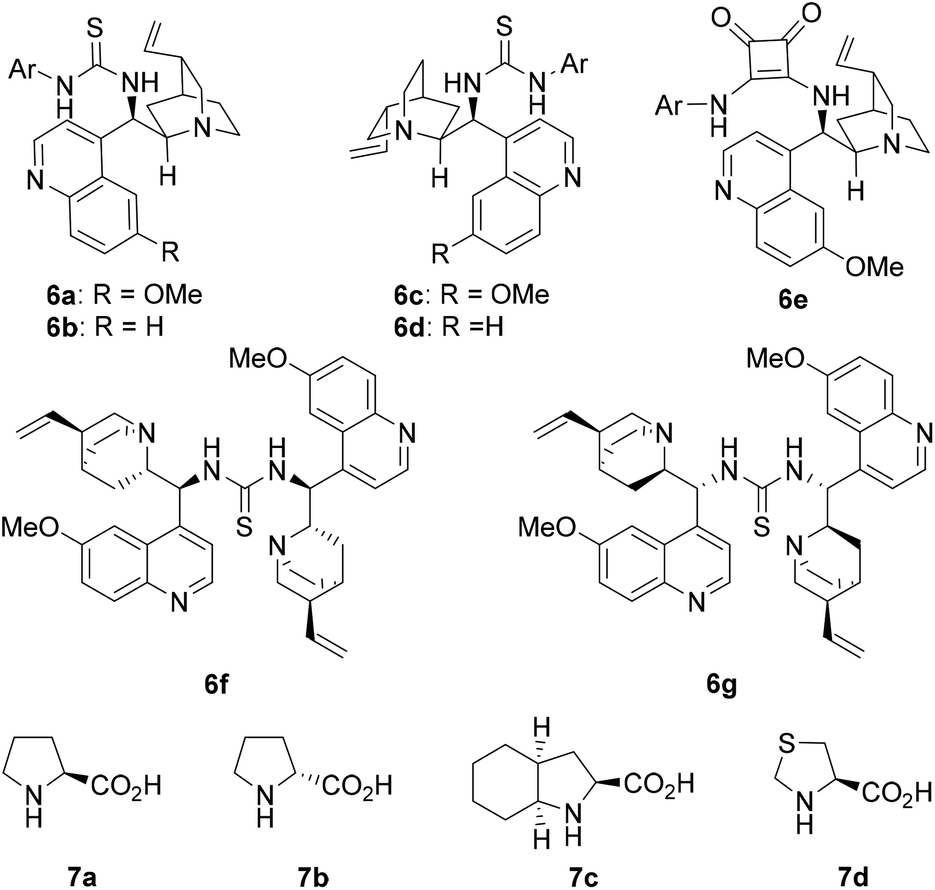 | ||
| Fig. 1 Structure of the precatalyst modules [Ar = 3,5-(CF3)2C6H3–]. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Modules | Solvent | Yieldb (%) | drc | eed (%) | |
| a Unless otherwise specified, all reactions were carried out with 1a (0.12 mmol), 2a (0.10 mmol), and the precatalyst modules (0.010 mmol each, 10 mol%) in dry toluene (1.0 mL) at room temperature for 16 h. Once the reaction was complete, the initial products were purified by flash column chromatography and then oxidized with PCC (3.0 equiv.) in CH2Cl2 at rt for 24 h. b Yield of the isolated product after flash column chromatography (overall yield after two steps of reactions). c Determined by 1H NMR analysis of the crude product. d Determined by HPLC analysis on a ChiralPak AD-H column. e The opposite enantiomer was obtained as the major product. f A mixture of all dimethylbenzene isomers. g Cyclopentyl methyl ether. h The loading of the precatalyst modules 6a and 7a was 5 mol% each. | ||||||
| 1 | 6a | 7a | Toluene | 94 | 96:4 |
99 |
| 2 | 6a | — | Toluene | — | — | — |
| 3 | — | 7a | Toluene | — | — | — |
| 4 | 6b | 7a | Toluene | 80 | 87:13 |
98 |
| 5 | 6c | 7a | Toluene | 80 | 81:19 |
78 |
| 6 | 6d | 7a | Toluene | 97 | 83:17 |
78 |
| 7 | 6e | 7a | Toluene | 90 | 85:15 |
87 |
| 8 | 6f | 7a | Toluene | 87 | 84:16 |
84 |
| 9 | 6g | 7a | Toluene | 80 | 80:20 |
32 |
| 10 | 6a | 7b | Toluene | 97 | 84:16 |
75e |
| 11 | 6a | 7c | Toluene | 94 | 88:12 |
98 |
| 12 | 6a | 7d | Toluene | <5 | — | — |
| 13 | 6a | 7a | Xylenesf | 99 | 88:12 |
99 |
| 14 | 6a | 7a | Benzene | 99 | 80:20 |
99 |
| 15 | 6a | 7a | CH2Cl2 | 87 | 82:18 |
96 |
| 16 | 6a | 7a | CPMEg | 70 | 95:5 |
91 |
| 17 | 6a | 7a | MeOH | 94 | 84:16 |
14 |
| 18 | 6a | 7a | THF | <5 | — | — |
| 19 | 6a | 7a | 1,4-Dioxane | — | — | — |
| 20 | 6a | 7a | CH3CN | — | — | — |
| 21h | 6a | 7a | Toluene | 89 | 93:7 |
98 |
