
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of protonation states of iminosugar–enzyme complexes using photoinduced electron transfer†
Bo
Wang
 ,
Jacob Ingemar
Olsen
,
Bo W.
Laursen
,
Jens Christian
Navarro Poulsen
and
Mikael
Bols
*
,
Jacob Ingemar
Olsen
,
Bo W.
Laursen
,
Jens Christian
Navarro Poulsen
and
Mikael
Bols
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: bols@chem.ku.dk; Tel: +45 35320160
First published on 14th September 2017
Abstract
A series of N-alkylated analogues of 1-deoxynojirimycin containing a fluorescent 10-chloro-9-anthracene group in the N-alkyl substituent were prepared. The anthracene group acted as a reporting group for protonation at the nitrogen in the iminosugar because an unprotonated amine was found to quench fluorescence by photoinduced electron transfer. The new compounds were found to inhibit β-glucosidase from Phanerochaete chrysosporium and α-glucosidase from Aspergillus niger, with Ki values in the low micro- to nanomolar range. Fluorescence and inhibition versus pH studies of the β-glucosidase–iminosugar complexes revealed that the amino group in the inhibitor is unprotonated when bound, while one of the active site carboxylates is protonated.
Introduction
Iminosugars, or monosaccharide analogues with a nitrogen atom in place of the oxygen atom in the ring,1 are natural products found in some plants and microorganisms.2 Many iminosugars are potent inhibitors of glycosidases, and the iminosugar structure has been used as a pharmacophore in drugs and drug candidates.3 Examples of such molecules are 1-deoxynojirimycin (1), which is present in mulberries and clearly mimics D-glucose in the pyranose form, and its derivatives miglitol (2) and miglustat (3), which are drugs that have been approved for the treatment of diabetes and Niemann–Pick disease, respectively (Fig. 1).4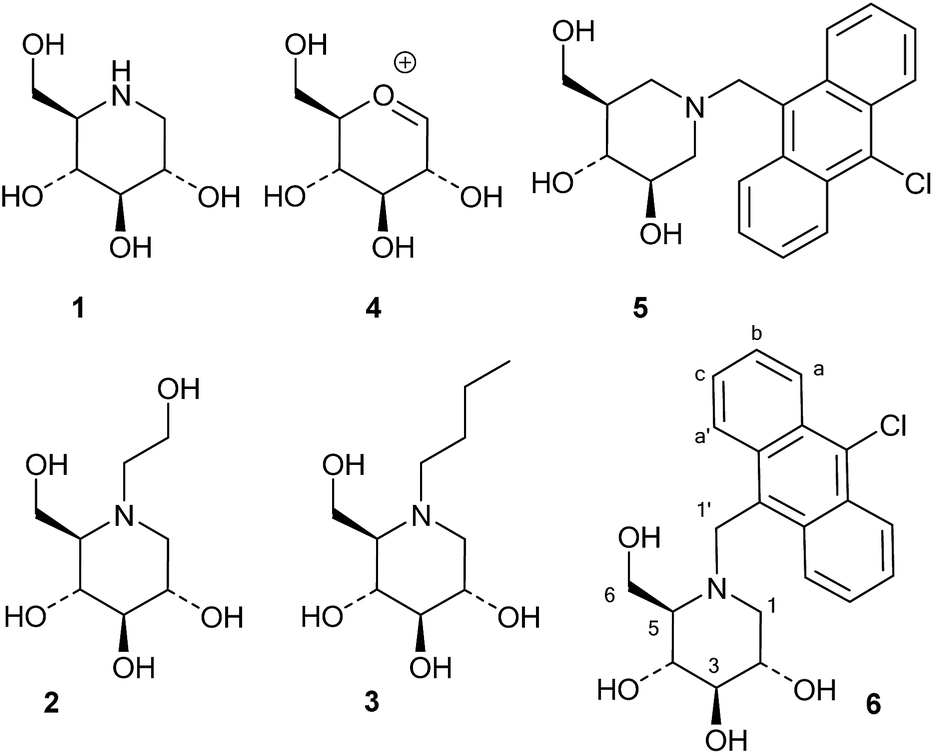 | ||
| Fig. 1 Iminosugars and oxocarbenium ion 4. | ||
In these compounds the nitrogen atom is typically considered to be crucial for inhibitory activity, as the corresponding oxygen,5 sulfur and carbon analogues are normally much weaker inhibitors.6 Wong and collaborators concluded from a QSAR-type analysis of iminosugars and other inhibitors of almond β-glucosidase and yeast α-glucosidase that a positive charge on the inhibitor was important for binding.6 Legler similarly concluded that basic sugar analogues were most likely protonated at the glycosidase active site, and he divided glycosidases into two classes based on their mechanism of binding basic inhibitors.7 The first class, which contains the family 1 enzyme almond β-glucosidase, binds the inhibitor in the neutral form and then uses an active site carboxylate to protonate it. The second group, which contains the GH3 family enzyme Aspergillus wentii β-glucosidase as an example, binds the ammonium form of the inhibitor directly.8 One way of distinguishing between these two modes of inhibitor binding is that only the second class bind inhibitors with a permanent positive charge, such as glycosyl pyridinium ions or quaternary ammonium compounds such as the dimethylated analogue of 1-deoxynojirimycin.9 Nojirimycins could be reasonably expected to be transition state (TS) analogue inhibitors, as 1, when protonated, clearly resembles the glucosylium ion 4 (Fig. 1), which is quite similar to the TS of acidic or spontaneous glycoside hydrolysis.10 The Withers group addressed the mimicry of iminosugar 1 and castanospermine to the TS of Agrobacterium β-glucosidase (GH1 family).11 Poor correlations in the double logarithmic plots of kcat/Kmversus the Ki values of similarly modified inhibitor and substrate analogues (free energy relationship plots) led to the conclusion that iminosugars are not transition state analogues, but rather charged compounds that just happen to bind tightly.12 Gloster and Davies also discussed the imperfections in the transition state mimicry of iminosugars when they were analysed with rigorous methods, such as free energy relationship plots or comparisons of the enzyme and inhibitor pH profiles.13 They suggested that these striking observations might be related to the cationic nature of iminosugars, which are frequently found to be very potent inhibitors.14
Whether iminosugars are TS mimics or not, it is apparent that it is the norm to assume that iminosugars are, or become, protonated at the nitrogen atom when they bind to an enzyme. However, because protonation within the active site is difficult to observe by X-ray or NMR,15 there is to our knowledge only one example in the literature where the protonation state of an iminosugar was definitively established.16 Davies and collaborators used synchrotron radiation to obtain a high resolution structure of an isofagomine derivative bound to a cellulase and found the iminosugar in the ammonium form.16 Since the pKa of isofagomine is very high (pKa 8.4), protonation is comparatively more likely in this case, and it is therefore far from certain whether protonation occurs with the many less basic iminosugars, such as 1, that have pKa values in the range of 3–7. We therefore speculated whether the principle behind de Silva’s fluorescent pH indicators17a could be used for a more general method to determine the protonation state of inhibitors in the glycosidase active site (Fig. 2). In these indicators, the emission from the fluorescent chloroanthracene group is quenched by photoinduced electron transfer (PET) from the lone pair of a covalently attached amino group.17 When the amino group is protonated, and the lone pair is involved in bonding, the PET process becomes unfavourable and fluorescence returns. So an iminosugar with a suitably attached chloroanthracene should work as a fluorescent indicator for the pH at the active site. In order to obtain a full inventory of the protons in the active site, it is necessary to combine these results with inhibition pH dependency studies. Using our recent method, one can determine whether the inhibitor binds to the glycosidase with 0, 1, 2 or 3 protons in the complex (EI, EHI, EH2I or EH3I).18 Recently, we tested the methodology with almond β-glucosidase and the isofagomine derivative 5 (Fig. 1).19 It was found that 5 was a good inhibitor and, surprisingly, bound to the enzyme as the EI complex, which was confirmed by both fluorescence and the pH dependence of inhibition. In this work we have studied 1-deoxynojirimycin derivatives such as 6 and their inhibition of glycosidases, and have elucidated the protonation behaviour during inhibition.
