
Zn and Co redox active coordination polymers as efficient electrocatalysts†
Ruslan
Shekurov
 a,
Vera
Khrizanforova
a,
Leysan
Gilmanova
a,
Mikhail
Khrizanforov
a,
Vasily
Miluykov
a,
Olga
Kataeva
ab,
Zilya
Yamaleeva
a,
Timur
Burganov
a,
Tatiana
Gerasimova
a,
Airat
Khamatgalimov
ac,
Sergey
Katsyuba
a,
Valeri
Kovalenko
ac,
Yulia
Krupskaya
d,
Vladislav
Kataev
d,
Bernd
Büchner
de,
Volodymyr
Bon
f,
Irena
Senkovska
f,
Stefan
Kaskel
f,
Aidar
Gubaidullin
a,
Oleg
Sinyashin
a and
Yulia
Budnikova
*a
a,
Vera
Khrizanforova
a,
Leysan
Gilmanova
a,
Mikhail
Khrizanforov
a,
Vasily
Miluykov
a,
Olga
Kataeva
ab,
Zilya
Yamaleeva
a,
Timur
Burganov
a,
Tatiana
Gerasimova
a,
Airat
Khamatgalimov
ac,
Sergey
Katsyuba
a,
Valeri
Kovalenko
ac,
Yulia
Krupskaya
d,
Vladislav
Kataev
d,
Bernd
Büchner
de,
Volodymyr
Bon
f,
Irena
Senkovska
f,
Stefan
Kaskel
f,
Aidar
Gubaidullin
a,
Oleg
Sinyashin
a and
Yulia
Budnikova
*a
aArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, Arbuzov Str. 8, 420088 Kazan, Russia. E-mail: yulia@iopc.ru
bA.M. Butlerov Chemistry Institute of the Kazan Federal University, Kremlevskaya str. 18, 420000, Russia
cKazan National Research Technological University, Karl Marx Str. 68, Kazan 420015, Russia
dIFW Dresden, Helmholtzstrasse 20, 01069 Dresden, Germany
eInstitute of Solid State and Materials Physics, Technical University Dresden, D-01062 Dresden, Germany
fChair of Inorganic Chemistry, Technische Universität Dresden, Bergstr. 66, 01062 Dresden, Germany
First published on 21st January 2019
Abstract
New redox active 1D helical coordination polymers M(fcdHp) (M(II) = Zn(1), Co(2)) have been obtained by utilizing the 1,1′-ferrocenylenbis(H-phosphinic) acid (H2fcdHp) ligand and Zn or Co nitrate salts. Complexes 1 and 2 are isomorphic, crystallizing in the chiral space groups P4122 and P4322, respectively. Their redox, electrocatalytic and other properties are described. These compounds incorporated into carbon paste electrodes and exhibited reversible redox reactions, arising from the ferrocenyl moiety. These coordination polymers are efficient as electrocatalysts for the reduction of protons to hydrogen. Using N,N-dimethylformamidium ([DMF(H)+]) as the acid in the acetonitrile solution, Co CP (2) displays a turnover frequency of 300 s−1, which is among the fastest rates reported for any CP electrocatalyst in CH3CN. This high rate of catalytic reaction comes at the cost of the 820–840 mV overpotential at the potential of catalysis. As the hydrogen evolution reaction (HER) catalysts, the CPs exhibited in 0.5 M H2SO4 the overpotential η10 of 340 or 450 mV, onset overpotential of 220 or 300 mV (vs. RHE), Tafel slope of 110 or 120 mV dec−1, correspondingly for 1 and 2, and considerable long-term stability for the HER.
1. Introduction
Coordination polymers (CPs) with a boundless range of metal centres and bridging ligand combinations, each of which may be chosen to be redox active or inactive and may be incorporated into 1D chains, 2D sheets or 3D networks, provide an ideal basis for redox modification in the solid state for different applications.1–12 The development of redox-active CPs is a highly sought-after goal: at a fundamental level these materials offer unprecedented insights into charge transfer in coordination space, and at an applied level, their properties may underpin the next generation of technologically useful devices. The synergetic effect between the electronic properties of the framework and the intrinsic porous properties has led to new applications of CPs in a wide range of important fields. In particular, the redox active CPs may be useful materials for electrochemical applications such as ion storage13 or electrocatalysis.7,14 Because of the many appealing advantages of electrocatalysis, fast development on improving chemical stabilities, and the urgent need for efficient energy storage/conversion technologies/materials, CPs and CP-based composite materials have experienced fast growth as electrocatalysts for the hydrogen evolution reaction (HER) and other reactions in the past few years.7 A principal advantage of metal–organic frameworks is their designable structure with clear chemistry. A new electroactive hybrid material based on the structural and functional [NiFe]-hydrogenase model complex incorporated into the Zr-based CPs has been synthesized, characterized, and its electrocatalytic capabilities for the H+ reduction on the cyclic voltammetry time scale was shown.14b A summary of CP-based HER electrocatalysts demonstrated the theoretical TOFs at η = 200 mV in acidic solutions (H2SO4 or HCl) from 5.7 × 10−4 to 34 s−1.7The chemical and hydrothermal stability of CPs is crucial for most potential applications. Although the judicious choice of the metal ion and organic linker provides the redox activity within CPs,15 the redox reaction often changes the coordination environment of metal ions, thus leading to the destruction of the framework. One way to overcome this issue is to construct the framework with stable ligands for the redox reaction. In this context, ferrocene-based coordination polymers are excellent candidates because they contain a stable ferrocene moiety and two carboxylate, phosphinates or other coordination sites.16 Some phosphonyl-substituted ferrocene derivatives have been reported and their complexation behavior towards transition metal cations has been explored.17 Some research groups have reported on the syntheses of ferrocene-based coordination polymers,18 but there are only a few reports of the solid-state electrochemical properties of coordination polymers containing the 1,1′-ferrocenedicarboxylate ligand,19 and the data for CPs with linkers based on phosphinate derivatives are absent. And, while the majority of carboxylate-based CPs display low stability in aqueous media,19 phosphinate derivatives give more hydrothermally stable CPs.20 Perhaps then surprisingly, phosphinates have obtained much less attention for the preparation of CPs,21 especially in comparison with carboxylic acid based linkers.19d Only monophosphinic acids,22–24 bisphosphinic acids with a short spacer,25,26 or bisphosphinic acids with a flexible spacer27 and recently phenylene-1,4-bis(methylphosphinic acid)20 have been used for the preparation of CPs. But the conformational lability of 1,1′-ferrocenediphosphinate can provide many interesting structures and allows the ligand to adopt a conformation most suitable for metal coordination.28
Thus, the synthesis of new ferrocene-based coordination polymers with phosphinate linkers is very important and can lead to new redox-active structures that are steady during redox processes in various environments, including acids and water, fundamentally interesting as electrocatalysts for hydrogen evolution, oxygen reduction, as electrochemical sensors and for other applications. The key to successful implementation is not only to construct the redox active framework itself, but also to hybridize the framework with an electrode, which allows the investigation of redox transformations of CPs and catalytic currents in the presence of a proton donor or other subjects of inquiry.
Herein we report on the synthesis, crystal structure, and the physico-chemical (redox, electrocatalytic, spectroscopic and magnetic) properties of redox-active Co(II)- and Zn(II)-based one-dimensional CPs with a ferrocene-containing diphosphinate ligand. The choice of cobalt and zinc derivatives is due to the importance of searching for catalysts or metal-based materials based on wide-spread non-platinum metals. An interesting additional observation has been found, that the enantiopure pairs of chiral CPs were obtained as a conglomerate by spontaneous resolution from the methanol/DMF solution of the ligand and metal nitrates.
2. Experimental section
2.1. Synthesis
Reagent 1,1′-ferrocenylenbis(H-phosphinic) acid (H2fcdHp) was prepared according to the literature procedure.29 All other chemicals and solvents purchased were reagent grade and used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v, 6 mL). The vial was placed into a preheated oven (80 °C) for 16 h. Orange crystals formed were washed with DMF/MeOH and dried in air. Yield: 77%. Anal. calcd for C10H10P2O4FeZn: C, 31.82; H, 2.65. Found: C, 31.25; H, 2.60.