Similar results were obtained when the MDO self-assembled from cinchonine thioureas 6b and 7a was applied, except that the obtained product yield (80%) and diastereoselectivity (87:13 dr) were slightly lower (entry 4). A much lower product ee value (78% ee) was obtained when the MDO 6c/7a was employed as the catalyst (entry 5). The MDO 6d/7a yielded very similar stereoselectivities to 6c/7a, but the product yield (97%) was much better (entry 6). Similar results were also obtained for the MDOs 6e/7a and 6f/7a (entries 7 and 8). In contrast, a poor product ee value (32% ee) was obtained when the MDO 6g/7a was applied (entry 9). This screening identified that the stereocontrolling module 6a is the best one for this reaction in terms of both the product yield and stereoselectivities (entry 1). Using 6a as the stereocontrolling module, we next screened several amino acids as the reaction-center module. The pseudo-diastereomeric MDO formed from 6a and D-proline (7b) led to the formation of the enantiomer of 4a in a high yield, but only moderate stereoselectivities (84:16 dr, 75% ee) (entry 10). Very good results were also obtained from the MDO 6a/7c (entry 11), which was only slightly inferior to that of 6a/7a (entry 1). However, almost no product could be isolated from the reaction catalyzed by the MDO self-assembled from 6a and L-thioproline (7d) (entry 12). Thus, the above screening identified MDO 6a/7a (entry 1) as the best catalyst for this domino Michael/hemiacetalization reaction. Next the solvent was screened for this best MDO. Common organic solvents, such as xylenes (entry 13), benzene (entry 14), and CH2Cl2 (entry 15) all yielded inferior diastereoselectivities. Slightly inferior results in terms of both yield and stereoselectivities were also obtained from the environmentally benign solvent cyclopentyl methyl ether (entry 16). On the other hand, a much poorer product ee value was obtained (14% ee) in MeOH (entry 17). THF (entry 18), 1,4-dioxane (entry 19), and CH3CN (entry 20) also turned out to be poor solvents for this reaction since either only a trace amount of product or no product could be obtained from these solvents. When the catalyst loading was reduced to 5 mol%, the yield and stereoselectivities obtained for 4a were only slightly lower (entry 21).