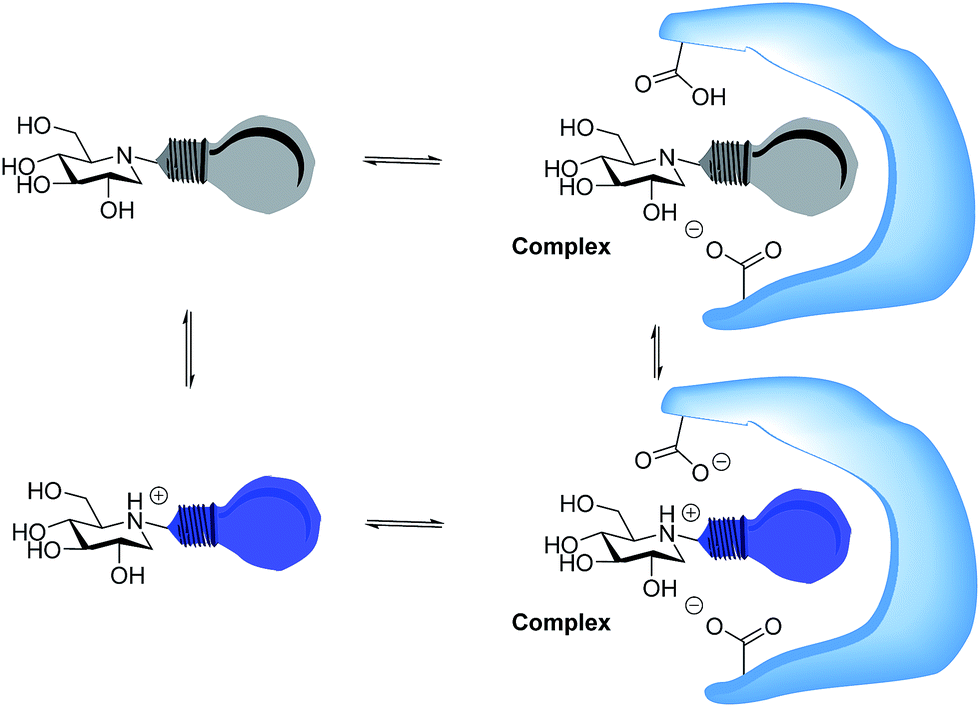 | ||
| Fig. 2 Iminosugar–enzyme complex formation with the inhibitor as a fluorescent indicator. The protonated inhibitors are fluorescent and readily detected. The protonation behaviour of the enzyme can be seen from the degree to which it changes the ratio between the fluorescent and non-fluorescent inhibitor. | ||
Results
Synthesis
A series of 10-chloroanthracenyl derivatives of 1-deoxynojirimycin with different chain lengths were prepared (6, 12 and 15) in order to maximise the chance of enzyme inhibition and to investigate the effective distance of PET in this system. We also needed a fluorescence inhibitor where PET quenching could not occur in order to check if the enzyme itself quenches fluorescence. For this we decided to make a 10-chloroanthracenyl derivative of D-glucono-1,5-lactam (19), as D-glucono-1,5-lactam is often a good glucosidase inhibitor1b,5 and the amide is too weak an electron donor to participate in PET.The compound with the shortest tether (6) was prepared by a different method than the reductive amination we had previously used,19 as that methodology appeared low yielding and difficult to reproduce. It was therefore made by alkylation (Scheme 1). Commercially available aldehyde 7 was reduced with NaBH4 in methanol to the alcohol 8 in a yield of 87%.
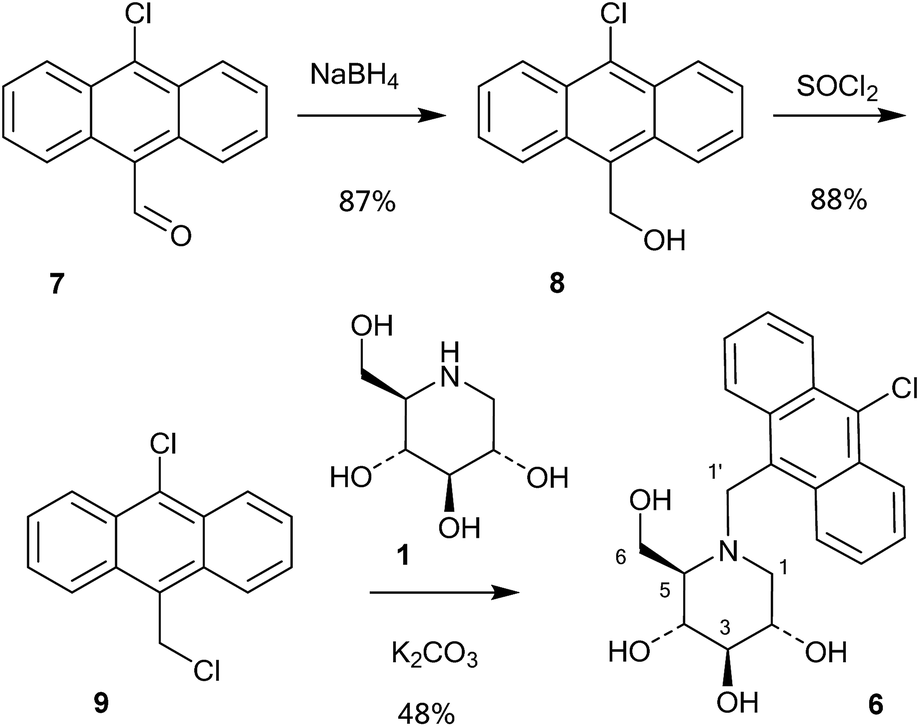 | ||
| Scheme 1 Synthesis of 6. | ||
This compound was then converted with thionyl chloride and pyridine to chloride 9 in 88% yield. The reaction of 1-deoxynojirimycin with 9 and anhydrous K2CO3 in DMF at 80 °C for 4 h gave the pure alkylation product 6 in 48% yield. The crude yield was higher and it is possible that some by-products were formed, though these were not identified. The homo-analogues of 6, however, were made by reductive amination. Thus aldehyde 7 was reacted with a Wittig ylide made from methoxymethyltriphenylphosphonium chloride and butyl lithium to afford the vinyl ether 10 in 60% yield (Scheme 2). The vinyl ether was converted to the aldehyde by hydrolysis with perchloric acid in refluxing aqueous ether, giving 11 in 89% yield. Finally, reductive amination of 1-deoxynojirimycin (1) with one equivalent of 11 and NaCNBH3 and acetic acid afforded the target conjugate 12 in 55% yield.