:3 v/v, 6 mL). The vial was placed into a preheated oven (80 °C) for 16 h. Orange crystals formed were washed with DMF/MeOH and dried in air. Yield: 77%. Anal. calcd for C10H10P2O4FeZn: C, 31.82; H, 2.65. Found: C, 31.25; H, 2.60.
2.2. Structure determination
The dataset from the single crystal of 2, prepared in a glass capillary, was collected at beamline BL14.2, Joint Berlin–MX Laboratory of Helmholtz Zentrum Berlin, equipped with a MX-225 CCD detector (Rayonics, Illinois) and a 1-circle goniometer.30 The data collection was performed at room temperature using monochromatic radiation with λ = 0.88561 Å. XDSAPP software was used for processing the diffraction images.31 Systematic absences were found for the screw axis 41, therefore suggesting the chiral space group P4122 (No. 91) for the crystal structure solution and refinement. The dataset for the single crystal of 1a was collected on a Bruker AXS Kappa APEX Duo and for crystal 1b on a Bruker AXS Smart APEX II diffractometers with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures of both compounds 1a and 1b were solved by direct methods using the following software: APEX332 for data collection, SAINT33 for data reduction, SHELXS34 for structure solution, SHELXL33 for structure refinement by full-matrix least-squares against F2, and SADABS35 for multi-scan absorption correction. CCDC 1861598; 1861599; and 1866808† contain the supplementary crystallographic data for this paper.1a: C10H10FeO4P2Zn, M = 377.34 g mol−1, crystal size (mm) 0.180 × 0.120 × 0.120, temperature 150(2) K, tetragonal, space group P4122 (No. 91), a = 8.254(2) Å, c = 18.660(4) Å, V = 1271.1(7) Å3, Z = 4, ρcalc = 1.972 g cm−3, μ = 3.279 mm−1, θ range: from 2.70° to 25.62°, 3369 reflection collected (−10 ≤ h ≤ 8, −10 ≤ k ≤ 7, −22 ≤ l ≤ 19), 1194 independent reflections (Rint = 0.0527), 1051 observed reflections with I ≥ 2σ(I), 86 refined parameters, R = 0.0302, wR2 = 0.0500, Flack parameter 0.03(2), and max. residual electron density 0.271 (−0.266) e Å−3.
1b: C10H10FeO4P2Zn, M = 377.34 g mol−1, crystal size (mm) 0.365 × 0.108 × 0.078, temperature 296(2) K, tetragonal, space group P4322 (No. 95), a = 8.250(2) Å, c = 18.655(4) Å, V = 1296.7(7) Å3, Z = 4, ρcalc = 1.974 g cm−3, μ = 3.283 mm−1, θ range: from 2.47° to 28.62°, 23622 reflection collected (−11 ≤ h ≤ 11, −11 ≤ k ≤ 11, −24 ≤ l ≤ 24), 1602 independent reflections (Rint = 0.0471), 1313 observed reflections with I ≥ 2σ(I), 86 refined parameters, R = 0.0320, wR2 = 0.0724, Flack parameter −0.01(1), and max. residual electron density 0.480 (−0.362) e Å−3.
2: C10H10FeO4P2Co, M = 370.90 g mol−1, tetragonal, P4122 (No. 91), a = 8.2200(12) Å, c = 18.840(4) Å, V = 1273.0(5) Å3, Z = 4, ρcalc = 1.935 g cm−3, λ = 0.88561 Å, T = 293 K, θmax = 34.1°, reflections collected/unique 1374/1341, Rint = 0.023, R1 = 0.0749, wR2 = 0.2287, S = 1.15, Flack parameter 0.23(3), and the largest diff. peak 1.23 e Å−3 and hole −0.70 e Å−3.
2.3. Thermogravimetry (TGA) and differential scanning calorimetry (DSC)
The thermal stabilities of solid samples were investigated by simultaneous thermogravimetry/differential scanning calorimetry (TG/DSC) analysis using NETZSCH (Selb, Germany) STA449 F3 instrument. Approximately 5–6 mg of samples were placed into an Al2O3 crucible with a pre-hole on the lid and heated from 30 to 1200 °C. The same empty crucible was used as the reference. High-purity argon was used with a gas flow rate of 50 mL min−1. TG/DSC measurements were performed at the heating rates of 10 °C min−1.2.4. IR spectroscopy
The IR spectra of samples were recorded using a Bruker Vector-27 FTIR spectrometer in the 400–4000 cm−1 range (optical resolution 4 cm−1). The samples were prepared as KBr pellets.2.5. Raman spectroscopy
Raman spectra were recorded at room temperature using a BRUKER RAM II module attached to a BRUKER VERTEX 70 FTIR spectrometer (excitation 1064 nm, Ge detector at liquid nitrogen temperature, back-scattering configuration; range 10–4000 cm−1, optical resolution 4 cm−1, and scan number 1024). The samples were inserted in a standard aluminum crucible.Powder samples were characterized by the UV-vis/DR technique using a Jasco V-650 spectrophotometer (Jasco International Co. Ltd., Hachioji, Tokyo, Japan) equipped with an integrating sphere accessory for diffuse reflectance spectra acquisition. BaSO4 powder was used as the reference for baseline correction.
2.6. Magnetic measurements
Static magnetization measurements were performed with a commercial SQUID (superconducting quantum interference device) VSM (vibrating sample magnetometer) from Quantum Design in static magnetic fields up to 7 T in the temperature range 1.8–300 K.2.7. Electrochemistry
Electrochemical measurements were taken on a BASiEpsilonE2P electrochemical analyzer (USA). The program concerned Epsilon-EC-USB-V200 waves. A conventional three-electrode system was used with glassy carbon for carbon paste electrode (CPE) solutions for powder samples as the working electrode, the An Fc+/Fc system served as the reference electrode, and a Pt wire as the counter electrode. 0.1 M Et4NBF4 was used as the supporting electrolyte to determine the current–voltage characteristics. To study the powder samples, a modified CPE working electrode was used, which was prepared as follows: the carbon particles/phosphonium salt (dodecyl(tri-tert-butyl)phosphonium tetrafluoroborate) composite electrode was prepared by grinding a mixture of graphite powder and phosphonium salt with a 90/10 (w/w) ratio in a mortar giving it a homogeneous mass. A modified electrode was also devised in a similar manner except that a portion (ca. 5%) of the graphite powder was replaced by the CPs under study. As a result, a portion of the resulting paste was packed firmly into (3 mm in diameter) a Teflon holder cavity. The performances of the electrocatalysts can be evaluated by many parameters, such as the onset potential (Eonset), overpotential (η) at a specific current density (j = 10 mA cm−2), Tafel slope (TS), and turnover frequency (TOF).3. Results and discussion
3.1. Synthesis and the crystal structure
CPs Zn(fcdHp) (1) and Co(fcdHp) (2) were synthesized utilizing metal nitrates of zinc or cobalt and the 1,1′-ferrocenylenbis(H-phosphinic) acid (H2fcdHp) at 80 °C (Scheme 1) by using a binary solvent mixture containing MeOH and N,N-dimethylformamide (DMF). The resulting materials are insoluble in common organic solvents.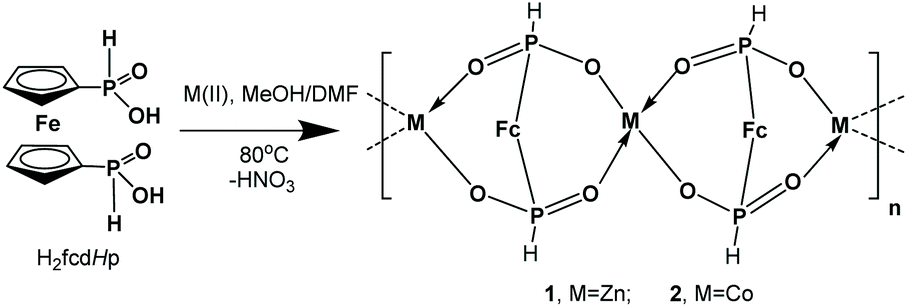 | ||
| Scheme 1 Synthesis of coordination polymers. | ||
The Zn and Co CPs are isotypic; hence the structural description is restricted to the Zn CPs. Both contain two crystallographically independent [ML]n entities with an identical coordination environment. The single crystal X-ray diffraction study showed that compounds 1 and 2 are one-dimensional coordination polymers with 8-membered macrocycles M–O–P–O–M–O–P–O, similar to that observed earlier for other metal phosphinates.36 Crystallizing in the chiral space groups P4122 and P4322, Zn-based coordination polymers 1a (left rotation) and 1b (right rotation) are axial enantiomers. The cobalt based CP 2a can be considered as an isomorphic structure to 1a. In all structures the ferrocene molecule is found in a slightly twisted conformation with a very small shift of the cyclopentadienyl rings, where phosphinic groups are located opposite one another. The zinc and cobalt ions in compounds 1a and 2a are coordinated by four oxygen atoms from symmetrically dependent phosphinic groups showing a tetrahedral coordination geometry (Fig. 1).