Once the reaction conditions were optimized, the scope of this reaction was studied and the results are collected in Table 2. As the results in Table 2 show, besides hydrocinnamaldehyde (1a, entry 1), other linear aldehydes, such as propanal (entry 2), butanal (entry 3), pentanal (entry 4), and heptanal (entry 5), also react with (E)-2-(2-nitrovinyl)phenol (2a) to give the desired chroman-2-ones 4b–e after oxidation in high yields (83–97%), good to excellent diastereoselectivities (81:19 to 98:2 dr), and excellent ee values (97–99% ee). In general, higher diastereoselectivities were obtained with longer chain aldehyde substrates. With the branched 3-methylbutanal, high diastereoselectivity of 99:1 dr and enantioselectivity of 93% ee were obtained for the corresponding chroman-2-one 4f (entry 6). Similarly, 2-methylpropanal also yielded the expected 4g after oxidation in 96% ee, although in a lower yield (69%, entry 7). Using pentanal as the aldehyde component, various substituted (E)-2-(2-nitrovinyl)phenols were then screened. It was found that these substituted (E)-2-(2-nitrovinyl)phenols usually led to slightly lower yields (65–87%) and diastereoselectivities (80:20 to 95:5 dr) of the corresponding chroman-2-ones (4h–n, entries 8–14) as compared to those obtained from the unsubstituted (E)-2-(2-nitrovinyl)phenol (entry 4). However, the product ee values remained high (entries 8–14). On the other hand, the electronic nature and the position of the substituent on the phenyl ring of (E)-2-(2-nitrovinyl)phenol had no significant effects on the diastereoselectivities or the product ee values (entries 8–14), except that a slightly lower ee value was obtained for the chroman-2-one product of the 4-nitro-substituted phenol (entry 10). Using the branched 3-methylbutanal as the aldehyde component yielded comparable results to those of pentanal (entries 15 and 16 vs. 8 and 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | R1 | X | 4/yieldb (%) | drc | eed (%) |
| a Unless otherwise specified, all reactions were carried out with 1 (0.12 mmol), 2 (0.10 mmol), and the precatalyst modules 6a and 7a (0.010 mmol each, 10 mol%) in dry toluene (1.0 mL) at room temperature for 16 h. Once the reaction was complete, the initial products were purified by flash column chromatography and then oxidized with PCC (3.0 equiv.) in CH2Cl2 at rt for 24 h. b Yield of the isolated product after flash column chromatography (overall yield after two steps of reactions). c Determined by 1H NMR analysis of the crude product. d Determined by HPLC analysis on ChiralPak AD-H, OD-H, or IC columns. The absolute stereochemistry was assigned by comparing the measured optical rotation of compound 4d with that reported in the literature (ref. 12). e The reaction time was 72 h. f The reaction time was 24 h. g Reaction performed at the 0.50 mmol scale. | ||||||
| 1 | Bn | H | H | 4a/94 | 96:4 |
99 |
| 2 | Me | H | H | 4b/86 | 89:11 |
99 |
| 3 | Et | H | H | 4c/97 | 81:19 |
96 |
| 4 | n-Pr | H | H | 4d/83 | 98:2 |
96 |
| 5 | n-Pent | H | H | 4e/90 | 95:5 |
97 |
| 6 | i-Pr | H | H | 4f/91 | 99:1 |
93 |
| 7e | Me | Me | H | 4g/69 | — | 96 |
| 8f | n-Pr | H | 4-Cl | 4h/74 | 84:16 |
98 |
| 9f | n-Pr | H | 4-Br | 4i/73 | 85:15 |
98 |
| 10f | n-Pr | H | 4-NO2 | 4j/68 | 80:20 |
87 |
| 11 | n-Pr | H | 4-Me | 4k/68 | 89:11 |
96 |
| 12f | n-Pr | H | 4-OMe | 4l/72 | 80:20 |
96 |
| 13f | n-Pr | H | 2-Me | 4m/65 | 95:5 |
98 |
| 14 | n-Pr | H | 3-Me | 4n/87 | 89:11 |
99 |
| 15f | i-Pr | H | 4-Cl | 4o/74 | 98:2 |
98 |
| 16f | i-Pr | H | 4-Br | 4p/73 | 87:13 |
89 |
| 17g | Bn | H | H | 4a/90 | 94:6 |
98 |
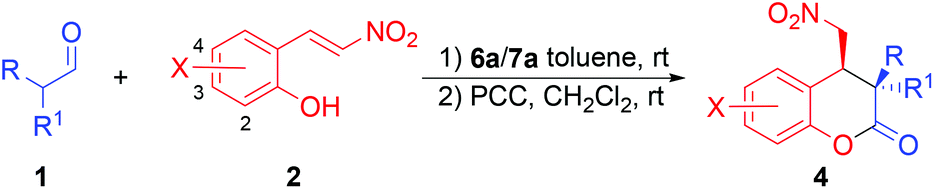
To demonstrate the synthetic utility of this method, the same reaction was also carried out at the 0.5 mmol scale of 1a and 2a. As the results in Table 2 show, product 4a was obtained in a comparable yield, diastereoselectivity, and an ee value to those of the small-scale reaction (entry 17 vs. entry 1).