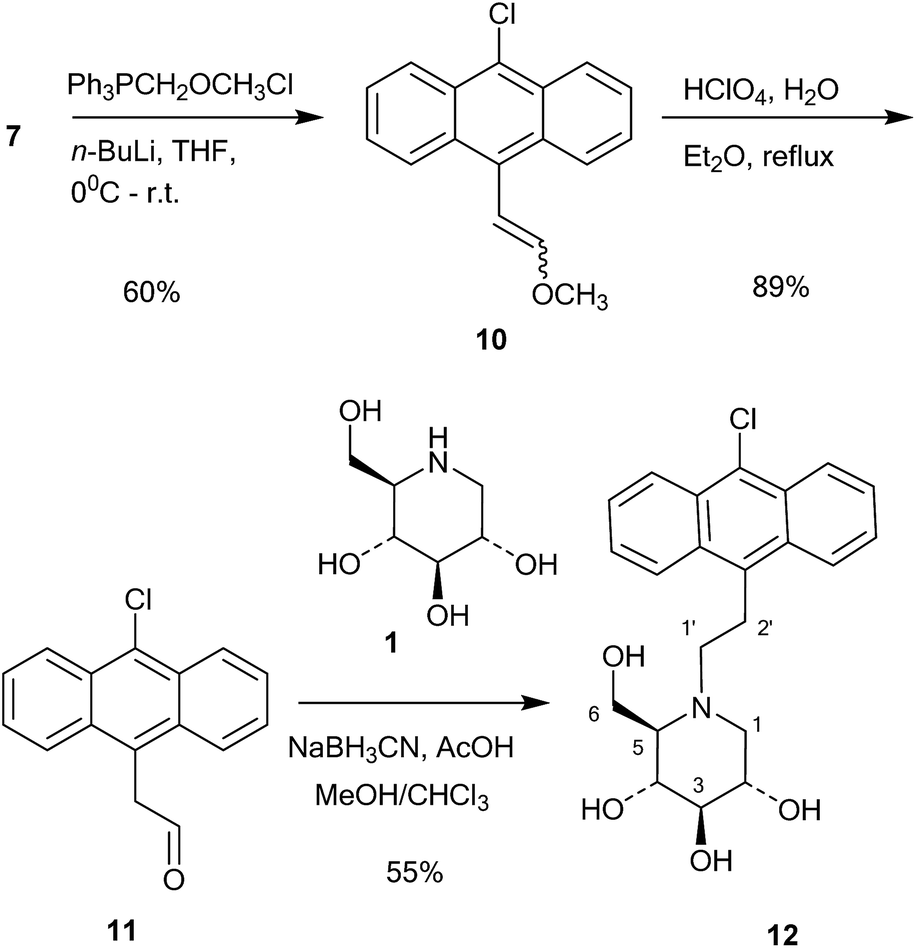 | ||
| Scheme 2 Synthesis of 12. | ||
Further chain elongation with another methylene group was performed by another round of Wittig homologation (Scheme 3). The homoaldehyde 11 was reacted with methoxymethyltriphenylphosphonium ylide and gave the vinyl ether 13 in 53% yield. Perchloric acid hydrolysis in refluxing aqueous ether gave the aldehyde 14 in 78% yield, which was then coupled with 1-deoxynojirimycin (1) by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reductive amination with NaCNBH3 and acetic acid. This gave the conjugate 15 in 82% yield. The reason why the reductive amination of 1 is more successful with aldehydes 11 and 14 is probably because they are aliphatic and more readily form imines or even enamines.
:1 reductive amination with NaCNBH3 and acetic acid. This gave the conjugate 15 in 82% yield. The reason why the reductive amination of 1 is more successful with aldehydes 11 and 14 is probably because they are aliphatic and more readily form imines or even enamines.
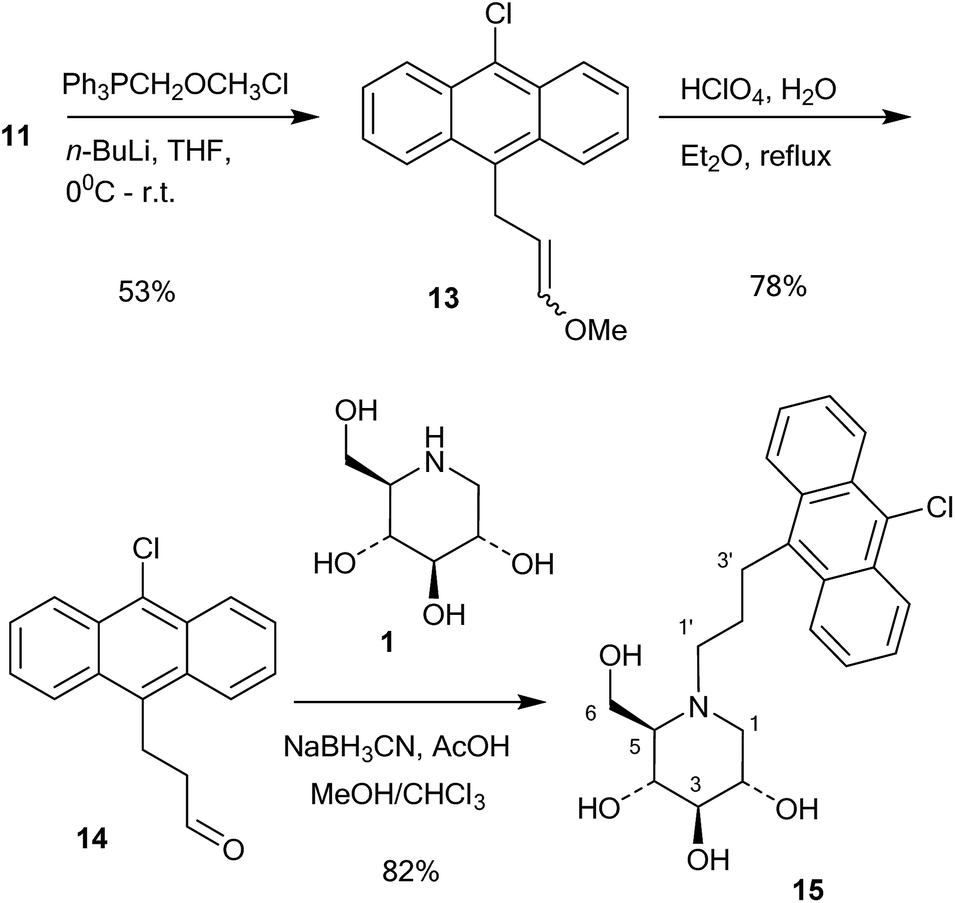 | ||
| Scheme 3 Synthesis of 15. | ||
The 10-chloroanthracenyl derivative of D-glucono-1,5-lactam (19) was more difficult to prepare, as reductive amination is not possible and alkylation of the amide requires a strong base and may not be selective for nitrogen. Also, the 10-chloroanthracene is sensitive to reduction, which precludes the use of benzyl groups and can make it complicated to introduce earlier in the synthesis. However, eventually the relatively straightforward procedure shown in Scheme 4 led to the compound: D-glucono-1,5-lactam 16 (ref. 20) was persilylated with excess tert-butyldimethylsilyl triflate in pyridine/DMAP at 60 °C for 18 h. This gave the product 17 in 60% yield in the familiar all-axial conformation caused by the bulky silyl groups. This can clearly be seen from the change of the 2,3 proton coupling constant from 8.5 Hz in 16 to 3.5 Hz in 17 (Scheme 4). N-Alkylation of 17 with the chloromethyl derivative 9 and sodium hydride in THF and tetrabutylammonium iodide as a nucleophilic catalyst gave product 18 in 49% yield. Finally, deprotection of 18 using tetrabutylammonium fluoride in THF gave 19 in 14% yield. The reason for the low yield in this reaction is not clear.
