
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical predictions suggest carbon dioxide phases III and VII are identical†
Watit
Sontising
,
Yonaton N.
Heit
 ,
Jessica L.
McKinley
and
Gregory J. O.
Beran
*
,
Jessica L.
McKinley
and
Gregory J. O.
Beran
*
Department of Chemistry, University of California, Riverside, California 92521, USA. E-mail: gregory.beran@ucr.edu
First published on 5th September 2017
Abstract
Solid carbon dioxide exhibits a rich phase diagram at high pressures. Metastable phase III is formed by compressing dry ice above ∼10–12 GPa. Phase VII occurs at similar pressures but higher temperatures, and its stability region is disconnected from III on the phase diagram. Comparison of large-basis-set quasi-harmonic second-order Møller–Plesset perturbation theory calculations and experiment suggests that the long-accepted structure of phase III is problematic. The experimental phase III and VII structures both relax to the same phase VII structure. Furthermore, Raman spectra predicted for phase VII are in good agreement with those observed experimentally for both phase III and VII, while those for the purported phase III structure agree poorly with experimental observations. Crystal structure prediction is employed to search for other potential structures which might account for phase III, but none are found. Together, these results suggest that phases III and VII are likely identical.
1 Introduction
Carbon dioxide is one of the most fundamental chemical species on earth, yet its solid-phase behavior at high pressures continues to confound. Starting with the 1994 powder X-ray diffraction structure of phase III,1 researchers have mapped out a rich phase diagram with 8–10 crystalline phases that range from molecular crystals at lower pressures to extended covalent and ionic phases at high pressures.2 Experimental characterization of these crystal structures and their solid–solid phase boundaries has often proved challenging, with considerable kinetic path-dependence and hysteresis in the phase transitions, difficulty in obtaining high-quality diffraction data, and sharp pressure gradients within samples that complicate spectroscopic measurements. As a result, the literature on high-pressure carbon dioxide contains numerous contradictory experimental interpretations. Fortunately, substantial advances in computational modeling of molecular materials3 mean that ab initio calculations can help resolve such controversies and play an integral role in molecular crystallography. Using high-level electronic structure calculations, the present study investigates several molecular crystal phases of carbon dioxide and demonstrates quantitative agreement between predicted and experimentally observed structural, mechanical, and spectroscopic properties for several of them. However, the same theoretical evidence indicates that the long-accepted structure of phase III carbon dioxide is inconsistent with spectroscopic data, and that phases III and VII are likely identical.Controversy has long surrounded the high-pressure phase diagram of carbon dioxide (Fig. 1). Substantial research developed a framework in which lower pressure molecular crystal phases I, III, and VII transition to “intermediate bonding” phases II and IV at moderate pressures (∼20–40 GPa), before eventually forming extended covalent or ionic phases at higher pressures.2 The intermediate bonding phases purportedly exhibit elongated and/or bent carbon dioxide molecules and abnormally large bulk moduli.5–7 However, subsequent experiments8,9 and density functional theory (DFT) calculations10,11 have challenged this interpretation, suggesting instead more traditional molecular crystal structures at intermediate pressures, with linear carbon dioxide molecules and typical bulk moduli.
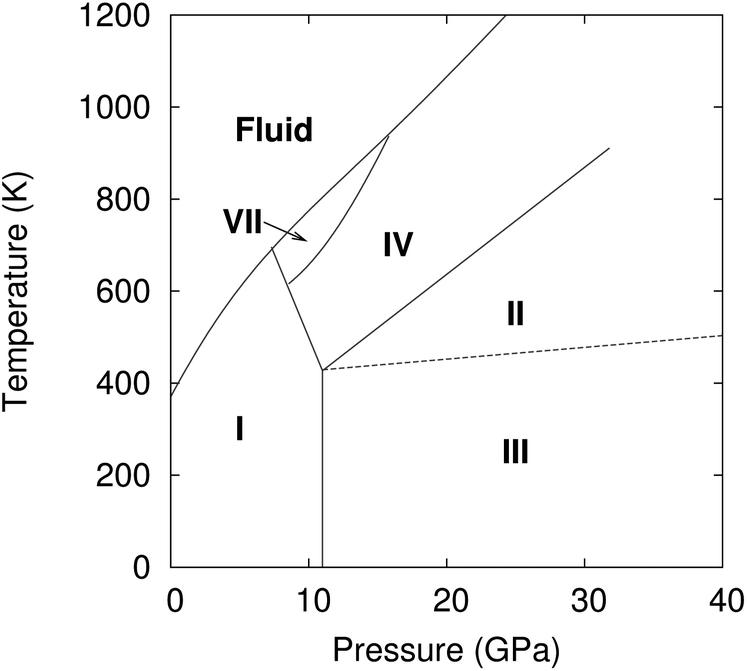 | ||
| Fig. 1 Phase diagram of carbon dioxide up to 40 GPa. | ||
Phases III and VII represent another conundrum. X-ray diffraction studies suggest that both phases adopt similar Cmca space group structures (Fig. 2).1,12 The primary differences lie in effectively swapping the a and b lattice constants and slightly altering the angle the molecule forms relative to the c crystallographic axis. Phase III can be formed at room temperature by compressing phase I (dry ice) to pressures above ∼12 GPa, though the precise phase boundary remains unclear due to the sluggish martensitic phase change.2 Obtaining high-quality diffraction data for phase III has proved challenging, and the currently accepted structure was extracted from powder X-ray diffraction on a sample believed to contain a mixture of phases I and III.1 Despite routinely being included in the low-temperature region of the phase diagram, phase III is actually believed to be metastable and monotropic relative to phase II. It converts to phase II upon annealing to ∼500 K at 12 GPa or above.5,9
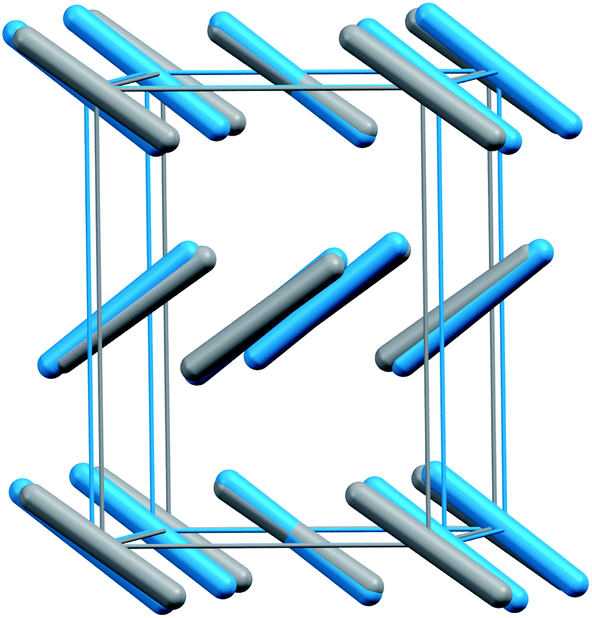 | ||
| Fig. 2 Structure overlay of the experimental crystal structures for phases III (blue) and VII (gray). Root-mean-square deviation4 = 0.24 Å. | ||