To obtain the 3,4-substituted chromanes 5, the primary domino Michael/hemiacetalization products 3 were dehydroxylated by treating with triethylsilane and boron trifluoride diethyl etherate in dichloromethane (Table 3). As shown in Table 3, the dehydroxylation reaction provided the desired products 5a–c in good to excellent yields (72–94%) with preservation of the diastereoselectivities (85:15 to 95:5 dr) and enantioselectivities of the domino reaction (92 to 98% ee).
|
|
||||
|---|---|---|---|---|
| Entry | R | 5/yieldb (%) | drc | eed (%) |
| a Unless otherwise specified, all reactions were carried out with 3 (0.10 mmol), triethylsilane (0.30 mmol), and boron trifluoride diethyl etherate (0.30 mmol) in CH2Cl2 (3.0 mL) at 0 °C to room temperature for 2 h. b Yield of the isolated product after flash column chromatography. c Determined by 1H NMR analysis of the crude product. d Determined by HPLC analysis on ChiralPak OD-H or IB columns. The absolute stereochemistry was assigned by comparing the measured optical rotation of compound 5a with that reported in the literature (ref. 12). | ||||
| 1 | Et | 5a/90 | 88:12 |
98 |
| 2 | n-Pr | 5b/94 | 85:15 |
98 |
| 3 | i-Pr | 5c/72 | 95:5 |
92 |
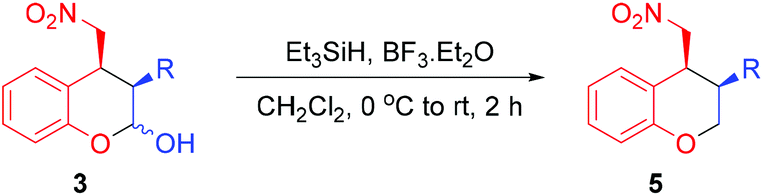
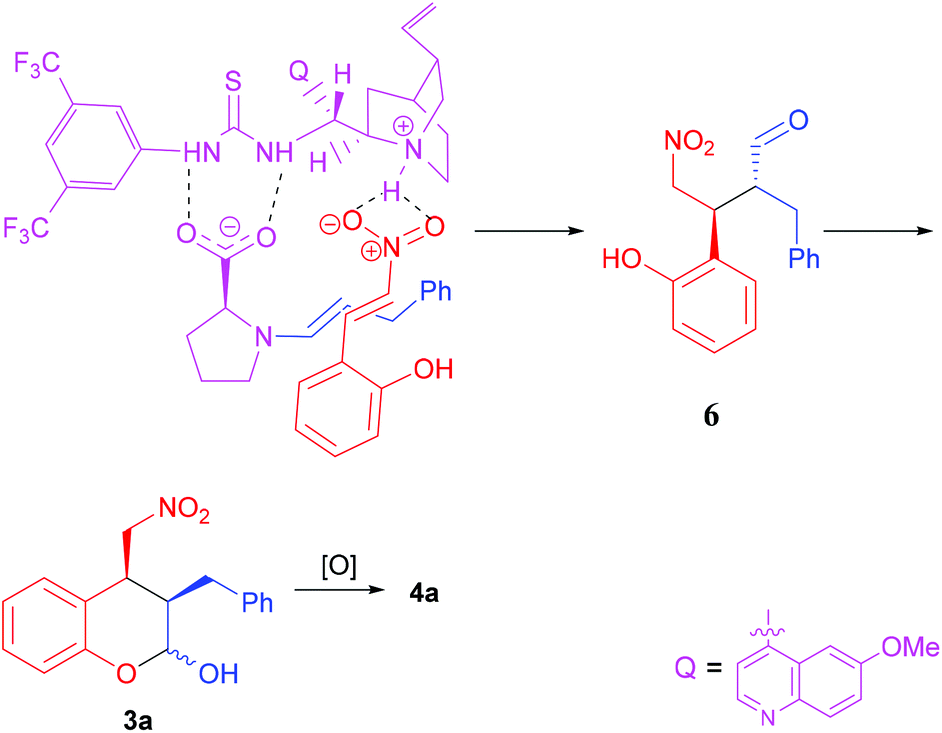
The absolute stereochemistry of the major enantiomeric products of compounds 4 and 5 was determined as shown in the tables by comparing the measured optical rotation of compounds 4d and 5a with those reported in the literature.12 Based on the product stereochemistry and a recent computational study of our MDO catalytic system,18 a plausible transition state is proposed to account for the formation of the major stereoisomer of the domino Michael/hemiacetalization reaction (Scheme 2). As shown in Scheme 2, the Si–Si attack of the preferred syn-(E)-enamine18 of hydrocinnamaldehyde onto the (E)-2-(2-nitrovinyl)phenol (2a) yields the Michael addition intermediate 6 with the expected stereochemistry of the two stereogenic centers, which, after an intramolecular hemiacetalization reaction, gives product 3a. Product 3a yields the expected 4a upon oxidation.