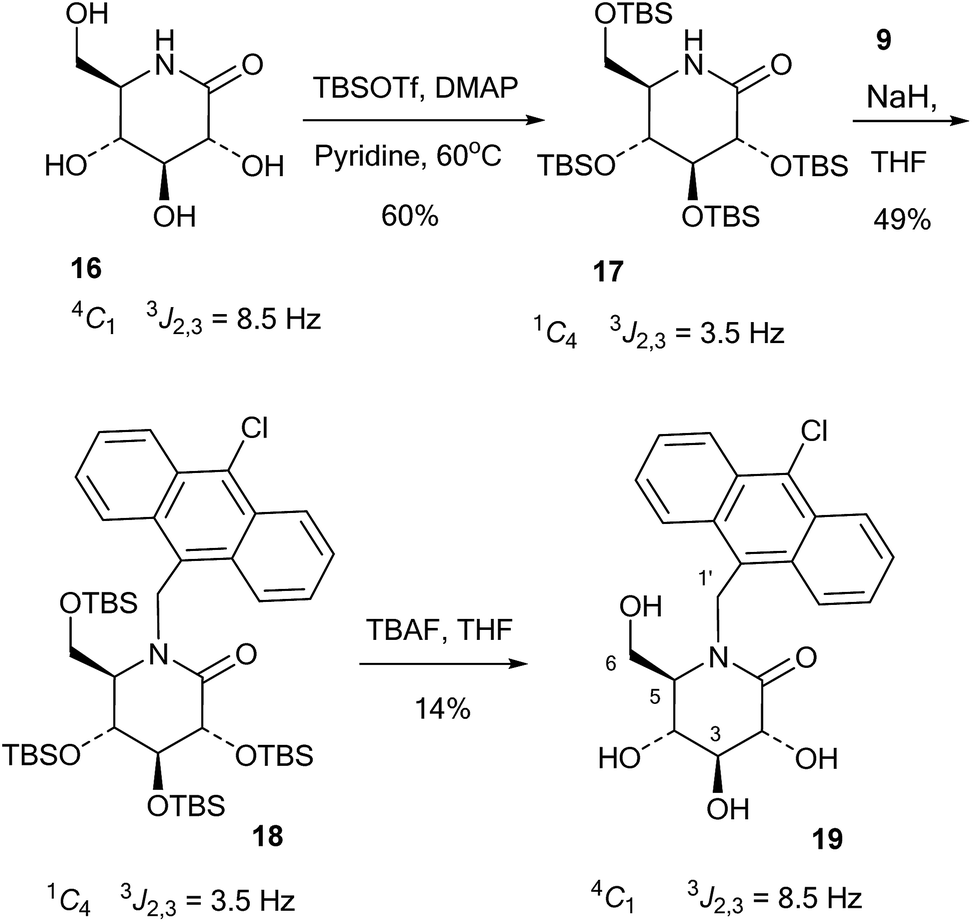 | ||
| Scheme 4 Synthesis of lactam 19. | ||
Acidity constants
The acidity constants of the 1-deoxy-nojirimycin derivatives were determined by measuring the relative fluorescence intensity as function of pH (Fig. 3). Fitting titration curves to the data gave the pKa values shown in Table 1.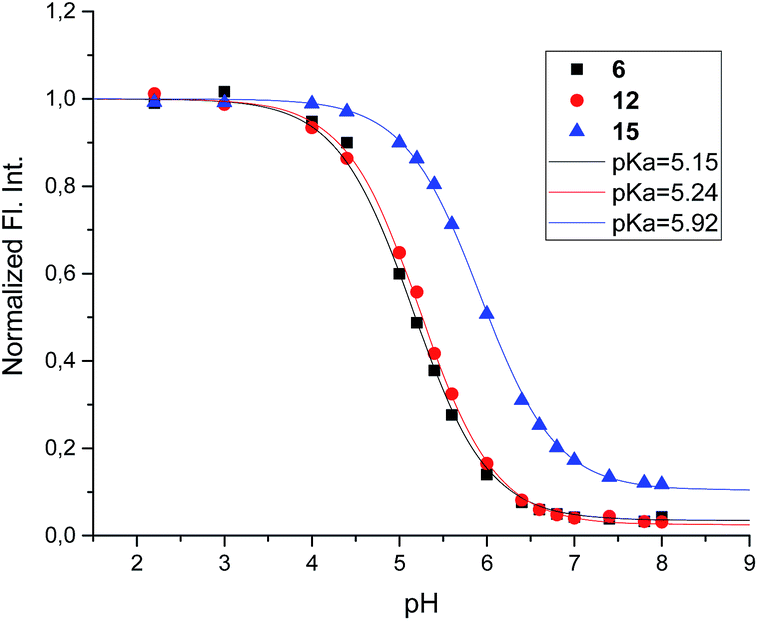 | ||
| Fig. 3 Fluorescence titration of 6 (black), 12 (red) and 15 (blue). Shown is the fluorescence intensity as a function of pH, normalized relative to pH = 2. | ||
| Compound | 6 | 12 | 15 | 1 |
| pKa | 5.15 | 5.24 | 5.92 | 6.7 |
Enzyme inhibition
Compounds 6, 12, 15 and 19 were tested for inhibition of the selected glycosidases. The enzyme substrates in these experiments were 4-nitrophenyl α-D-glucopyranoside for the α-glucosidases and 4-nitrophenyl β-D-glucopyranoside for the β-glucosidases, the solvent was 50 mM phosphate buffer containing 10% DMSO, and the reactions were monitored by measuring the absorption at 400 nm. 6 was a competitive inhibitor of one β-glucosidase and two α-glucosidases (Table 2), while 12 and 15 only inhibited the β-glucosidase from Phanerochaete chrysosporium. However, they were 300–500 times more potent than 6, and 12 was the strongest inhibitor.| Enzyme/Ki (μM) | 6 | 12 | 15 | 19 |
|---|---|---|---|---|
| β-Glucosidase (T. maritima) | NI | NI | NI | — |
| β-Glucosidase (P. chrysosp.) | 2.7 | 0.0062 | 0.0103 | 24.3 |
| β-Glucosidase (A. oryzae) | NI | NI | NI | 169 |
| α-Glucosidase (A. niger) | 1.04 | NI | NI | 14.4 |
| α-Glucosidase (S. cerevisiae) | NI | NI | NI | — |
| α-Glucosidase (T. maritima) | NI | NI | NI | — |
| α-Glucosidase (Bacillus sp.) | NI | NI | NI | — |
| α-Glucosidase (B. stearoth.) | 93 | NI | NI | — |
pH dependence of enzyme activity and inhibition
The pKa values of the enzyme active site residues were determined from plots of the enzyme activity at different pH values. Fitting the theoretical function for the divalent acid that is catalytic in the monoprotonated form gave the pKa values shown in Table 3.| Enzyme | Source | pKa(1) | pKa(2) |
|---|---|---|---|
| β-Glucosidase | Phanerochaete chrysosp. | 4.0 | 5.9 |
| α-Glucosidase | Aspergillus niger | 3.2 | 6.4 |
The pH dependence of inhibition was examined for 12 and 15 against β-glucosidase from Phanerochaete chrysosporium, and for 6 against α-glucosidase from Aspergillus niger. These data are plotted as 1/Kiversus pH in Fig. 4 and 5.