Phase VII occurs in a narrow pressure and temperature region around 15–17 GPa and 750 K,2 and producing it experimentally can also be challenging.12,13 Nevertheless, its structure was determined via X-ray diffraction on single crystals grown from the melt. Given the difficulty of obtaining quality diffraction data for phase III and the correspondingly poor constraints on its structure,12,14,15 the possibility that phases III and VII were actually the same phase was raised immediately.12 However, the non-contiguous existence domains for III and VII in the phase diagram and subtle differences in the Raman spectra were cited in favor of there being two distinct phases.12
Resolving these issues experimentally has proved challenging. Ab initio crystallography plays an increasingly important role in molecular crystals, materials, and even biological systems. Computational refinement of experimental crystal structures has long been integral in many studies, and advances in crystal structure prediction16,17 have made ab initio structure determination even more viable. Unfortunately, energy alone is often an insufficient descriptor—one commonly predicts multiple potential structures whose energies are sufficiently close so as to prevent clear discrimination. By predicting and comparing additional spectroscopic observables such as infrared, Raman, or nuclear magnetic resonance spectra, however, one can markedly increase confidence in the structural assignments.3,18–21
Here, several molecular crystalline phases of carbon dioxide are revisited with large basis, quasi-harmonic second-order Møller–Plesset perturbation theory (MP2) electronic structure theory calculations.22,23 These high-level calculations quantitatively reproduce structures, mechanical properties, and Raman spectra across most of the phases considered. However, these models do not predict a distinct phase III structure whatsoever. Moreover, even if the experimental structure were correct, the predictions here indicate that its Raman spectrum would differ from the experimentally observed spectra. In contrast, the predictions for phase VII are consistent with those observed experimentally for phase III.
These high-level ab initio calculations are made feasible for crystalline carbon dioxide using the fragment-based hybrid many-body interaction (HMBI) model.24,25 The HMBI model partitions the crystal into molecular fragments. Unit cell monomers and short-range dimers are treated quantum mechanically, while longer-range dimers and many-body effects are approximated with a classical AMOEBA26,27 polarizable force field. A quasi-harmonic phonon treatment was employed to incorporate zero-point vibrational energy and thermal vibrational effects computed at the same MP2 level of theory into the model. See Section 4 for more details.
Previous quasi-harmonic MP2 and coupled cluster singles, doubles, and perturbative triples (CCSD(T)) HMBI calculations on phase I carbon dioxide predicted the experimental thermal volume expansion within 2%, the experimental sublimation enthalpy within 1.5 kJ mol−1, and the experimental sublimation entropy within 2 J mol−1 K−1 between 0–200 K.22 The sublimation point was predicted within 3 degrees Celsius.23 Differences between MP2 and CCSD(T) for phase I in those studies were small: 0.1 cm3 mol−1 (less than 1%) in molar volume and 0.3 kJ mol−1 (1%) in lattice energy. Given the high computational cost of coupled cluster calculations compared to MP2 ones and their small marginal impact on the results in those earlier studies, MP2 calculations are used here.
2 Results and discussion
To begin, compare the predicted and experimental structures for several different molecular crystalline phases. As can be seen in Fig. 3 and 4, complete basis set quasi-harmonic MP2 predicts the phase I, II, and VII lattice parameters and unit cell volumes in excellent agreement with experiment, with root-mean-square deviations of only 0.01–0.05 Å. The errors in the predicted room-temperature lattice constants never exceed 1% across a broad pressure range. The phase II predictions indicate a normal 1.155 Å C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length at 25.8 GPa, in excellent agreement with the 2014 crystal structure9 and DFT calculations,10,11 and contrary to earlier suggestions of an “intermediate bonding” structure with extended 1.33 Å bonds.6 Only one experimental crystal structure has been reported for phase VII (at 726 K and 12.1 GPa). Despite the high temperature which is more challenging for a quasi-harmonic approximation, only the 1.4% error in the a lattice constant exceeds this 1% error threshold.
O bond length at 25.8 GPa, in excellent agreement with the 2014 crystal structure9 and DFT calculations,10,11 and contrary to earlier suggestions of an “intermediate bonding” structure with extended 1.33 Å bonds.6 Only one experimental crystal structure has been reported for phase VII (at 726 K and 12.1 GPa). Despite the high temperature which is more challenging for a quasi-harmonic approximation, only the 1.4% error in the a lattice constant exceeds this 1% error threshold.