 | ||
| Scheme 2 Proposed transition state that accounts for the formation of the major stereoisomer. | ||
Experimental
Representative procedure for the synthesis of chroman-2-ones via the domino Michael/hemiacetalization followed by an oxidation reaction
To a vial were added sequentially the precatalyst modules 6a (5.9 mg, 0.010 mmol, 10.0 mol%) and 7a (1.1 mg, 0.010 mmol, 10.0 mol%) and dry toluene (1.0 mL). The resulting mixture was stirred at room temperature for 15 min. Compound 1a (16.0 mg, 0.12 mmol, 1.2 equiv.) was then added and the mixture was further stirred for 5 min before the addition of compound 2a (16.5 mg, 0.1 mmol, 1.0 equiv.). The resulting solution was stirred at room temperature for 16 h until the reaction was complete (monitored by TLC). Then the reaction mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography to give the chroman-2-ol 3a as a colorless oil (29.9 mg). A solution of the chroman-2-ol 3a (29.9 mg, 0.10 mmol) in CH2Cl2 (3.0 mL) and PCC (64.5 mg, 0.30 mmol, 3.0 equiv.) was stirred at room temperature for 24 h until the completion of the reaction (monitored by TLC). The suspension was filtered through a short pad of silica gel and washed with ethyl acetate. Removing the solvents under reduced pressure afforded the crude product 4a, which was then purified by flash chromatography (30:70 EtOAc/hexane as the eluent) to afford product 4a (28.0 mg, 94%) as a colorless oil.
General procedure of the dehydroxylation reaction10,12
To a solution of chroman-2-ol 3 (0.10 mmol, 1.0 equiv.) in CH2Cl2 (3.0 mL) at 0 °C were added triethylsilane (34.9 mg, 0.30 mmol, 3.0 equiv.) and boron trifluoride etherate (42.6 mg, 0.30 mmol, 3.0 equiv.) with stirring. The ice bath was removed after 15 min and the mixture was further stirred for 2 h. Then the reaction was quenched with saturated aqueous NaHCO3 solution (3 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography on silica gel to afford the corresponding chromane 5.Conclusions
In summary, we have developed a highly stereoselective synthesis of cis-3,4-disubstituted chroman-2-ones and chromanes using a domino Michael/hemiacetalization reaction of aliphatic aldehydes and (E)-2-(2-nitrovinyl)phenols catalyzed by modularly designed organocatalysts (MDOs) followed by a PCC oxidation or dehydroxylation. The corresponding chroman-2-ones and chromanes were obtained in good to excellent yields and diastereomeric ratios and high ee values.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The generous financial support of this research from the National Science Foundation (grant No. CHE 1664278) and the Welch Foundation (Grant No. AX-1593) is gratefully acknowledged. Some of the NMR data reported in this paper were collected on an NMR spectrometer acquired with the funding from the NSF (Grant No. CHE-1625963). The HRMS used in this research was supported by a grant from the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health. The authors also thank Dr Wendell P. Griffith for help with the HRMS analysis of the samples.Notes and references
- For reviews, see: (a) D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 11058–11076 CrossRef CAS PubMed; (b) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701–1716 CrossRef CAS; (c) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877–1894 CrossRef CAS; (d) S. Sulzer-Mossé and A. Alexakis, Chem. Commun., 2007, 3123–3135 RSC; (e) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065–2092 CrossRef CAS; (f) D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299–365 CrossRef; (g) A. M. Faisca Phillips, Curr. Org. Synth., 2016, 13, 687–725 CrossRef CAS.