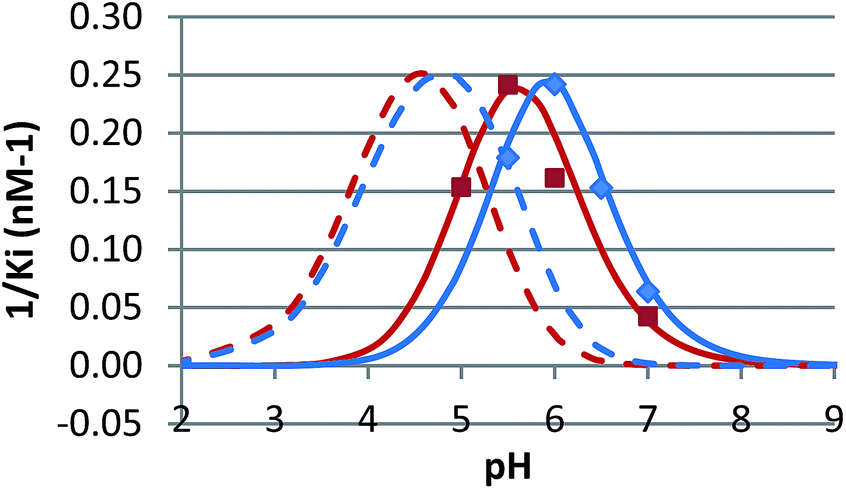 | ||
| Fig. 4 1/Kiversus pH for the inhibition of β-glucosidase from Phanerochaete chrysosporium by 12 (red, nM−1) and 15 (blue, multiplied by 2.5, nM−1), both in phosphate buffer containing 10% DMSO. The data is compared with the calculated curves for EHI binding (solid) and EH2I binding (dashed). | ||
 | ||
| Fig. 5 1/Kiversus pH for the inhibition of α-glucosidase from Aspergillus niger by 6 (red, μM−1) in phosphate buffer containing 10% DMSO. The data is compared with the calculated curves for EHI binding (solid) and EH2I binding (dashed). | ||
Fluorescence measurements
The compounds (6, 12, 15 and 19) all gave the characteristic fluorescence spectra of the 10-chloroanthracenes when excited at 358 nm. As anticipated, lactam 19 displayed an unchanged spectrum at the full range of pH values, while the basic inhibitors 6, 12 and 15 displayed pH dependent fluorescence as anticipated (Fig. 3, ESI S1 & S3A†).To check if the enzymes had a fluorescence quenching effect, β-glucosidase from Phanerochaete chrysosporium and α-glucosidase from Aspergillus niger were mixed with 19 at enzyme concentrations above Ki (Fig. 6). The fluorescence intensity from 19 showed minor decreases (10–20%) with both enzymes, but confirmed that no significant fluorescence quenching from the enzymes took place.
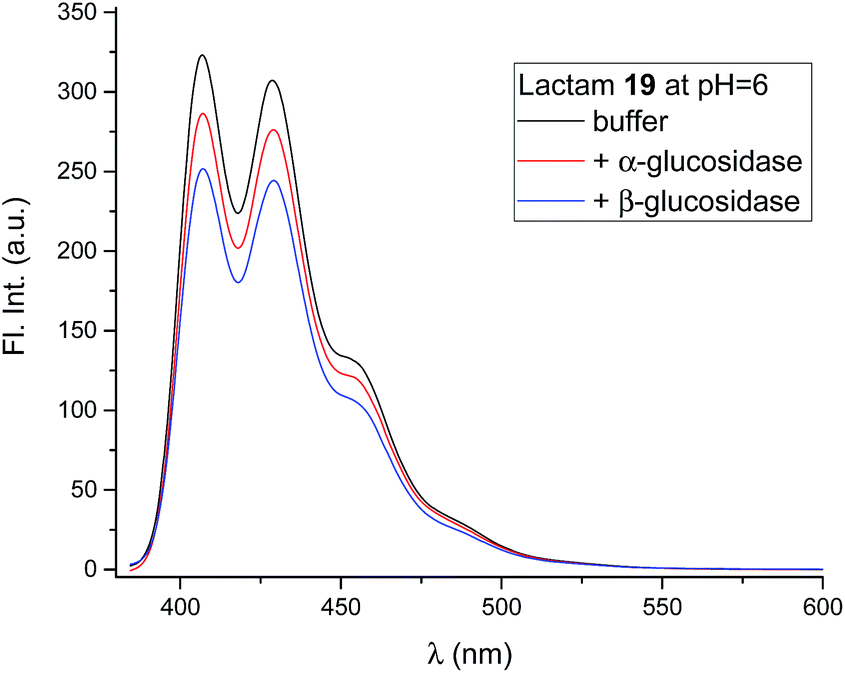 | ||
| Fig. 6 Fluorescence spectra of 19 (0.8 μM, black), in the presence of β-glucosidase from P. chrysosporium (63 μM, blue) and in the presence of α-glucosidase from A. niger (54 μM, red). All solutions were at pH 5.86 in 90% phosphate buffer and 10% DMSO. | ||
Mixing the strong binding deoxynojirimycin analogues 12 and 15 with β-glucosidase from Phanerochaete chrysosporium led to a large decrease in fluorescence for both inhibitors (Fig. 7 and S4C–F†), independent of pH. Inhibitor 6 was only measured at pH 6.0, where the fluorescence intensity was nearly fully quenched (Fig. S4A and B†).
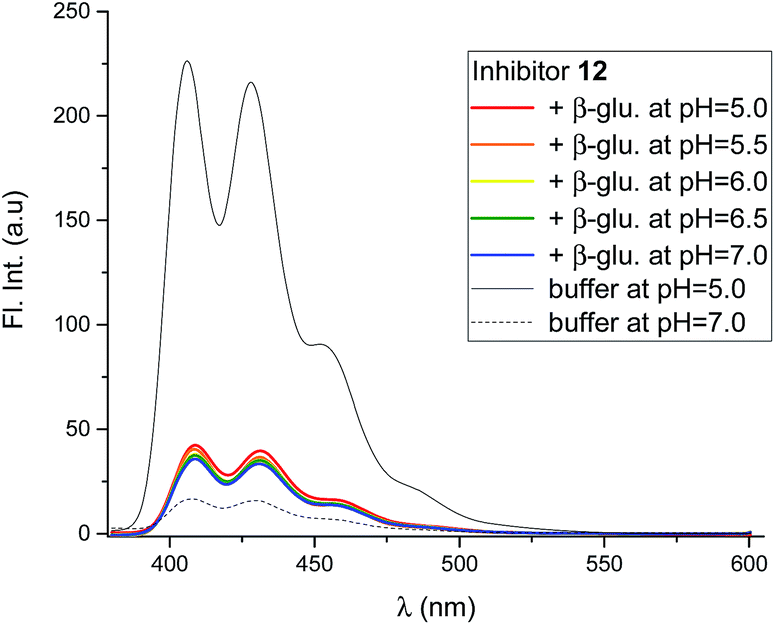 | ||
| Fig. 7 Fluorescence spectra of 12 (0.8 μM), in the presence of β-glucosidase from P. chrysosporium (5.8 μM, blue) at 5 different pH values, and without the enzyme at two pH values. All the solutions were in 90% phosphate buffer and 10% DMSO. | ||
Using 12 as the titrant, a fluorescence titration of β-glucosidase was conducted (Fig. S5†). This showed that the loss of fluorescence is associated with specific binding to the binding site. When the binding site is full, no further loss of fluorescence takes place.