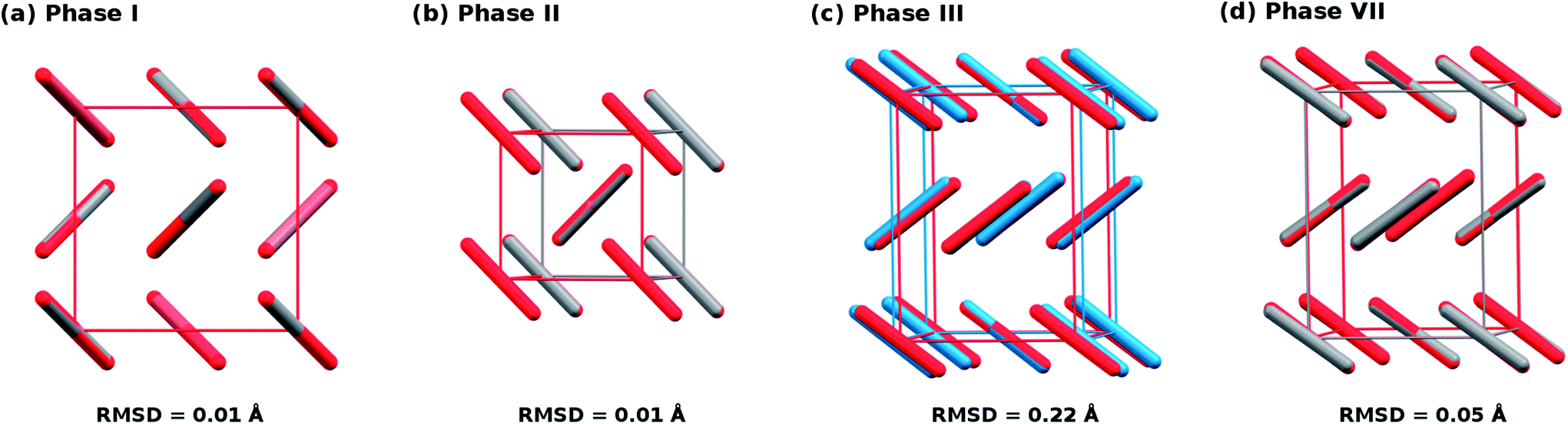 | ||
| Fig. 3 Overlays and root-mean-square deviations (RMSD) between MP2-predicted (red) and experimental crystal structures for (a) phase I at 7.46 GPa and room temperature,1 (b) phase II at 25.8 GPa and room temperature,9 (c) phase III at 11.8 GPa and room temperature,1 and (d) phase VII at 12.1 GPa and 726 K.12 Note the discrepancy between theory and experiment for the unit cell of phase III. | ||
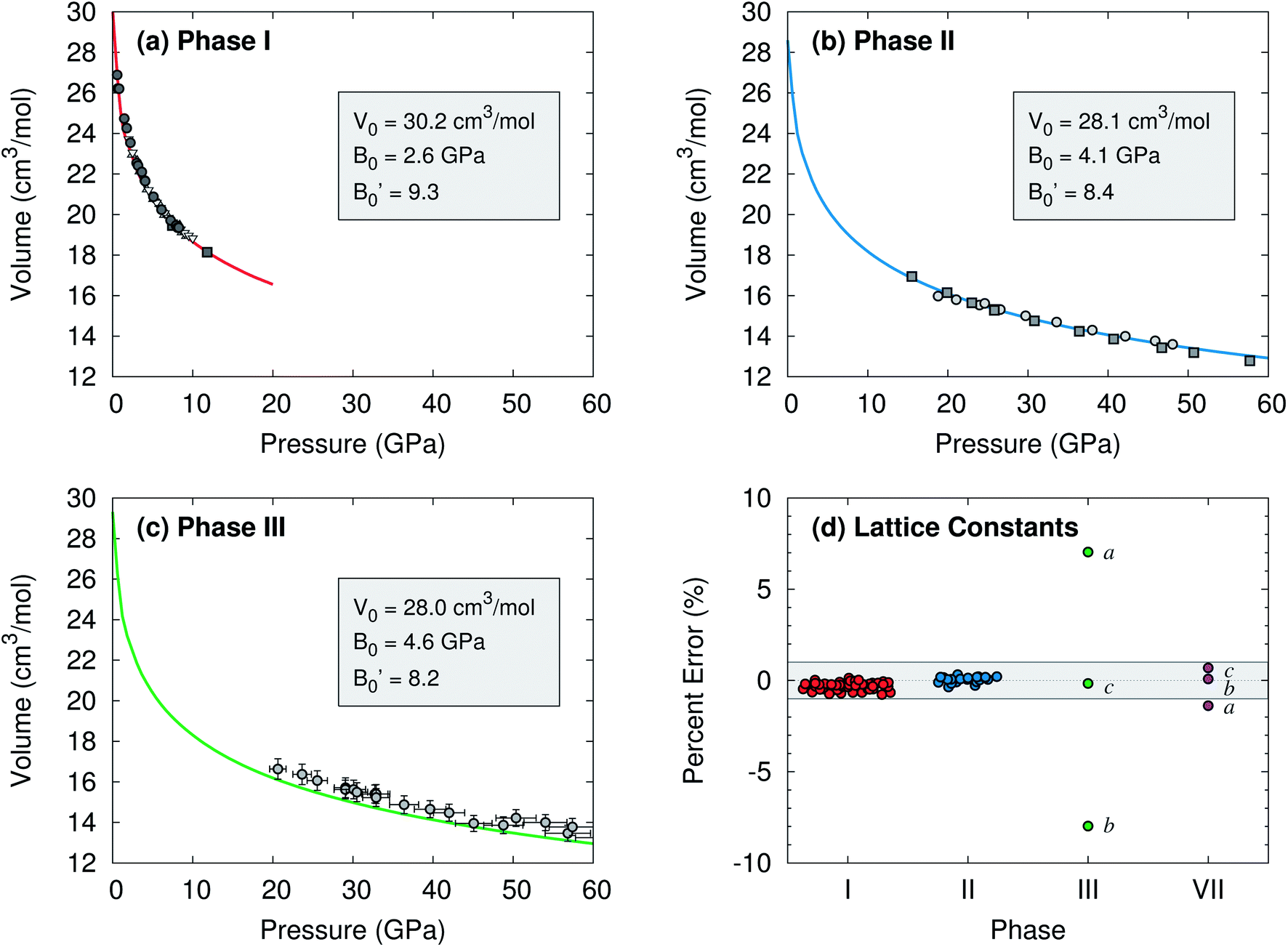 | ||
| Fig. 4 Comparison of predicted complete-basis-set MP2 (lines) and experimental (points) room temperature isotherms for (a) phase I,1,28–30 (b) phase II,6,9 and (c) phase III15 carbon dioxide. Vinet equation of state31 parameters fitted to the MP2 volumes are listed. ESI Tables S1–S4† show good agreement between the predicted and literature values for specific structures and the equation of state data. (d) Errors in the predicted lattice constants versus experiment over various pressures at room temperature, except for phase VII which is at 726 K. The shaded band indicates ±1% error. | ||
The situation for phase III is completely different. As shown in Fig. 4c and d, the predicted volumes are modestly smaller than the experimentally reported values, and the errors in the a and b lattice parameters relative to the experimental crystal structure are 5–10 fold larger than those for the other phases. The 0.22 Å RMSD between the experimental and predicted phase III structure is twenty-times larger than that for phases I and II.