 | ||
| Fig. 1 A fragment of a polymeric chain of compound 2 showing spiro-fused 8-membered rings and the coordination geometry of Co(II). | ||
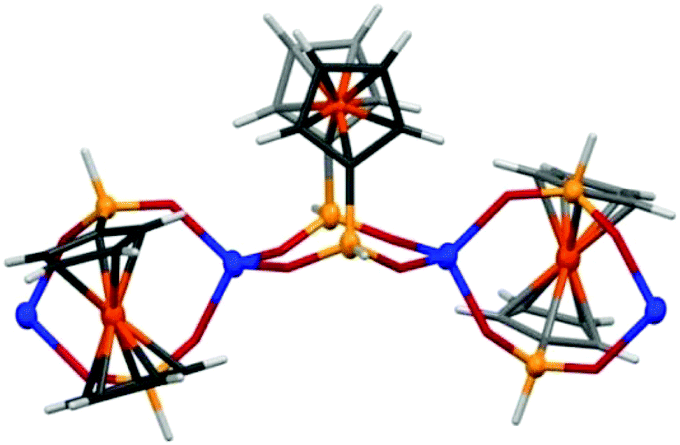
The M–O bond lengths range from 1.926(3) to 1.936(4) Å for the phosphinate oxygen atoms in 1a. The result is a 4-fold helix, in which the Co⋯Co distance between the adjacent metal centers in the chain is 4.867 Å, while the pitch of the helix is given by the unit cell c parameter, 18.659(4) for 1a and 18.840(4) Å for 2a. In such a manner each metal ion is interconnected by two ligand molecules forming 8-membered macrocycles M–O–P–O–M–O–P–O. This leads to the formation of infinite 1D helical chains along the [001] direction that generates the axial chirality in the structures. The length of the helix loop corresponds to the length of the c axis, namely 18.6 Å (Fig. 2). In the case of 1a and 2a, the dextrorotatory R-enantiomers were isolated and in the case of 1b, a levorotatory S-enantiomer was confirmed by the single crystal X-ray diffraction study.
 | ||
| Fig. 2 Helical chains of opposite chirality for 1a (R-enantiomer) and 1b (S-enantiomer). | ||
Each crystal was found to be enantiomerically pure. All the helices in a given crystal have the same handedness, either all having a right-handed thread or all being left-handed.
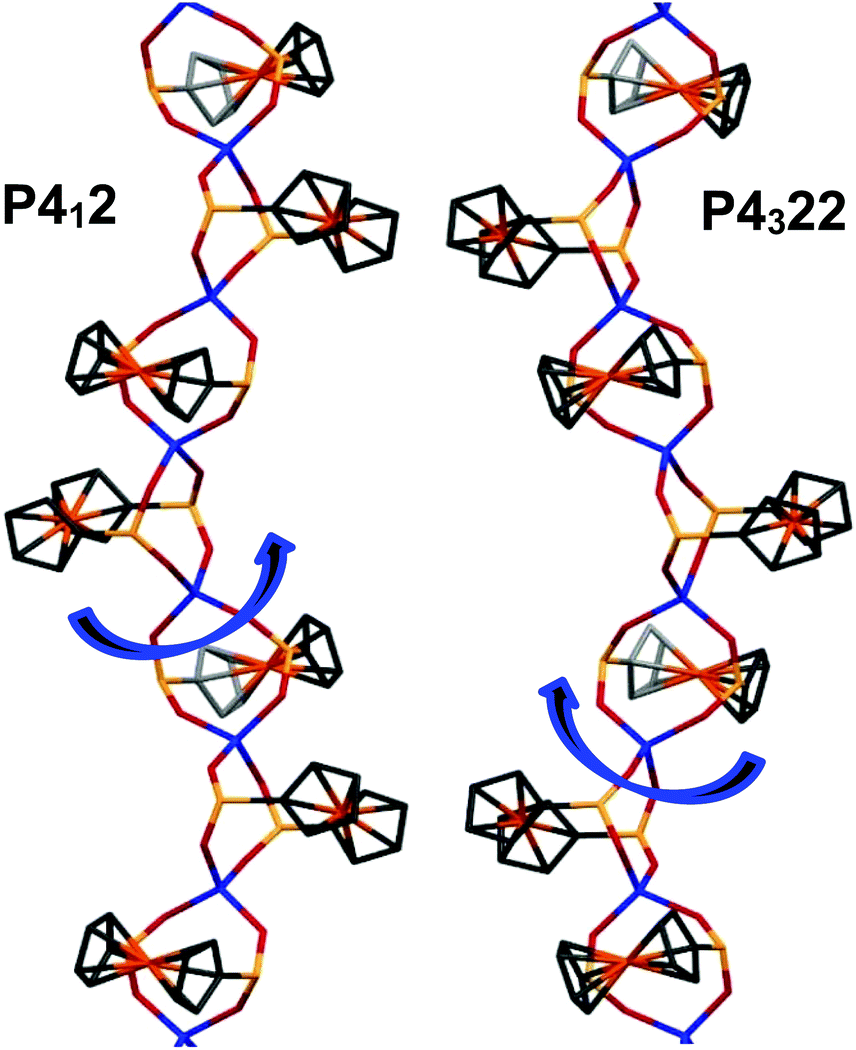
The space groups P4122 and P4322 form an enantiomeric pair, and within a given batch of crystals, there is a statistical 1:1 mixture of crystals corresponding to each of the two space groups. In the crystal structure, helixes are interacting via weak C–H⋯O bonds and van der Waals interactions, as well as P–H⋯O hydrogen bonds (see the discussion of IR and Raman spectra below). The crystal structure is stabilized by a number of C–H ⋯O interchain interactions. These, involving phosphinate and Cp from adjacent chains, have C⋯O distances in the range of 3.111–3.435 Å. This results in a dense packing of the chains, with no space for any lattice solvent molecules between the chains (Fig. 3). By analyzing the large number of reported chiral CPs that exhibit helix architecture, it can be observed that the structures of most of them (around 64%) exhibit only 2- and 3-fold screw axes; the CPs exhibiting high symmetry 4-fold screw axes are rather rare (about 22%).