Mixing 6 with α-glucosidase from Aspergillus niger had no influence on the fluorescence from the inhibitor. Fluorescence in the presence of this enzyme varied with pH in the same manner as it did without (Fig. 8).
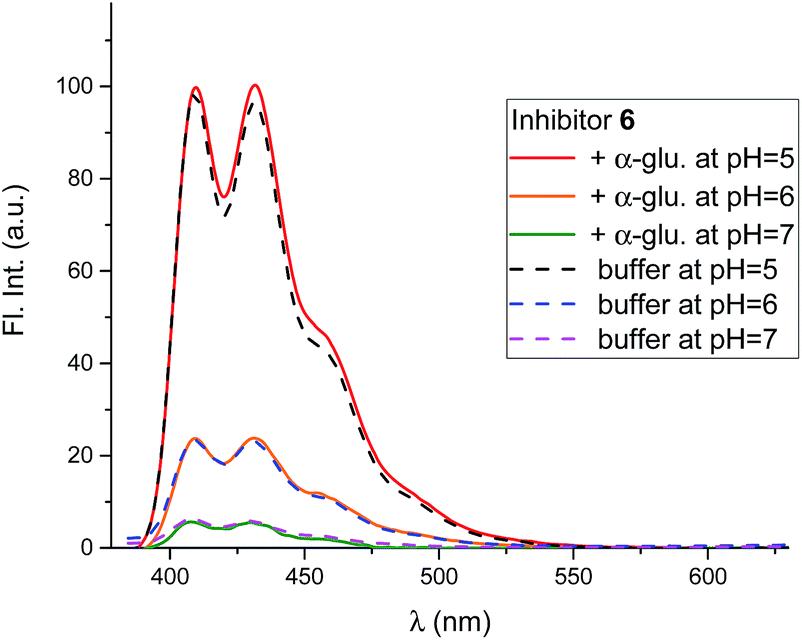 | ||
| Fig. 8 Fluorescence spectra of 6 (0.8 μM) at 3 different pH values in the presence and absence of α-glucosidase from A. niger (54 μM). All the solutions were in 90% phosphate buffer and 10% DMSO. | ||
Discussion
Photoinduced electron transfer has been reported to occur up to 10 Å (ref. 17), and in accordance with this we see effective fluorescence quenching at basic pH for all the 1-deoxynojirimycin analogues (Fig. 3). However, the rate of the PET process is also strongly distance dependent and for 15 the non-protonated inhibitor displays approximately 10% residual fluorescence. Time-resolved fluorescence measurements confirm that this residual fluorescence at high pH originates from the deprotonated inhibitor (Fig. S2†). On the other hand, no fluorescence quenching was observed for lactam 19 and, as anticipated, it displayed full fluorescence over the entire pH range (not shown).The pKa values of 6, 12 and 15 of 5.15, 5.24 and 5.92 are rather low compared to 1 (pKa 6.7). This is caused by steric hindrance versus protonation in the tertiary amine, and the inductive effect from the aromatic system (the electron withdrawing influence of a benzyl group is 1.3 pH units21), which is obviously larger when the chloroanthracene is closer to the amine.
N-Alkylated iminosugar analogues are sometimes more potent glycosidase inhibitors than their parent structures,1 but this depends very much on the enzyme.9,22 In line with this, we see that 6, 12, 15 and 19 only effectively inhibit the β-glucosidase from Phanerochaete chrysosporium and α-glucosidase from Aspergillus niger (Table 2). Inhibition of the other enzymes is non-existent or too weak to be practically useful in fluorescence measurements. The β-glucosidase from Phanerochaete chrysosporium (white rot fungus) is an extracellular enzyme which participates in the degradation of cellulose.23 It has been classified as a GH3 family enzyme.23 Inhibitors of this specific enzyme have not been reported, but iminosugars such as 1 are known to inhibit other GH3 β-glucosidases, such as the β-glucosidase from Aspergillus wentii.1b,9a The α-glucosidase from Aspergillus niger is an enzyme with high transglucosidase activity.24 It is classified as a GH31 family enzyme. The chain-extended analogues 12 and 15 are evidently particularly useful for studying the inhibition of Phanerochaete chrysosporium β-glucosidase as they are 3–500 times more potent than 6. On the other hand, they display a complete loss of inhibition of Aspergillus niger α-glucosidase.
In competitive inhibition, the pH dependence of inhibition means that the inhibitor prefers to bind to the active site with a specific number of protons. For a normal glycosidase with two active site carboxylates and a monobasic iminosugar, four complexes are possible (i.e. EI, EHI, EH2I or EH3I) and a general binding scheme as outlined in Scheme 5 is expected. While some of the pathways may be kinetically impossible for certain enzymes, as Legler suggested, virtual equilibria will still exist between the different species due to the possibility of interchange by a different pathway. The mutual interdependence of the various equilibria means that it is not necessary to know the pKa values of the various complexes. When the enzyme and inhibitor pKa values are known, the pH-stability curves (Ki−1vs. pH) can easily be calculated using a spreadsheet (Fig. 9).18 In the present cases, the data clearly fit EHI binding for 6, 12 and 15 inhibiting β-glucosidase (Fig. 4) and for 6 inhibiting α-glucosidase (Fig. 5). This means that one proton is bound either to the amino group or to one of the carboxylate groups. Each of the inhibitors has an optimum pH, as shown in Table 4. This binding mode is the most common for iminosugar glycosidase inhibitors in the literature,13,18 but there are exceptions and more may emerge as only a fraction of inhibitors have been analysed this way. The isofagomine derivative 5 binds to almond β-glucosidase as the EI complex,19 and L-fuco-1-deoxynojirimycin (FNJ, the L-fucose analogue of 1) appears to bind to Bacteroides thetaiotaomicron α-fucosidase as the EH3I complex, according the published binding data (Fig. 9).3c In those two cases the existence of nitrogen protonation is evident, as 5 must be unprotonated and FNJ must be protonated. This can, to some extent, be explained by the pKa values of the inhibitors. L-Fuco-1-deoxynojirimycin is very basic (pKa 8.4) and is thus readily protonated, while 5 has a surprisingly low pKa (5.1) and is thus more difficult to protonate.