Both conventional electronic and quasi-harmonic MP2 free energy relaxation of the experimental phase III structure always converge to the phase VII structure, regardless of temperature or pressure. In fact, no reported electronic structure calculation on phase III predicts a distinct phase III structure.10,11,32–34 Even if phase III is only metastable relative to phase II as inferred experimentally, it should exist as a local minimum on the free energy surface that is distinct from phase VII.
Raman spectroscopy provides further evidence that the phase III structure is problematic. Consider the librational phonons, which are sensitive to crystal packing and do not suffer from the anharmonic modeling complexities35 of the Fermi resonance that occurs at higher frequencies. Fig. 5 compares room-temperature experimental Raman spectra against those computed at the MP2/aug-cc-pVDZ level using unit cell parameters from the complete basis set quasi-harmonic MP2 calculations. Note that switching to the larger aug-cc-pVTZ basis has only a small effect on the predicted spectrum (ESI Fig. S2†).
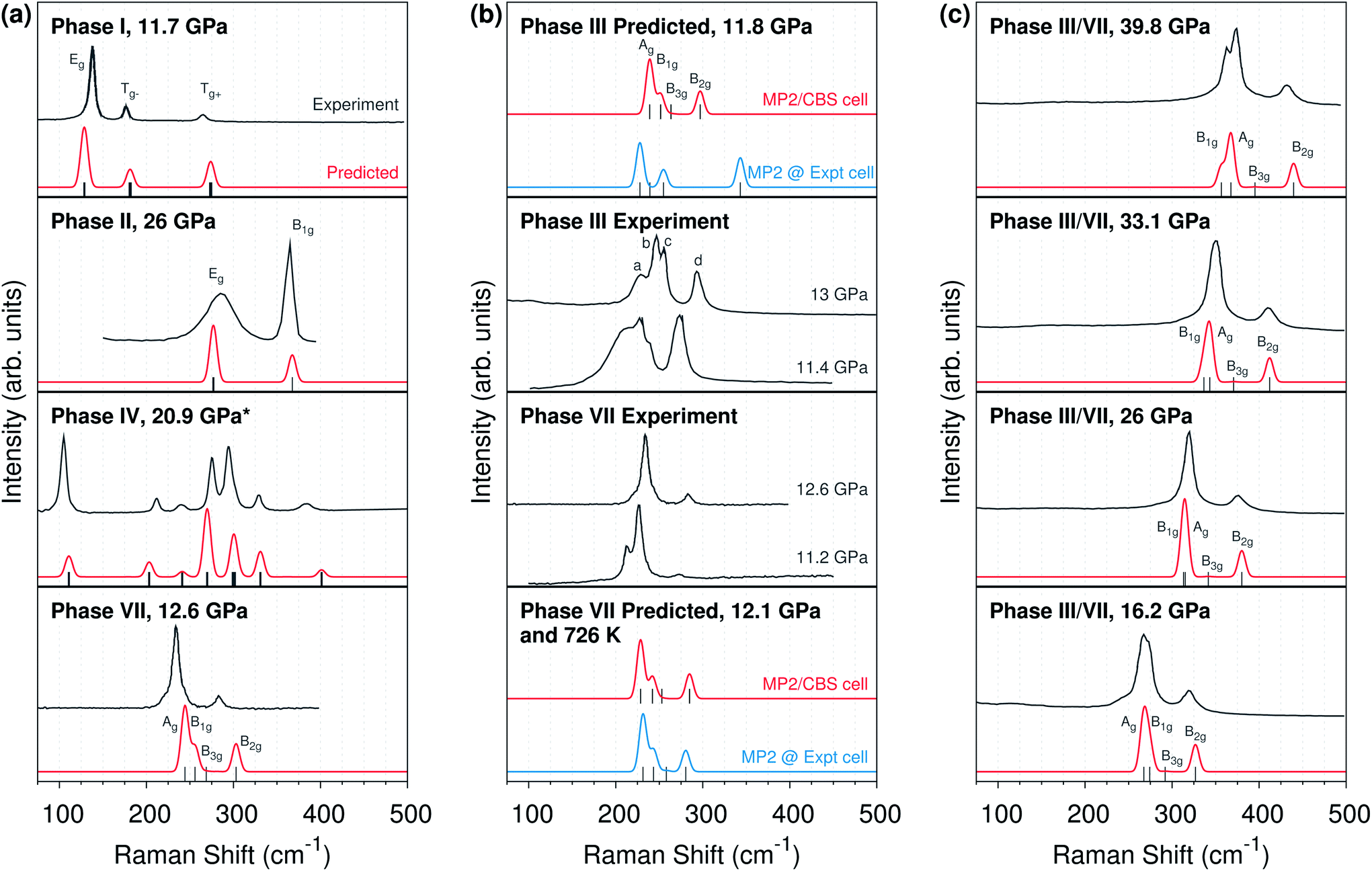 | ||
| Fig. 5 (a) Comparison of MP2-predicted and experimental lattice phonon Raman spectra for phases I,14 II,9 IV,8 and VII12 carbon dioxide. (b) Comparison of Raman spectra for phases III and VII,12,14 including predicted spectra using either quasi-harmonic complete basis set MP2 (“MP2/CBS cell”) or experimentally determined unit cell parameters (“Expt cell”). (c) Pressure dependence of the experimental phase III14 and predicted phase VII Raman spectra. All spectra are at room temperature unless otherwise indicated. *The phase IV spectrum employs an MP2/aug-cc-pVDZ optimized cell instead of a complete basis set quasi-harmonic MP2 one. | ||
Fig. 5a shows that the predicted Raman peak positions and intensities generally agree very well with experiment for several different molecular phases of carbon dioxide. For phases I and II, the predicted peaks lie within ∼10 cm−1 of experiment. Similarly good results are obtained at other pressures as well (ESI Fig. S4 and S5†). The phase II calculations confirm the assignments9,10 of the broad band near 300 cm−1 to the doubly-degenerate Eg mode, and the band near 365 cm−1 to the B1g mode, contrary to earlier reports.6 Note that the experimental broadening of the Eg mode is sample dependent, and it has been attributed to microscopic strains which may lift the two-fold degeneracy via orthorhombic distortion9,10—factors which are not present in the modeling.