 | ||
| Fig. 3 A fragment of the crystal structure of 2a (viewed along the c axis). | ||
It is well-known that spontaneous resolution generally yields a conglomerate (racemic mixture of chiral crystals). Although the ligand chirality is not a critical prerequisite for the formation of homochiral CPs and several publications report the synthesis of chiral CPs starting from achiral ligands,37 it should be noted that in many reports the yielded crystals are separated from the reaction mixtures in the form of racemates or conglomerates.38
The combination of a labile ferrocene fragment in the ligand together with five-valent phosphorus used in this work allows obtaining coordination polymers demonstrating optical activity. The synergism of both fragments leads to the formation of axial coordination polymers demonstrating spontaneous resolution during crystallization. Earlier in our studies, it was demonstrated that ferrocenylphosphinic acids themselves also demonstrate the formation of supramolecular polymeric structures. We have shown that the phosphorus atom in this series of monosubstituted ferrocenylphosphinic acids is chiral and the formation of enantiomorphous hydrogen-bonded chains with chiral recognition occurs in these compounds.29
3.2. IR and Raman spectra
Vibrational spectra of crystals are well known to be sensitive to very tiny details of the structure, which none of the other structural methods can detect.39 Below we discuss the variations of frequencies and intensities of some vibrational bands of both CPs by comparing them with the bands of compounds that are parts of CP molecules i.e. ferrocene and acid H2fcdHp to assign their structural variations. The vibrational spectra of ferrocenes are reported in the literature, see e.g.,40 including the detailed analysis of crystalline Fc.41The IR and Raman spectra of the two CPs are very similar, both in terms of frequencies and intensities (Fig. S1–S3†). The positions of νP–H bands in the spectra are red-shifted by ca. 44 cm−1 with respect to the spectrum of H2fcdHp (Fig. S1, Table S1†), suggesting that P–H⋯O short contacts revealed by the X-ray study (vide supra) represent hydrogen bonding. Red shifts are also found for the Raman bands of stretching vibration ν(Fe–C) of ferrocenyl fragments: from 310 and 316 cm−1 for H2fcdHp to 280 and 290 cm−1 for 2 and to 283 and 290 cm−1 for 1. These red shifts, as well as the blue shift of the ν(C–H) bands of ferrocenyl fragments both in the IR and Raman spectra of 1D-CPs, indicate a redistribution of electron density in ferrocenyl moieties caused by the formation of 1D-chains of the CPs.
The comparison of experimental X-ray diffraction and vibrational spectroscopy data on 1 and 2 shows the similarity of the 1D coordination polymers, nonetheless there are differences in the UV-Vis spectra of their crystals. Due to the structural similarity of both CPs, it is possible to evaluate the role of metal nature on the electronic properties of 1 and 2.
3.3. UV-Vis absorption spectra
Differences in the electronic structures of Zn(II) and Co(II) metal centers result in different colors of the corresponding CPs: Zn(II) based 1 is orange, while Co(II) based CP 2 is dark blue. These differences between CPs 1 and 2 are reflected in their UV-Vis spectra, which are compared in Fig. 4 with the spectra of initial H2fcdHp acid and ferrocene.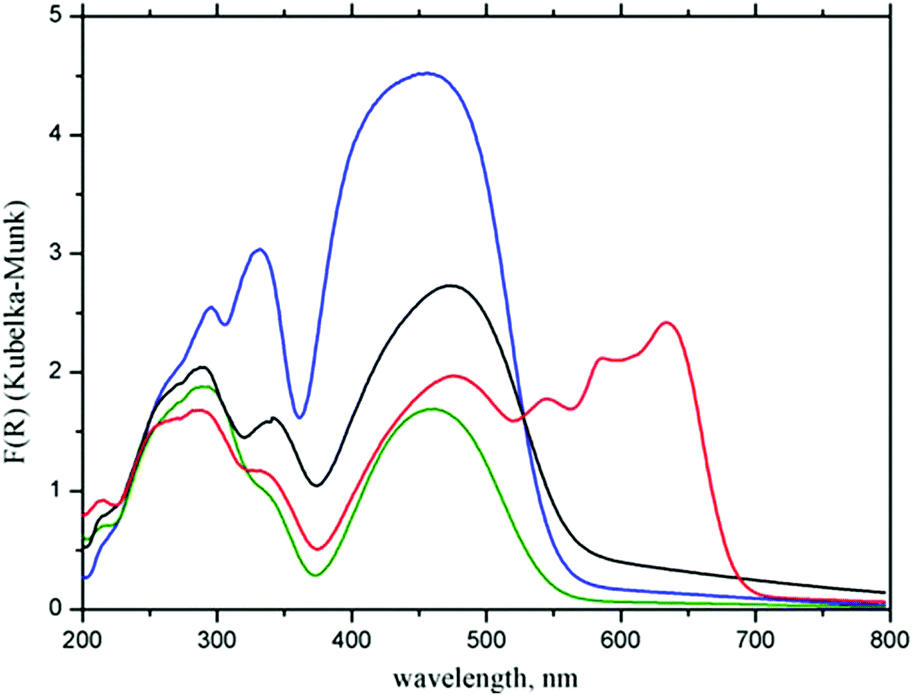 | ||
| Fig. 4 Experimental UV-Vis spectra of ferrocene (blue), H2fcdHp (green), complex 1 (black) and complex 2 (red). | ||
Bands below 500 nm in the UV-Vis spectra of the CPs and the acid are attributed to the Fc moiety.42 The formation of CP leads to the appearance of strong bands in the spectrum of complex 2 at 547, 585, and 633 nm associated with the 4A2(F) → 4T1(P) transitions specific for tetrahedral Co2+ chromophores.43
Complex 1 does not exhibit the d–d electronic transition because the d-orbitals of Zn(II) are completely occupied. Interesting, that in the spectra of complexes 1 and 2 the Fc bands are shifted to 477 nm relative to 454 nm in the spectrum of ferrocene and initial acid. The reason for this effect may be a transformation of the Fc moiety from a staggered conformation adopted in solid ferrocene at room temperature44 to an eclipsed conformation found in the crystals of complexes 1 and 2.
3.4. Thermal properties
The thermogravimetric curves of the Zn(II) and Co(II) based CPs shown in Fig. 5a indicate that the thermal properties of both samples are approximately identical: there are two mass loss steps upon heating of both samples. The first step occurs at 453 °C (2.9% mass loss) and 487 °C (1.7% mass loss) for the Zn(II) and Co(II) based CPs, respectively. The second one occurs at 951 °C (42.2% mass loss) and at 875 °C (38.3% mass loss) for the Zn(II) and Co(II) based CPs, respectively, corresponding to the decomposition.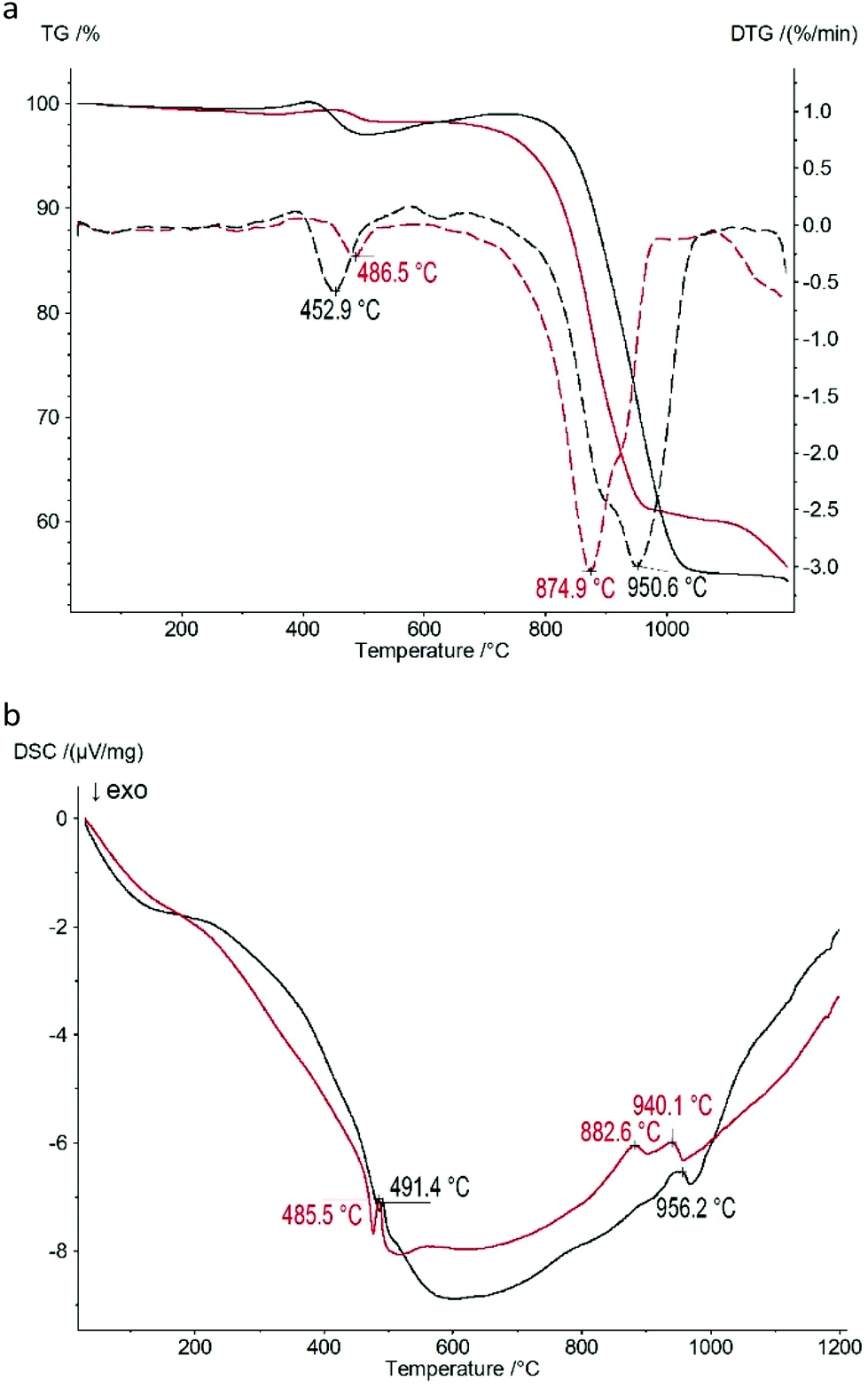 | ||
| Fig. 5 Thermal analysis data of 1 (black) and 2 (red) CPs: (a) thermogravimetry (TG, solid lines) curves and curves of the first derivative of TG (DTG, dashed lines); (b) DSC curves. | ||
One of the possible explanations of the weight increase in the ranges of 350–400 °C and 500–750 °C is the initial oxidation of P–H bonds in Zn(II) and Co(II) phosphinates to P–OH bonds and the following oxide formation during further oxidation. The corresponding thermal effects on the DSC curves (Fig. 5b) accompany these two mass loss steps: there are two endothermic peaks at 485–491 °C and 883–956 °C.