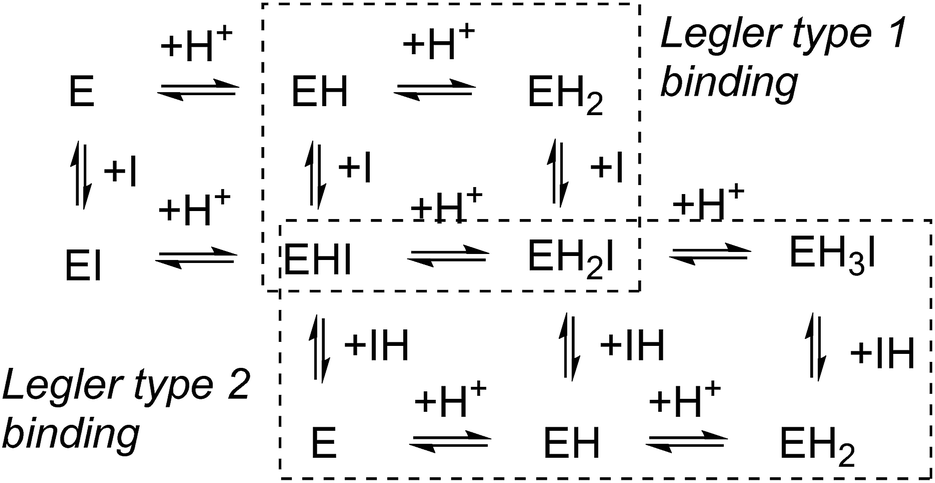 | ||
| Scheme 5 pH dependent inhibition of a divalent glycosidase by a monobasic inhibitor. Legler’s two binding mechanisms are shown: either binding of the unprotonated inhibitor (type 1) or binding of the protonated inhibitor (type 2). | ||
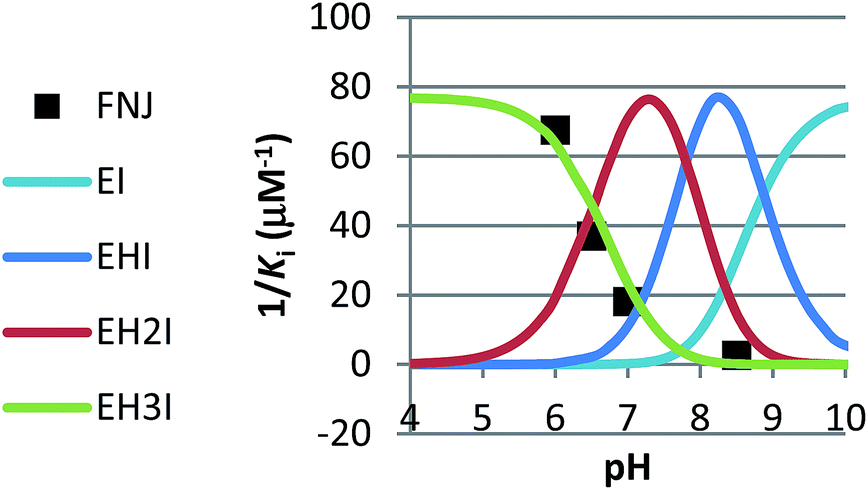 | ||
| Fig. 9 Curves for the pH dependence of the selective binding of a monobasic inhibitor to a diacidic enzyme with 0, 1, 2 or 3 protons when the pKa values of the enzyme (Bacteroides thetaiotaomicron α-fucosidase) are 6.7 and 8.1 and the inhibitor pKa is 8.4 (FNJ). The actual binding data for FNJ (black dots3c) show binding with the inhibitor and enzyme fully protonated. | ||
| Enzyme | 6 | 12 | 15 | 19 |
|---|---|---|---|---|
| β-Glucosidase (P. chrysosporium) | 5.55 | 5.60 | 5.93 | 4.95 |
| α-Glucosidase (A. niger) | 5.78 | — | — | 4.8 |
The disappearance of fluorescence when 6, 12 and 15 bind to P. chrysosporium β-glucosidase at pH 5 can only be interpreted as the inhibitors being bound in the amine form. The proton that we know is present must be bound to one of the carboxylates, presumably the more basic residue (Fig. 10C). The possibility that the observed fluorescence quenching is caused by an amino group in the protein (lysine or tryptophan) is eliminated by the fact that only a small decrease in fluorescence is observed when lactam 19 is bound to the enzyme. For the strong inhibitors 12 and 15, where essentially all the inhibitor becomes bound, we see a residual fluorescence of 10–15% from the bound inhibitor. This could be consistent with a small degree of protonation or could be due to incomplete PET quenching in the specific geometry of the EI complexes, similar to what is observed for inhibitor 15 in solution. It is noteworthy here that the fluorescence from the bound inhibitor is totally independent of the solution pH and also of the pKa of the inhibitor.
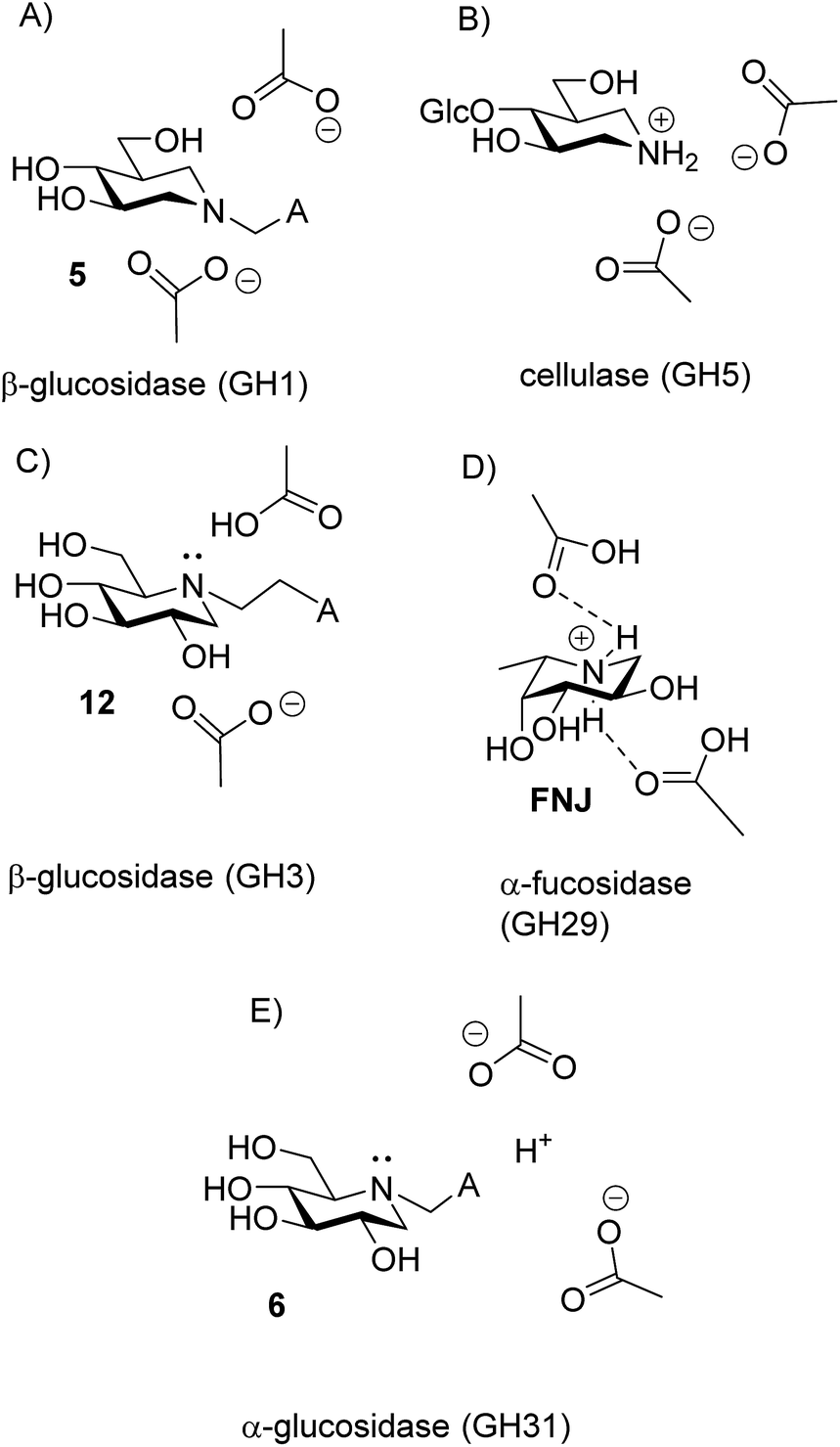 | ||
| Fig. 10 Protonation states of iminosugar–enzyme complexes. (A) EI complex of isofagomine derivative 5 and almond β-glucosidase. (B) EHI complex of 4-O-β-D-glucosyl isofagomine and cellulase from Bacillus agaradhaerens, showing protonation on N. (C) EHI complex of 12 and β-glucosidase from P. chrysosporium, showing unprotonated N. (D) EH3I complex of L-fuco-1-deoxynojirimycin with α-fucosidase from Bacteroides thetaiotaomicron. (E) EHI complex of 6 and α-glucosidase from A. Niger showing the amino-group exposed to the solvent. | ||
The finding that iminosugars bind to a GH3 β-glucosidase in the amine form appears to contrast with Legler’s findings that a GH3 α-glucosidase binds the dimethylammonium analogue of 1, which suggests that this family of enzymes likes to bind cations.9 However, it could be that Legler’s α-glucosidase binds 1 as the amine, even though it binds the quaternary amine as a cation, as this would also be fully consistent with the data.