For phase VII, agreement between the predicted and experimental structures is also reasonably good, with the predicted frequencies ∼10–15 cm−1 too high. Although this study does not perform complete-basis MP2 quasi-harmonic calculations for the larger phase IV unit cell, even the Raman spectrum predicted for the MP2/aug-cc-pVDZ optimized phase IV cell is in good agreement with the experiment spectrum (due to error cancellation between the small basis and the neglect of thermal expansion; ESI Fig. S3†).
Consider next the comparison of phase III and VII spectra in Fig. 5b. The Cmca structures should exhibit four Raman-active lattice modes. Experimentally, these modes have been assigned to the four peaks which are labeled a–d in the phase III spectrum at 13 GPa. The MP2 predictions concur with the three higher-frequency modes, b–d. However, they suggest the fourth Raman-active mode is not a, but rather a low-intensity B3g mode in between the c and d modes.
Focus first on the three major peaks b–d which are clearly present in both the experimental and predicted spectra. The positions and intensities of these peaks in the complete basis set MP2 phase VII structure are consistent with both the experimental phase III and VII spectra. Of course, that consistency alone is insufficient to rule out the phase III structure. However, theory allows one to predict what the Raman spectrum would look like if the phase III structure were correct. Although the phase III structure is not a stationary point on the free energy surface, one can start with the purported experimental structure of phase III, freeze the lattice constants, relax the atomic positions, and predict the Raman frequencies and intensities. This differs from the other Raman calculations here only in obtaining the lattice constants from the experimental structure instead of from complete basis set quasi-harmonic MP2 calculations. The phase III structure optimized with fixed experimental lattice constants reproduces the claimed experimental structure very well (RMSD 0.03 Å, ESI Fig. S1†). Strikingly, adopting the purported experimental structure shifts the highest-frequency band ∼50 cm−1 to almost 350 cm−1 at 11–12 GPa (Fig. 5b, top panel), versus below 300 cm−1 experimentally. That ∼50 cm−1 disagreement between theory and experiment is several-fold larger than the disagreements observed between theory and experiment for any of the other phases examined here.
For comparison, performing the same procedure on the 726 K experimental phase VII structure12 results in a predicted spectrum that is in excellent agreement with both the complete basis set MP2 cell Raman spectrum at the same temperature and the experimental room-temperature Raman at ∼11–13 GPa. In other words, while the experimentally reported phase VII structure is consistent with the predicted one in terms of both structure and Raman activity, the putative phase III structure is neither a stationary point on the free energy surface, nor is its predicted Raman spectrum compatible with the experimentally observed one.
If the phase III structure is incorrect, might some other unknown structure account for the experimental data? To investigate this possibility, evolutionary algorithm-driven crystal structure prediction was performed with the OPLS-AA force field36 to generate potential carbon dioxide crystal structures with either two or four molecules in the unit cell at ambient pressure, followed by subsequent structural relaxation at 11.8 GPa with planewave PBE-D2. The crystal structure prediction generated phase I, II, and VII (phase IV has more than four molecules in the unit cell), along with 22 other potential structures within 10 kJ mol−1 of the most stable one (Fig. 6). However, none of the other structures has a simulated powder X-ray diffraction pattern that is plausibly consistent with the experimental phase III one (ESI Section S6†). Of course, the potential for structures with a different number of molecules in the cell or otherwise missed by this crystal structure prediction cannot be ruled out.