3.5. Magnetic properties of 2
The static magnetic properties of compound 2 were investigated in the temperature range of 1.8–300 K in magnetic fields up to 7 T. The temperature dependence of the static magnetic susceptibility χ(T) of 2 and the corresponding inverse susceptibility curve χ−1 are plotted in Fig. 6. The observed downturn of the susceptibility by lowering the temperature below Tmax ∼ 4.5 K (see the upper insert in Fig. 6) indicates low-dimensional antiferromagnetic interactions between the Co spins. The high-temperature (T > 150 K) data were fitted to the Curie–Weiss law, χ = C/(T − θ) + χ0, where C is the Curie constant, θ is the Curie–Weiss temperature, and χ0 is the temperature independent contribution to the magnetic susceptibility (e.g. diamagnetic). The fit yields C = 2.4erg per G2 per mole which corresponds to the high spin state of Co(II) and θ = −3 K. The negative sign of θ confirms the presence of antiferromagnetic interactions and its value relates well with the temperature Tmax at which the maximum of χ(T) is observed. Besides, the magnetic field dependence of the static magnetization M(H) was measured at 1.8 K (see the lower insert in Fig. 6). A characteristic S-shape form of the M(H) curve indicates a spin-flop transition (SPT) at around μ0HSPT ∼ 4 T typical of low-dimensional antiferromagnetic interactions. Indeed, a small magnetic energy scale of the SPT corresponding to a few Kelvin is consistent with the small values of Tmax and θ.
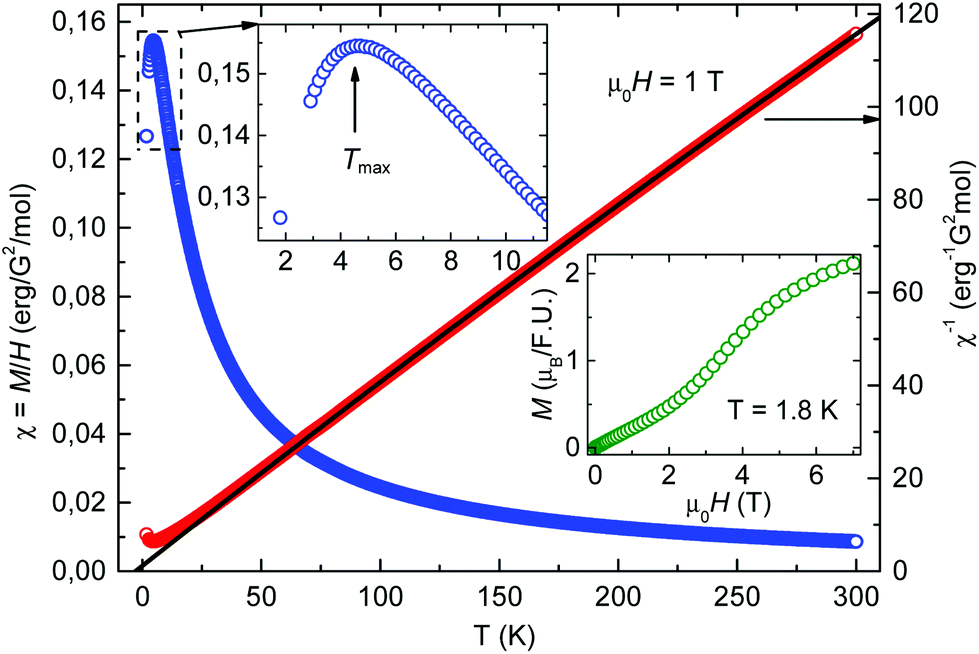 | ||
| Fig. 6 Temperature (T) dependence of the static magnetic susceptibility χ = M(T)/H of 2 and the corresponding inverse susceptibility χ−1. The black solid line represents the Curie–Weiss fit. Upper insert: χ in the low temperature range. Lower insert: Magnetic field H dependence of the static magnetization M at T = 1.8 K. | ||
3.6. Electrochemical and electrocatalytic properties of Zn and Co CPs
An electrochemical study of the redox properties of polymers 1 and 2 was carried out using a carbon paste electrode (CPE) based on a phosphonium salt as a binder, which has proven itself for this purpose.45The ferrocene fragment of 1 and 2 in the solid state was found to reversibly oxidize at 0.43–0.53 V vs. Fc+/Fc with ΔE = 105–110 mV (Table 1), that is, at potentials comparable to the ferrocene-1,10-diyl-bis(H-phosphinic acid precursor (E1/2 = 0.35 V in MeOH)29 and was much more difficult compared to unsubstituted ferrocene (0 V and ΔEp = (Eap − Ecp) = 60 mV.45 That is, the oxidation potential is determined primarily by the influence of the acceptor nature of the phosphinate group. Polymer 1 has one oxidation peak and one pronounced reduction peak at high negative potentials (Fig. 7, Table 1). Polymer 2 has two pronounced oxidation peaks (Fig. 7) and one reduction peak. The second oxidation peak of 2 can be attributed to the oxidation of a CoII/III couple; it is not observed for the zinc analog.