- (a) R. D. H. Murray, J. Mendez and S. A. Brown, The Natural Coumarins: Occurrence, Chemistry, and Biochemistry; Wiley, New York, 1982 Search PubMed; (b) R. O'Kennedy and R. D. Thornes, Coumarins: Biology, Applications, and Mode of Action, 1st edn., Wiley, New York, 1997 Search PubMed; (c) K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G. Q. Cao, S. Barluenga and H. J. Mitchell, J. Am. Chem. Soc., 2000, 122, 9939–9953 CrossRef CAS; (d) E. E. Schweizer and O. Meeder-Nycz, in Chromenes, Chromanes, Chromones, ed. G. P. Ellis, Wiley-Interscience, New York, 1977 Search PubMed; (e) A. Kumar, B. K. Singh, R. Tyagi, S. K. Jain, S. K. Sharma, A. K. Prasad, H. G. Raj, R. C. Rastogi, A. C. Watterson and V. S. Parmar, Bioorg. Med. Chem., 2005, 13, 4300–4305 CrossRef CAS PubMed; (f) J. Posakony, M. Hirao, S. Stevens, J. A. Simon and A. Bedalov, J. Med. Chem., 2004, 47, 2635–2644 CrossRef CAS PubMed; (g) X.-F. Zhang, H.-M. Wang, Y.-L. Song, L.-H. Nie, L.-F. Wang, B. Liu, P.-P. Shen and Y. Liu, Bioorg. Med. Chem. Lett., 2006, 16, 949–953 CrossRef CAS PubMed.
- L. Jurd, J. Heterocycl. Chem., 1997, 34, 601–604 CrossRef CAS.
- M. Takechi, Y. Tanaka, M. Takehara, G.-I. Nonaka and I. Nishioka, Phytochemistry, 1985, 24, 2245–2250 CrossRef CAS.
- F. Hsu, G. Nonaka and I. Nishioka, Chem. Pharm. Bull., 1985, 33, 3142–3152 CrossRef CAS.
- M. Iinuma, T. Tanaka, M. Mizuno, T. Katsuzaki and H. Ogawa, Chem. Pharm. Bull., 1989, 37, 1813–1815 CrossRef CAS PubMed.
- L. M. V. Tillekeratne, A. Sherette, P. Grossman, L. Hupe, D. Hupe and R. A. Hudson, Bioorg. Med. Chem. Lett., 2001, 11, 2763–2767 CrossRef CAS PubMed.
- (a) Z.-P. Zhang, L. Chen, X. Li and J.-P. Cheng, J. Org. Chem., 2018, 83, 2714–2724 CrossRef CAS PubMed; (b) L. Cui, D. Lv, Y. Wang, Z. Fan, Z. Li and Z. Zhou, J. Org. Chem., 2018, 83, 4221–4228 CrossRef CAS PubMed; (c) H. Kim and J. Yun, Adv. Synth. Catal., 2010, 352, 1881–1885 CrossRef CAS; (d) T. Matsuda, M. Shigeno and M. Murakami, J. Am. Chem. Soc., 2007, 129, 12086–12087 CrossRef CAS PubMed; (e) J.-L. Li, S.-L. Zhou, B. Han, L. Wu and Y.-C. Chen, Chem. Commun., 2010, 46, 2665–2667 RSC; (f) X. Han and X. Lu, Org. Lett., 2010, 12, 108–111 CrossRef CAS PubMed; (g) E. Alden-Danforth, M. T. Scerba and T. Lectka, Org. Lett., 2008, 10, 4951–4953 CrossRef CAS PubMed.
- (a) D. B. Ramachary and R. Sakthidevi, Org. Biomol. Chem., 2010, 8, 4259–4265 RSC; (b) D. B. Ramachary and R. Sakthidevi, Chem. – Eur. J., 2009, 15, 4516–4522 CrossRef CAS PubMed.
- (a) D. Enders, X. Yang, C. Wang, G. Raabe and J. Runsik, Chem. - Asian J., 2011, 6, 2255–2259 CrossRef CAS PubMed; (b) D. Enders, C. Wang, X. Yang and G. Raabe, Adv. Synth. Catal., 2010, 352, 2869–2874 CrossRef CAS.