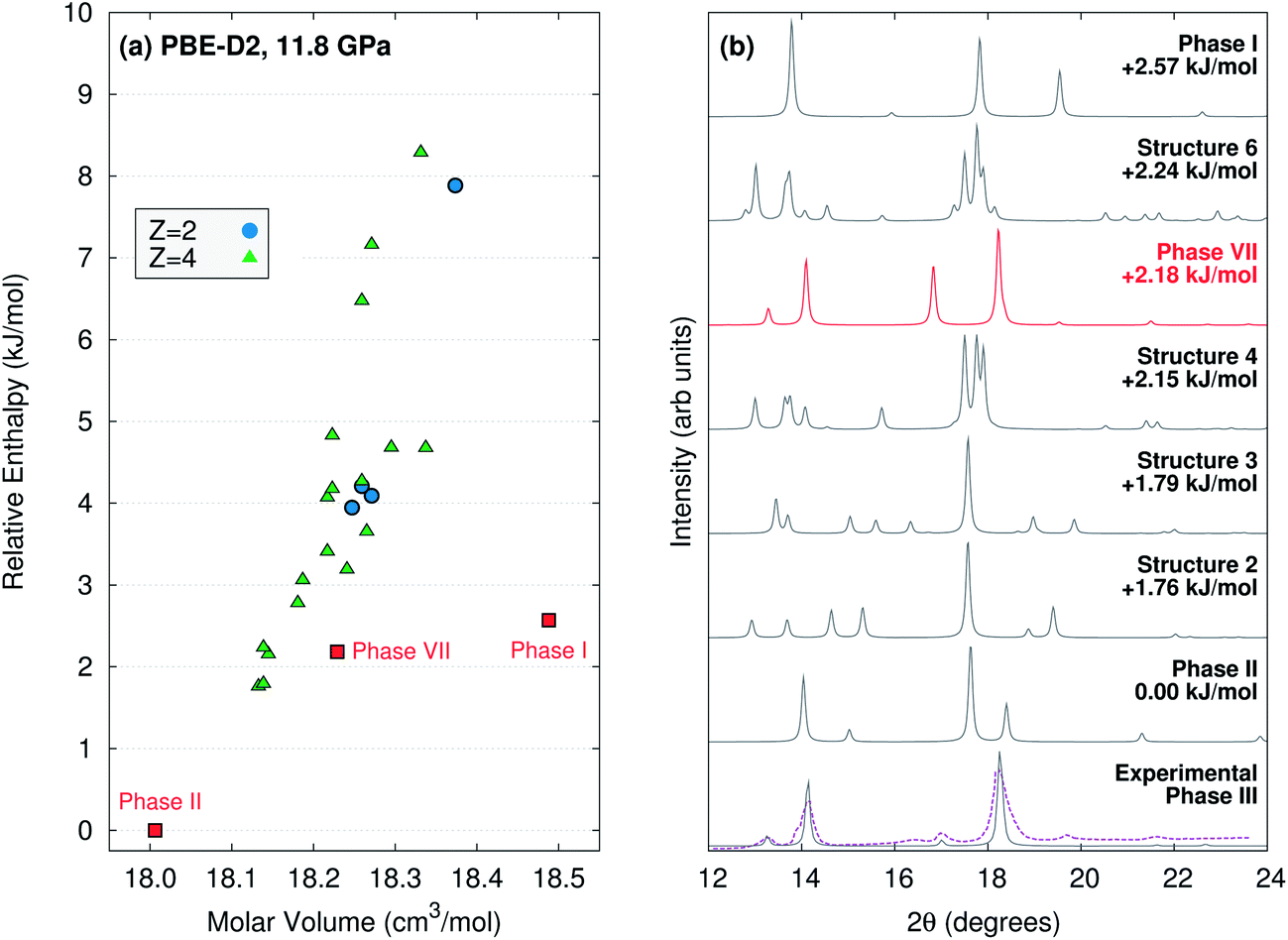 | ||
| Fig. 6 (a) PBE-D2 crystal energy landscape for potential carbon dioxide crystal structures at 11.8 GPa with Z = 2 or 4 molecules in the unit cell. (b) Comparison for simulated powder X-ray diffraction patterns for the lowest-lying PBE-D2 predicted structures against the actual1 (purple) and simulated experimental phase III (using the purported structure) ones. Aside from the purported phase III structure, only the phase VII structure plausibly corresponds to the experimental data. Simulated diffraction patterns for all 25 low-energy structures are provided in ESI Section S6.† | ||
Nevertheless, in the absence of other viable phase III structures, the most obvious alternative is that phase III and VII are in fact the same, as was first raised (and subsequently discounted) by Giordano and Datchi.12 Raman spectroscopic evidence supports this hypothesis. The phase VII predictions quantitatively reproduce the pressure dependence of the phase III Raman spectra over tens of GPa (Fig. 5c). The predicted phase VII Ag and B1g modes cross at 25 GPa, just like experimentally observed b and c modes for phase III (ESI Fig. S6†).
The most significant disagreement between theory and experiment for phases III and VII stems from mode a, which appears as a lower-frequency shoulder on mode b in some experiments. Neither our calculations nor previous MP2 calculations33 reproduce this shoulder. Instead, they predict the low-intensity fourth B3g librational mode between experimental modes c and d. Although the DFT frequencies predicted by Bonev et al.10 provide nominal agreement with the experimental frequencies for modes a–d, closer inspection of their symmetry character and the errors expected from the neglect of van der Waals dispersion in those calculations suggests they are actually more consistent with the MP2 results here (ESI Section S5†).
Several experimental details also support the possibility that the peak ascribed to mode a has erroneously been attributed to phase III. First, Raman spectra in the phase I–III transition region cannot always be described as a simple combination of the modes for the two phases14 The transition between phases I and III is notoriously sluggish over a broad pressure range, and other difficult-to-assign peaks are common during the transition. The Raman spectra is sensitive to strain and behaves very differently upon pressure loading and unloading. For example, the intensity of mode a is considerably stronger upon loading to 16.8 GPa than it is upon unloading to 16.2 GPa.14 See also the significant differences in the experimental intensity of the mode a peak for phase VII at 11.2 and 12.6 GPa (Fig. 5b).12
Second, the intensity of mode a decreases rapidly as pressure is increased beyond the phase transition region, and it is difficult to observe above ∼20 GPa.14 Assigning it to one of the four Raman-active librational modes would require that its intensity exhibit substantially larger pressure dependence than is exhibited by any of the other modes experimentally or theoretically. Third, experimental Raman spectrum of the analogous Cmca phases of CS2 (ref. 37) and C2H2 (ref. 38) support the theoretical assignment for CO2. In both species, only three of the expected four librational modes exhibit appreciable intensity, and the lowest two frequency modes are the ones that cross as a function of pressure (instead of the middle two modes b and c according to the experimental assignments for CO2). MP2/aug-cc-pVDZ calculations on CS2 suggest the fourth unobserved mode is indeed a weak-intensity one, just as predicted for CO2. Taken together, this evidence bolsters the case that mode a is an artifact of the phase transition rather than a librational mode of phase III, and that the experimental phase III Raman spectra are well-reproduced by the phase VII structure instead of the phase III one.
3 Conclusions
To summarize, large-basis, quasi-harmonic MP2 electronic structure calculations accurately reproduce experimentally observed structural, mechanical, and spectroscopic properties for several different molecular crystal phases of carbon dioxide across broad pressure range. However, theoretical optimization of the purported structure for phase III relaxes directly to phase VII. Even if phase III is only metastable, the existence of two distinct phases should translate to two separate free energy basins, but only one is found computationally. Furthermore, the claimed experimental phase III structure would produce a Raman spectrum in the librational region that disagrees with the experimentally observed ones. In contrast, Raman spectra predicted for phase VII agree well with the experimentally observed phase III and VII ones over a broad pressure range.Based on the above results and the failure to identify a plausible alternative structure, we propose that phases III and VII are in fact the same. Although this hypothesis accounts for the data discussed above, questions remain. While the subtle differences in the experimental phase III and VII Raman spectra might be attributed to the variations arising from microstrain or other experimental complications, the apparent disconnect between the phase III and VII regions in the phase diagram is more difficult to rationalize. On the one hand, there is no contradiction in having phase III/VII be kinetically accessible in the phase III region and thermodynamically stable in the phase VII region. Instead of phase III being monotropically related to phases II and/or IV, phase III/VII would be enantiotropically related to them.