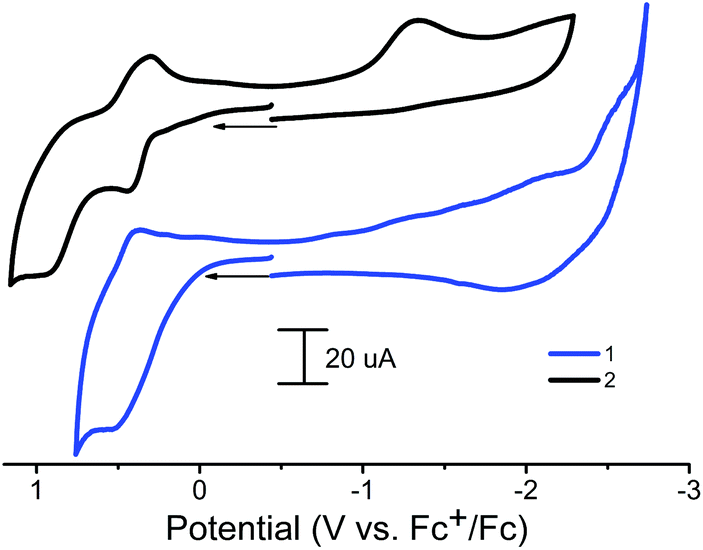 | ||
| Fig. 7 Cyclic voltammograms obtained with a modified CPE of polymers 1 and 2 (paste composition: graphite + ionic liquid + MOF) in 2 ml CH3CN (0.1 M Bu4NBF4). Potentials vs. Fc+/Fc. | ||
| Compound | Oxidation | Reduction | ||
|---|---|---|---|---|
| E p, V | E 1/2, V | ΔEf–bp, mV | E p, V | |
| 1 | 0.53 | 0.43 | 105 | −2.57 |
| 2 | 0.43 | 0.36 | 110 | −1.33 |
1 and 2 were found to be active electrocatalysts for hydrogen production using [(DMF)H]OTf46 as the proton source in acetonitrile solution (Fig. 8). At a potential of approximately −1.3 V (ref. Fc+/Fc), for both CPs, significant increases in the reduction current are observed in the presence of proton donors. The direct acid reduction on the surface of modified electrodes was also observed at −1.95 V (Fig. 8). Thus the new catalytic peak at less negative potential provides 650 mV of voltage economy. The overpotential (difference between the standard potential of the acid reduction46 and the potential of the catalytic process) for 1 and 2 is 820–840 mV. When using N,N-dimethylformamidium ([DMF(H)+]) as the acid in the acetonitrile solution, 2 CP displays a turnover frequency of 300 s−1, calculated from the catalytic current47 measured in the presence of the acid, which is among the fastest rates reported for any CP electrocatalysts in non-aqueous solutions. For 1, such a TOF calculation cannot be applied, since there is an increase in the current of the new peak.
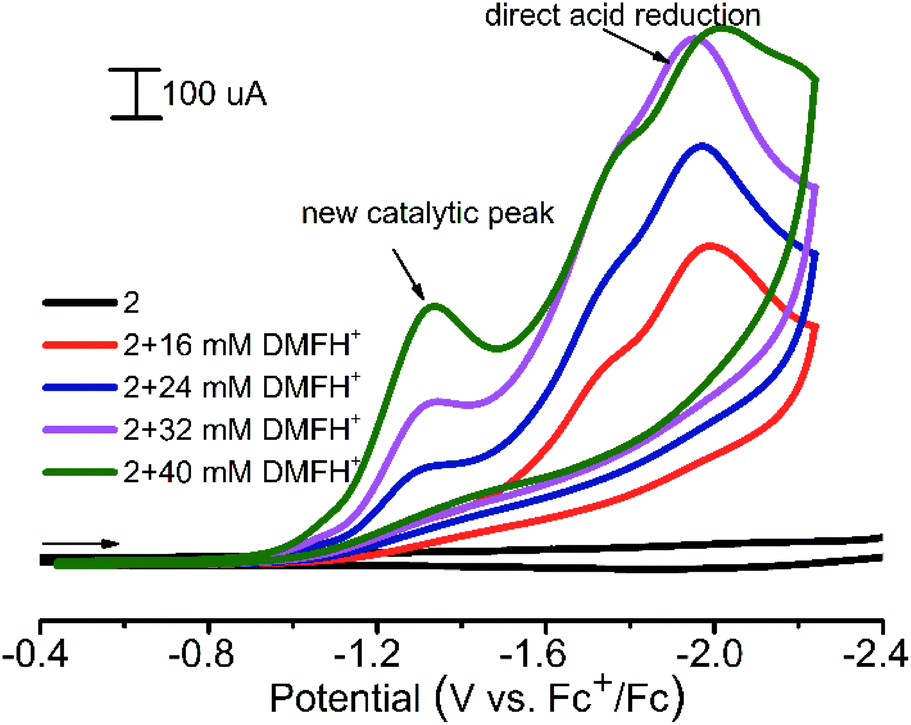 | ||
| Fig. 8 Cyclic voltammograms obtained with a modified CPE for 2 in the presence of various amounts of [(DMF)H]OTf in acetonitrile. Conditions: 0.1 V s−1 scan rate, 0.1 M Bu4NBF4. | ||
These catalysts were found to be stable in water and under strongly acidic conditions, with only trace decomposition observed over 2 weeks in the presence of 0.5 M H2SO4 or [DMF(H)+] (in the absence of current).
The electrocatalytic HER activities of CPs were investigated in the 0.5 M H2SO4 solution by linear sweep voltammetry (LSV) (Fig. 9, Table 2). As the hydrogen evolution reaction (HER) catalysts, the CPs exhibited in 0.5 M H2SO4 the overpotential η10 (at 10 mA cm−2 current density) of 340 or 450 mV, the onset overpotential of 220 or 300 mV (vs. RHE), the Tafel slope of 110 or 120 mV dec−1, correspondingly, high cycle durability and considerable long-term stability for the HER. Thus, these CPs can continuously work for 1000 cycles with negligible activity loss. TOFs in 0.5 M H2SO4 were evaluated by the literature method.7 For 1 and 2 TOFs (at 300 mV) were 4.5 10−3 and 1.5 10−3 s−1 respectively. Although this is not a record activity in the aqueous solutions of H2SO4, it is quite satisfactory among the known organometallic structures. And for the Zn and Co redox active coordination polymers based on the ferrocene-containing diphosphinate ligand, such activity is observed for the first time.
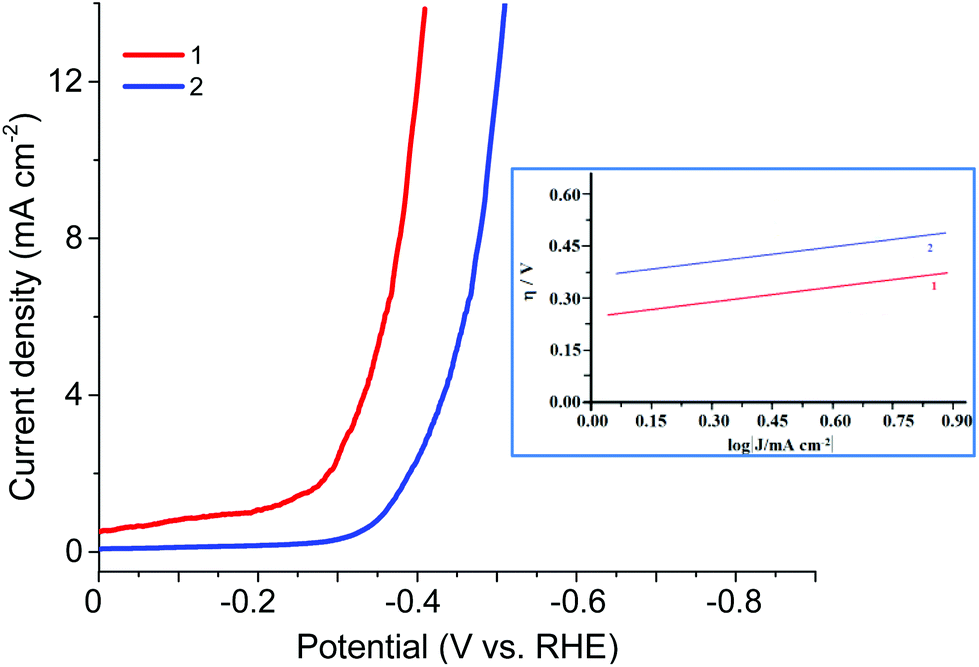 | ||
| Fig. 9 Electrochemical characterization of the prepared catalysts: LCV polarization curves obtained with a modified CPE for 1 and 2 in 0.5 M H2SO4 at 0.05 V s−1 and the corresponding Tafel plots (insertion). | ||
| −Eonset, mV | η (at 10 mA cm−2), mV | Tafel slope, mV dec−1 | |
|---|---|---|---|
| 1 | 220 | 340 | 110 |
| 2 | 300 | 450 | 120 |
We conducted the dynamic study of the coordination polymer state in water and acids over time. To control the state, we used both voltammetry and powder XRD vs. time. This allowed us to assert that in aqueous neutral media, polymers did not change their structure over a week, at least. And in acidic media (0.5 M H2SO4) the zinc polymer showed traces of decomposition after a week, and the cobalt polymer after one day (color change, the disappearance of peaks or the presence of new peaks in voltammograms and in the powder diffraction pattern, the drop of catalytic current gains) (see ESI†).