- (a) B.-C. Hong, P. Kotame and J.-H. Liao, Org. Biomol. Chem., 2011, 9, 382–386 RSC; (b) B.-C. Hong, P. Kotame, C.-W. Tsai and J.-H. Liao, Org. Lett., 2010, 12, 776–779 CrossRef CAS PubMed; (c) P. Kotame, B.-C. Hong and J.-H. Liao, Tetrahedron Lett., 2009, 50, 704–707 CrossRef CAS.
- D. Lu, Y. Li and Y. Gong, J. Org. Chem., 2010, 75, 6900–6907 CrossRef CAS PubMed.
- (a) T. Zhang, C. Ma, J.-Y. Zhou, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2018, 360, 1128–1137 CrossRef CAS; (b) X. Yang, Y.-C. Zhang, Q.-N. Zhu, M.-S. Tu and F. Shi, J. Org. Chem., 2016, 81, 5056–5065 CrossRef CAS PubMed; (c) Y.-C. Zhang, Q.-N. Zhu, X. Yang, L.-J. Zhou and F. Shi, J. Org. Chem., 2016, 81, 1681–1688 CrossRef CAS PubMed; (d) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS PubMed; (e) X.-L. Jiang, S.-F. Wu, J.-R. Wang, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2018, 360, 4225–4235 CrossRef CAS.
- For reviews, see: (a) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237–294 CrossRef CAS; (b) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS; (c) T. Chanda and J. C.-G. Zhao, Adv. Synth. Catal., 2018, 360, 2–79 CrossRef CAS; (d) C. Wang, X. Yang, G. Raabe and D. Enders, Adv. Synth. Catal., 2012, 354, 2629–2634 CrossRef CAS; (e) Z. Wu, X. Wang, F. Li, J. Wu and J. Wang, Org. Lett., 2015, 17, 3588–3591 CrossRef CAS PubMed; (f) E. Massolo, S. Palmieri, M. Benaglia, V. Capriati and F. M. Perna, Green Chem., 2016, 18, 792–797 RSC.
- (a) N. K. Rana, H. Huang and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2014, 53, 7619–7623 CrossRef CAS PubMed; (b) H. Huang, S. Konda and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 2213–2216 CrossRef CAS PubMed; (c) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS.
- (a) T. Mandal and C.-G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714–7717 CrossRef CAS PubMed; (b) S. Muramulla and C.-G. Zhao, Tetrahedron Lett., 2011, 52, 3905–3908 CrossRef CAS; (c) D. Sinha, T. Mandal, S. Gogoi, J. J. Goldman and J. C.-G. Zhao, Chin. J. Chem., 2012, 30, 2624–2630 CAS; (d) S. Muramulla, J.-A. Ma and J. C.-G. Zhao, Adv. Synth. Catal., 2013, 355, 1260–1264 CrossRef CAS; (e) D. Sinha, S. Perera and J. C.-G. Zhao, Chem. – Eur. J., 2013, 19, 6976–6979 CrossRef CAS; (f) S. Perera, D. Sinha, N. K. Rana, V. Trieu-Do and J. C.-G. Zhao, J. Org. Chem., 2013, 78, 10947–10953 CrossRef CAS PubMed; (g) S. Konda and J. C.-G. Zhao, Tetrahedron, 2018, 74, 6166–6172 CrossRef CAS; (h) R. Parella, S. Jakkampudi, H. Arman and J. C.-G. Zhao, Adv. Synth. Catal. DOI:10.1002/adsc.201800987 , in press.
- Additional examples of using MDOs as the catalysts: (a) D. B. Ramachary, R. Sakthidevi and K. S. Shruthi, Chem. – Eur. J., 2012, 18, 8008–8012 CrossRef CAS PubMed; (b) D. B. Ramachary and K. S. Shruthi, Org. Biomol. Chem., 2014, 12, 4300–4304 RSC; (c) D. B. Ramachary, K. S. Shruthi and R. Madhavachary, Eur. J. Org. Chem., 2015, 6413–6418 CrossRef CAS; (d) Z. Hang, J. Zhu, X. Lian, P. Xu, H. Yu and S. Han, Chem. Commun., 2016, 52, 80–83 RSC.
- B. Bhaskararao and R. B. Sunoj, Chem. Sci., 2018, 9, 8738–8747 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of NMR spectra and HPLC chromatograms. See DOI: 10.1039/c8ob02677g |
| This journal is © The Royal Society of Chemistry 2019 |