On the other hand, if the two phases are the same, why is phase VII seemingly difficult to form from phase I (it is typically formed from the melt instead),13 while phase III forms readily? Perhaps given the sluggish nature of the phase I → III/VII transition and the narrow region of phase VII stability, the transformation from I → VII upon isothermal compression near 725 K is incomplete before one enters the region of phase IV stability. Similarly, why can one not form phase III kinetically via isothermal compression of phase I, then heat it to the phase VII region of thermodynamic stability without it transforming to phase II? If the kinetic barrier to transforming phase III → II is relatively small, maybe heating metastable phase III/VII from ambient temperatures provides sufficient thermal energy to convert to the more stable phase II before one reaches the regime of phase III/VII thermodynamic stability. Interestingly, one can quench phase VII down to room temperature, suggesting that the rate of heating may be significant. New experiments that investigate the crystal structure of phase III and its relationship to phase VII are clearly needed.
4 Methods
Quasi-harmonic structure optimizations
Crystal structure optimizations were performed by minimizing the free energy as a function of temperature and pressure, G(T,P):| G(T,P) = Uel + PV + Fvib(T) | (1) |
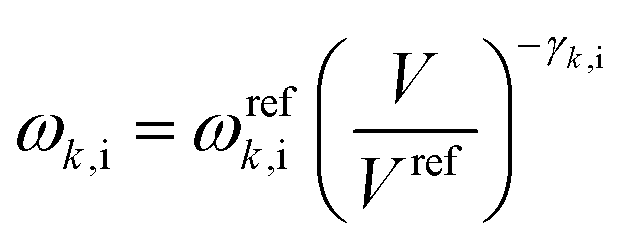 | (2) |
See ref. 22 and 23 for details.
Electronic structure calculations
The electronic energy calculations were performed using the fragment-based HMBI model.24,39 One-body and short-range two-body contributions were computed at the density-fitted complete basis set MP2 level using Molpro 2012,40 while the long-range and many-body contributions were evaluated using the AMOEBA force field, as implemented in Tinker 6.3.41 Complete basis set extrapolation of the energies, forces, and Hessians was performed via standard two-point extrapolation of the aug-cc-pVTZ and aug-cc-pVQZ42 results. Carbon dioxide force field parameters were generated using Poltype 1.1.3.27 Space group symmetry is exploited throughout to reduce the number of monomer and dimer fragments that need to be computed.43Raman spectra predictions
To compute Raman spectra, the complete basis set quasi-harmonic HMBI MP2 structures (or the experimental structures in selected cases) were electronic energy-minimized at the HMBI MP2/aug-cc-pVDZ level with lattice parameters held fixed. Zone-center (k = 0) phonons were computed at the same level of theory. Raman intensities were then approximated at the 1- and 2-body level.44 Many-body effects were neglected in computing the polarizability derivatives that are used to evaluate the Raman intensities, but these effects are generally small in carbon dioxide. Space group symmetry was again employed. To minimize numerical noise associated with finite differencing the polarizability derivatives, individual fragment MP2 frequency calculations were performed via analytic second derivatives as implemented in Gaussian 09.45 The polarizability derivatives were evaluated via finite difference of the polarizabilities. Simulated spectra were plotted as a sum of Gaussian functions with a full width at half maximum of 10 cm−1.Crystal structure prediction
Potential carbon dioxide crystal structures were generated via the evolutionary algorithms implemented in USPEX.46 Each of six runs was seeded with ten random structures from randomly chosen space groups, containing either two or four molecules in the unit cell (the unit cell sizes for phases I, II, and III/VII). Structures were relaxed at ambient pressure and energies computed using Tinker and the OPLS-AA force field.36 New structures were constructed for 15–20 generations via heredity, coordinate/rotational mutations, or lattice mutation.46 This process generated 660 structures with Z = 2 and 1083 structures with Z = 4 were generated, though many of these were redundant or clearly energetically unfavorable.After removal of obvious duplicates, the most stable 91 structures were then refined under 11.8 GPa of pressure (the pressure for the experimental powder X-ray diffraction of phase III) in Quantum Espresso47 using periodic PBE-D2,48,49 an 80 Ry planewave cutoff, a 7 × 7 × 7 Monkhorst–Pack k-point grid, and ultrasoft pseudopotentials C.pbe-rrkjus.UPF and O.pbe-rrkjus.UPF from http://www.quantum-espresso.org. Structures were analyzed in terms of energy, unit cell volume, root-mean-square deviations in atomic positions, and by comparing simulated powder X-ray diffraction patterns (wavelength 0.6888 Å) generated by Mercury.50 These were then compared against the experimental phase III diffraction pattern and the simulated pattern for the experimentally reported phase III crystal structure.1
Data analysis
Experimental crystal structures and Raman data was taken from the literature. When tabulated data was unavailable, Raman spectra and equation of state data were digitized from published figures. Root-mean-square deviations between predicted and optimized structures employ 15-molecule clusters,4 as implemented in Mercury.50) Bulk moduli were computed by fitting to the Vinet equation of state,31 which proves more numerically robust than the Birch–Murnaghan one (ESI Section S2†).22Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work from the National Science Foundation (CHE-1362465 and CHE-1665212) and supercomputer time from XSEDE (TG-CHE110064) are gratefully acknowledged. We thank Prof. Eric Chronister for helpful discussions.References
- K. Aoki, H. Yamawaki, M. Sakashita, Y. Gotoh and K. Takemura, Science, 1994, 263, 356–358 CAS.