4. Conclusions
Development of efficient noble metal-free electrocatalysts for accelerating the sluggish kinetics in the hydrogen evolution reaction (HER) has received a great deal of attention. Two novel Zn and Co redox active coordination polymers based on a ferrocene-containing diphosphinate ligand as efficient electrocatalysts for the hydrogen evolution reaction with high stability have been elaborated. The catalytic activity for hydrogen evolution was discovered in both acetonitrile and water solutions with overpotentials of 820–840 and 340–450 mV correspondingly.These discovered electrocatalytic properties of redox active coordination polymers based on the ferrocene-containing diphosphinate ligand make them a potential matrix for cost-effective catalysts in electrochemical hydrogen production. These electrocatalysts consisting of widely distributed metal (Fe, Zn, Co) are promising alternatives to high-cost Pt catalysts for the HER. Purposeful modification of the structure and ligand environment of the metals in these bimetallic coordination polymers will, one may hope, increase the efficiency of catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The partial financial support from the Russian Foundation for Basic Research (project 18-29-04004) is gratefully acknowledged. The X-ray structure determination of 1a was supported by the Russian Government Program of Competitive Growth of Kazan Federal University. The authors are grateful to the Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for technical assistance in research. We thank Helmholtz-Zentrum Berlin für Materialien und Energie for allocated beam time at MX Beamline BL14.2 and travel funding.Notes and references
- L.-J. Chen and H.-B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- G. Lan, Z. Li, S. S. Veroneau, Y.-Y. Zhu, Z. Xu, C. Wang and W. Lin, J. Am. Chem. Soc., 2018, 140, 12369–12373 CrossRef CAS PubMed.
- R. Tong, Y. Zhao, L. Wang, H. Yu, F. Ren, M. Saleem and W. A. Amer, J. Organomet. Chem., 2014, 755, 16–32 CrossRef CAS.
- J. Han, M. Zhang, G. Chen, Y. Zhang, Q. Wei, Y. Zhuo, G. Xie, R. Yuan and S. Chen, J. Mater. Chem. B, 2017, 5, 8330 RSC.
- K. Hirai, H. Uehara, S. Kitagawa and S. Furukawa, Dalton Trans., 2012, 41, 3924 RSC.
- B. F. Abrahams, A. M. Bond, T. H. Le, L. J. McCormick, A. Nafady, R. Robson and N. Vo, Chem. Commun., 2012, 48, 11422–11424 RSC.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- V. Chandrasekhar, A. Chakraborty and E. Carolina Sañudo, Dalton Trans., 2013, 42, 13436 RSC.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J. M. Grenéche and J. M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259 CrossRef PubMed.
- (a) L. Yang, S. Kinoshita, T. Yamada, S. Kanda, H. Kitagawa, M. Tokunaga, T. Ishimoto, T. Ogura, R. Nagumo, A. Miyamoto and M. Koyama, Angew. Chem., Int. Ed., 2010, 49, 5348 CrossRef CAS PubMed; (b) D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò and M. Gennari, Inorg. Chem., 2017, 56, 14801–14808 CrossRef CAS PubMed.
- (a) H. R. Moon, J. H. Kim and M. P. Suh, Angew. Chem., Int. Ed., 2005, 44, 1261 CrossRef CAS PubMed; (b) L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, A. Sampieri, D. W. Lewis, A. Gómez, B. Nohra, P. Mialane, J. Marrot, F. Sécheresse, C. Mellot-Draznieks, R. N. Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078 CrossRef PubMed; (c) Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120 CrossRef CAS.
- (a) R. Horikoshi and T. Mochida, Eur. J. Inorg. Chem., 2010, 5355 CrossRef CAS; (b) V. Chandrasekhar and R. Thirumoorthi, Dalton Trans., 2010, 39, 2684 RSC; (c) L. Wang, X. Meng, E. Zhang, H. Hou and Y. Fan, J. Organomet. Chem., 2007, 692, 4367 CrossRef CAS.
- M. Gawron, C. Dietz, M. Lutter, A. Duthie, V. Jouikov and K. Jurkschat, Chem. – Eur.J., 2015, 21, 16609–16622 CrossRef CAS PubMed.
- (a) M. Meilikhov, K. Yusenko and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 9644 CrossRef CAS PubMed; (b) J. E. Halls, A. Hernán-Gómez, A. D. Burrows and F. Marken, Dalton Trans., 2012, 41, 1475 RSC; (c) K. J. Wei, J. Ni and Y. Liu, Inorg. Chem., 2010, 49, 1834 CrossRef CAS PubMed.
- (a) J. Yang, J. F. Ma, Y. Y. Liu, S. L. Li and G. L. Zheng, Eur. J. Inorg. Chem., 2005, 2174 CrossRef CAS; (b) Y. Xu, C. Ran, L. Zhu and Y. Fan, J. Coord. Chem., 2009, 62, 410 CrossRef CAS; (c) K. Hirai, H. Uehara, S. Kitagawa and S. Furukawa, Dalton Trans., 2012, 41, 3924 RSC; (d) D. J. Tranchemontagne, J. L. Mendoza-Cort8s, M. OQKeeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- J. Hynek, P. Brázda, J. Rohlìček, M. G. S. Londesborough and J. Demel, Angew. Chem., Int. Ed., 2018, 57, 5016–5019 CrossRef CAS PubMed.
- I. Carson, M. R. Healy, E. D. Doidge, J. B. Love, C. A. Morrison and P. A. Tasker, Coord. Chem. Rev., 2017, 335, 150 CrossRef CAS.
- J.-L. Du, S. J. Rettig, R. C. Thompson and J. Trotter, Can. J. Chem., 1991, 69, 277 CrossRef CAS.
- E. Oleshkevich, C. ViÇas, I. Romero, D. Choquesillo-Lazarte, M. Haukka and F. Teixidor, Inorg. Chem., 2017, 56, 5502 CrossRef CAS PubMed.
- W. Yang, H. Wang, W.-G. Tian, J. Li and Z.-M. Sun, Eur. J. Inorg. Chem., 2014, 5378 CrossRef CAS.
- F. Costantino, A. Ienco, S. Midollini, A. Orlandini, L. Sorace and A. Vacca, Eur. J. Inorg. Chem., 2008, 3046 CrossRef CAS.
- F. Cecconi, D. Dakternieks, A. Duthie, C. A. Ghilardi, P. Gili, P. A. Lorenzo-Luis, S. Midollini and A. Orlandini, J. Solid State Chem., 2004, 177, 786 CrossRef CAS.
- R. Shekurov, V. Miluykov, O. Kataeva, D. Krivolapov, O. Sinyashin, T. Gerasimova, S. Katsyuba, V. Kovalenko, Y. Krupskaya, V. Kataev, B. Bgchner, I. Senkovska and S. Kaskel, Cryst. Growth Des., 2016, 16, 5084 CrossRef CAS.