- C. S. Yoo, Phys. Chem. Chem. Phys., 2013, 15, 7949–7966 RSC.
- G. J. O. Beran, Chem. Rev., 2016, 116, 5567–5613 CrossRef CAS PubMed.
- J. A. Chisholm and W. D. S. Motherwell, J. Appl. Crystallogr., 2005, 38, 228–231 CrossRef.
- V. Iota and C. S. Yoo, Phys. Rev. Lett., 2001, 86, 5922–5925 CrossRef CAS PubMed.
- C. Yoo, H. Kohlmann, H. Cynn, M. Nicol, V. Iota and T. LeBihan, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 1–6 Search PubMed.
- J.-H. Park, C. S. Yoo, V. Iota, H. Cynn, M. F. Nicol and T. Le Bihan, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 014107 CrossRef.
- F. Datchi, V. M. Giordano, P. Munsch and A. M. Saitta, Phys. Rev. Lett., 2009, 103, 185701 CrossRef PubMed.
- F. Datchi, B. Mallick, A. Salamat, G. Rousse, S. Ninet, G. Garbarino, P. Bouvier and M. Mezouar, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 144101 CrossRef.
- S. A. Bonev, F. Gygi, T. Ogitsu and G. Galli, Phys. Rev. Lett., 2003, 91, 065501 CrossRef CAS PubMed.
- S. Gohr, S. Grimme, T. Söhnel, B. Paulus and P. Schwerdtfeger, J. Chem. Phys., 2013, 139, 174501 CrossRef PubMed.
- V. M. Giordano and F. Datchi, Europhys. Lett., 2007, 77, 46002 CrossRef.
- C.-S. Yoo, A. Sengupta and M. Kim, High Pres. Res., 2011, 31, 68–74 CrossRef CAS.
- H. Olijnyk and A. P. Jephcoat, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 879–888 CrossRef CAS.
- C. S. Yoo, H. Cynn, F. Gygi, G. Galli, V. Iota, M. Nicol, S. Carlson, D. Häusermann and C. Mailhiot, Phys. Rev. Lett., 1999, 83, 5527–5530 CrossRef CAS.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CAS.
- A. R. Oganov, J. C. Schön, M. Jansen, S. M. Woodley, W. W. Tipton and R. G. Hennig, in Modern Methods of Crystal Structure Prediction, ed. A. R. Oganov, Wiley, Weinheim, Germany, 2011, pp. 223–231 Search PubMed.
- E. Salager, G. M. Day, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, J. Am. Chem. Soc., 2010, 132, 2564–2566 CrossRef CAS PubMed.
- C. Martineau, J. Senker and F. Taulelle, Annu. Rep. NMR Spectrosc., 2014, 82, 1–57 CrossRef.
- M. D. Eddleston, K. E. Hejczyk, E. G. Bithell, G. M. Day and W. Jones, Chem.–Eur. J., 2013, 19, 7874–7882 CrossRef CAS PubMed.
- S. Hirata, K. Gilliard, X. He, J. Li and O. Sode, Acc. Chem. Res., 2014, 47, 2721–2730 CrossRef CAS PubMed.
- Y. N. Heit, K. D. Nanda and G. J. O. Beran, Chem. Sci., 2016, 7, 246–255 RSC.
- Y. N. Heit and G. J. O. Beran, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 514–529 CAS.
- G. J. O. Beran and K. Nanda, J. Phys. Chem. Lett., 2010, 1, 3480–3487 CrossRef CAS.
- S. Wen and G. J. O. Beran, J. Chem. Theory Comput., 2011, 7, 3733–3742 CrossRef CAS PubMed.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. DiStasio, M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- J. C. Wu, G. Chattree and P. Ren, Theor. Chem. Acc., 2012, 131, 1138 CrossRef PubMed.
- B. Olinger, J. Chem. Phys., 1982, 77, 6255–6258 CrossRef CAS.
- L. Liu, Earth Planet. Sci. Lett., 1984, 71, 104–110 CrossRef CAS.
- V. M. Giordano, F. Datchi, F. A. Gorelli and R. Bini, J. Chem. Phys., 2010, 133, 144501 CrossRef PubMed.
- P. Vinet, J. R. Smith, J. Ferrante and J. H. Rose, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 1945–1953 CrossRef CAS.
- F. Gygi, Comput. Mater. Sci., 1998, 10, 63–66 CrossRef CAS.
- J. Li, O. Sode, G. A. Voth and S. Hirata, Nat. Commun., 2013, 4, 2647 Search PubMed.
- J. Li, O. Sode, G. A. Voth and S. Hirata, Nat. Commun., 2015, 6, 8907 CrossRef CAS PubMed.
- O. Sode, M. Keceli, K. Yagi and S. Hirata, J. Chem. Phys., 2013, 138, 074501 CrossRef PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- F. Bolduan, H. D. Hochheimer and H. J. Jodl, J. Chem. Phys., 1986, 84, 6997–7004 CrossRef CAS.
- K. Aoki, Y. Kakudate, M. Yoshida, S. Usuba, K. Tanaka and S. Fujiwara, Solid State Commun., 1987, 64, 1329–1331 CrossRef CAS.
- K. Nanda and G. J. O. Beran, J. Chem. Phys., 2012, 137, 174106 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, see http://www.molpro.net Search PubMed.
- J. W. Ponder, TINKER v6.3, 2014, http://dasher.wustl.edu/tinker/, accessed August 10, 2015.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- Y. Heit and G. J. O. Beran, J. Comput. Chem., 2014, 35, 2205–2214 CrossRef CAS PubMed.
- X. He, O. Sode, S. S. Xantheas and S. Hirata, J. Chem. Phys., 2012, 137, 204505 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- Q. Zhu, A. R. Oganov, C. W. Glass and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 215–226 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 5287 RSC.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 455–470 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Comparisons between predicted and experimental structures, bulk modulus data, assessment of modeling choices on the predicted Raman spectra, pressure-dependent Raman data, and crystal structure prediction results. See DOI: 10.1039/c7sc03267f |
| This journal is © The Royal Society of Chemistry 2017 |