- (a) R. P. Shekurov, M. N. Khrizanforov, Y. H. Budnikova, V. V. Khrizanforova, V. A. Miluykov and O. N. Kataeva, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1551 CrossRef CAS; (b) R. P. Shekurov, A. I. Tufatullin, V. A. Milyukov, O. N. Kataeva and O. G. Sinyashin, Russ. Chem. Bull., 2014, 63, 178 CrossRef CAS; (c) V. V. Yanilkin, G. R. Nasybullin, N. V. Nastapova, T. I. Madzhidov, L. Z. Latypova, V. I. Morozov, R. P. Shekurov and V. A. Milyukov, Russ. J. Electrochem., 2015, 51, 645 CrossRef CAS; (d) R. P. Shekurov, D. R. Islamov, D. B. Krivolapov, V. A. Miluykov, O. N. Kataeva, T. P. Gerasimova and O. G. Sinyashin, Russ. Chem. Bull., 2015, 64, 1819 CrossRef CAS.
- R. P. Shekurov, V. A. Miluykov, D. R. Islamov, D. B. Krivolapov, O. N. Kataeva, T. P. Gerasimova, S. A. Katsyuba, G. R. Nasybullina, V. V. Yanilkin and O. G. Sinyashin, J. Organomet. Chem., 2014, 766, 40 CrossRef CAS.
- U. Mueller, N. Darowski, M. R. Fuchs, R. Förster, M. Hellmig, K. S. Paithankar, S. Pühringer, M. Steffien, G. Zocher and M. S. Weiss, J. Synchrotron Radiat., 2012, 19, 442 CrossRef CAS PubMed.
- M. Krug, M. S. Weiss, U. Heinemann and U. Mueller, J. Appl. Crystallogr., 2012, 45, 568 CrossRef CAS.
- A. X. S. Bruker, APEX2-Software Suite for Crystallographic Programs, Bruker AXS, Inc., Madison, WS, USA, 2009 Search PubMed.
- Bruker, SMART and SAINT Version 6.0. in Area detector control and integration software, Wisconsin Bruker Analytical X-ray Instruments Inc., Madison, USA, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS. Program for absorption correction, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- I. Carson, M. R. Healy, E. D. Doidge, J. B. Love, C. A. Morrison and P. A. Tasker, Coord. Chem. Rev., 2017, 335, 150 CrossRef CAS.
- (a) A. Verma, K. Tomar and P. K. Bharadwaj, Inorg. Chem., 2017, 56, 13629 CrossRef CAS PubMed; (b) R. Carballo, A. Castineiras, B. Covelo, A. B. Lago, E. M. Vazquez-Lopez, O. Gómez-Paz, S. Balboa and P. Inmaculada, CrystEngComm, 2018, 20, 2455 RSC; (c) S. Tripathi, R. Srirambalaji, N. Singh and G. Anantharaman, J. Chem. Sci., 2014, 126, 1423 CrossRef CAS.
- (a) K. K. Bisht and E. Suresh, J. Am. Chem. Soc., 2013, 135, 15690 CrossRef CAS PubMed; (b) I. Mihalcea, N. Zill, V. Mereacre, C. E. Anson and A. K. Powell, Cryst. Growth Des., 2014, 14, 4729 CrossRef CAS; (c) J. W. Sun, J. Zhu, H. F. Song, G. M. Li, X. Yao and P. F. Yan, Cryst. Growth Des., 2014, 14, 5356 CrossRef CAS.
- G. N. Zhizhin, B. N. Mavrin and V. F. Shabanov, The optical vibrational spectra of crystals, Moscow-Nauka, 1984 Search PubMed.
- (a) E. R. Lippincott and R. D. Nelson, Spectrochim. Acta, 1958, 10, 307 CrossRef CAS; (b) N. Mohammadi, A. Ganesan, C. T. Chantler and F. Wang, J. Organomet. Chem., 2012, 713, 51 CrossRef CAS; (c) T. P. Gryaznova, S. A. Katsyuba, V. A. Milyukov and O. G. Sinyashin, J. Organomet. Chem., 2010, 695, 2586 CrossRef CAS; (d) W. K. Winter, B. Curnutte Jr. and S. E. Whitcomb, Spectrochim. Acta, 1959, 15, 1085 CrossRef CAS; (e) I. J. Hyams, Spectrochim. Acta, Part A, 1973, 29, 839 CrossRef CAS; (f) E. Kemner, I. M. De Schepper, G. J. Kearley and U. A. Jayasooriya, J. Chem. Phys., 2000, 112, 10926 CrossRef CAS.
- (a) K. Chhor, G. Lucazeau and C. Sourisseau, J. Raman Spectrosc., 1981, 11, 183 CrossRef CAS; (b) E. M. Smirnova, V. T. Aleksanyan and A. A. Lubovich, J. Struct. Chem., 1973, 14, 772 Search PubMed.
- D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 516 CrossRef CAS.
- (a) M. G. Alexandru, T. C. Velickovic, M. Krstic, M. M. Hrubaru and C. Draghici, J. Mol. Struct., 2013, 1041, 55 CrossRef CAS; (b) A. A. Verberckmoes, B. M. Weckhuysen and R. A. Schoonheydt, Microporous Mesoporous Mater., 1998, 22, 165 CrossRef CAS.
- (a) E. O. Fischer and W. Pfab, Z. Naturforsch., B: J. Chem. Sci., 1952, 7, 377 Search PubMed; (b) P. F. Eiland and R. Pepinsky, J. Am. Chem. Soc., 1952, 74, 4971 CrossRef CAS; (c) J. D. Dunitz and L. Orgel, Nature, 1953, 171, 121 CrossRef CAS; (d) J. D. Dunitz, L. E. Orgel and A. Rich, Acta Crystallogr., 1956, 9, 373 CrossRef CAS.
- (a) O. Kataeva, K. Metlushka, K. Ivshin, A. Kiiamov, V. Alfonsov, M. Khrizanforov, Y. Budnikova, O. Sinyashin, Y. Krupskaya, V. Kataev, B. Büchner and M. Knupfer, Eur. J. Inorg. Chem., 2018, 28, 3344 CrossRef; (b) I. Taidakov, S. Ambrozevich, R. Saifutyarov, K. Lyssenko, R. Avetisov, E. Mozhevitina, A. Khomyakov, M. Khrizanforov, Y. Budnikova and I. Avetissov, J. Organomet. Chem., 2018, 867, 253 CrossRef CAS; (c) T. V. Gryaznova, M. N. Khrizanforov, K. V. Kholin, M. A. Vorotyntsev, K. V. Gor'kov, N. V. Talagaeva, M. V. Dmitrieva, E. V. Zolotukhina and Y. H. Budnikova, Catal. Lett., 2018, 148(10), 3119 CrossRef CAS; (d) M. N. Khrizanforov, S. V. Fedorenko, A. R. Mustafina, K. V. Kholin, I. R. Nizameev, S. O. Strekalova, V. V. Grinenko, T. V. Gryaznova, R. R. Zairov, R. Mazzaro, V. Morandi, A. Vomiero and Y. H. Budnikova, Dalton Trans., 2018, 47, 9608 RSC; (e) M. K. Kadirov, S. T. Minzanova, I. R. Nizameev, L. G. Mironova, I. F. Gilmutdinov, M. N. Khrizanforov, K. V. Kholin, A. R. Khamatgalimov, V. A. Semyonov, V. I. Morozov and D. M. Kadirov, Inorg. Chem. Front., 2018, 5(4), 780 RSC; (f) T. Gryaznova, Y. Dudkina, M. Khrizanforov, O. Sinyashin, O. Kataeva and Y. Budnikova, J. Solid State Electrochem., 2015, 19(9), 2665 CrossRef CAS; (g) O. Kataeva, M. Khrizanforov, Y. Budnikova, D. Islamov, T. Burganov, A. Vandyukov, K. Lyssenko, B. Mahns, M. Nohr, S. Hampel and M. Knupfer, Cryst. Growth Des., 2016, 16(1), 331–338 CrossRef CAS.
- B. D. McCarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Inorg. Chem., 2014, 53, 8350–8361 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD, IR, Raman, and other information for the coordination polymers. CCDC 1861598, 1861599 and 1866808. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04618b |
| This journal is © The Royal Society of Chemistry 2019 |