The fluorescence behaviour of 6 bound to α-glucosidase from A. niger is surprisingly simple but is more enigmatic to interpret. The fluorescence of enzyme-bound 6, and thereby its degree of protonation, follows the pH of the solution whether it is bound to the enzyme or not. The simplest interpretation of this is that the amino group is somehow exposed to the aqueous medium (Fig. 10E), but since the inhibition pH dependence shows a clear preference for EHI binding, the bound inhibitor must be protonated at the same time as the mean deprotonation of the enzyme.
There are now 5 cases where we know the degree of protonation in iminosugar–glycosidase binding, and they are remarkably different (Fig. 10). In addition to the cellobioisofagomine–cellulase complex, where the iminosugar is protonated in an EHI complex (Fig. 10B),16 we also now know that FNJ must be protonated in the α-fucosidase binding site since it binds as the EH3I complex (Fig. 10D). At the other end of the spectrum, 5, 6, 12 and 15 do not bind to β-glucosidase as ammonium compounds. Isofagomine 5 binds to almond β-glucosidase as the EI complex (Fig. 10A),19 while 12 binds to P. chrysosporium β-glucosidase as the EHI complex, but as the amine (as do 6 and 15) (Fig. 10D). So why these differences? One significant difference between the protonated cases (Fig. 10B and D) and the non-protonated cases (Fig. 10A and C) is that the inhibitors are much more basic in the former (pKa 8.4) than in the latter (pKa 5.1 and 5.25). More basic inhibitors are obviously more likely to be protonated, as this will be energetically more favourable. When the inhibitor is a weak base, it appears that hydrogen bonding with the comparatively weak acid in the enzyme active site is more favourable.
Finally, we have special case of 6 binding to α-glucosidase, where the amine protonation is not fixed but follows the pH (Fig. 10E). The obvious explanation for this behaviour is that the amino group of the inhibitor is exposed to the solvent. The enzyme is known to be a trans-glucosidase, which could be an indication of an open or spatially active site since the enzyme has to accommodate large nucleophiles.
In any case, the findings in this paper show that the protonation of iminosugar inhibitors in glycosidase active sites can by no means be anticipated.25 We observe nanomolar inhibition of the iminosugars 12 and 15, where the amino group stays unprotonated but presumably participates in hydrogen bonding. The inhibitor is not transition state-like in terms of charge, and any potential hydrogen bond between the amino group and the enzyme does not resemble the hydrogen bonding involved in substrate conversion. With the many iminosugar inhibitors that are comparatively weak bases, it is likely that the behaviour we see here is widespread.
Conclusions
Glycosidases are sophisticated acid catalysts of glycoside hydrolysis. It would therefore seem likely that basic inhibitors that resemble the substrate would become protonated when bound to the active site of the highly efficient acid catalyst. However, considering the results in this paper it is evident that this is not always the case. In a number of cases the inhibitor binds in the neutral form. This dispels the view of the interaction of iminosugars with glycosidases as a base–acid interaction in the traditional sense.Experimental
General information
Dry solvents were trapped from a solvent purification system. Reactants were purchased from commercial sources and used without further purification. HRMS were recorded on a Bruker Solarix XR mass spectrometer analyzing TOF. Optical rotations were measured on a Perkin-Elmer 341 digital polarimeter or a Jasco P-2000 polarimeter with a path length of 1 dm. NMR spectra were recorded on a Bruker 500 MHz spectrometer. Chemical shifts (δ) are reported in ppm relative to the residual solvent signals (CDCl3: δ = 7.26 for 1H-NMR and 77.16 ppm for 13C-NMR. DMSO-d6: δ = 2.50 for 1H-NMR and 39.52 ppm for 13C-NMR). Assignments were aided by COSY and HSQC experiments.The enzymes were obtained from Megazyme. For the kinetic studies the enzymes were simply diluted and used directly. For fluorescence studies the enzyme preparations from Megazyme were added to 10 mM Na-phosphate buffer at pH 6.0 to solubilize the crystals, then further dialyzed three times against 0.5 liters of 10 mM Na-phosphate buffer at pH 6.0, one time overnight using a slide-a-lyzer (MWCO 10 kDa, 5–15 mL). After harvesting the dialyzed protein sample, the protein concentration was determined by measuring the absorbance at 280 nm with a Nanodrop ND-1000. Concentrations were calculated using Mw 84500 for β-glucosidase from P. chrysosporium23 and 116000 for α-glucosidase from A. niger.24
:4, then 1:3 and finally 9:1, a fraction of the target alcohol was collected (110 mg, 87%). 1H NMR (CDCl3, 500 MHz): δ 8.60 (m, 2H, 2 × H-a), 8.46 (m, 2H, 2 × H-a′), 7.64–7.61 (m, 4H, 2 × H-b, 2 × H-c), 5.68 (s, 2H, 2 × C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) OH). 13C NMR (CDCl3, 125 MHz): δ 130.86(3C), 128.90(3C) (6 × Cquat), 126.88(2C) (2 × C-b), 126.70(2C) (2 × C-c), 125.81(2C) (2 × C-a), 124.39(2C) (2 × C-a′), 57.57 (
OH). 13C NMR (CDCl3, 125 MHz): δ 130.86(3C), 128.90(3C) (6 × Cquat), 126.88(2C) (2 × C-b), 126.70(2C) (2 × C-c), 125.81(2C) (2 × C-a), 124.39(2C) (2 × C-a′), 57.57 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2OH). HRMS (MALDI) calcd for C15H11ClO 242.0498, found 242.0497.
Cl). 13C NMR (CDCl3, 125 MHz): δ 130.52(3C), 128.90(3C) (6 × Cquat), 127.34(2C) (2 × C-b), 126.83(2C) (2 × C-c), 126.00(2C) (2 × C-a), 123.93(2C) (2 × C-a′), 38.99 (H2Cl). HRMS (MALDI) calcd for C15H10Cl2 260.0160, found 260.0159.
:1, then 7:3, then 1:4 and finally ethyl acetate–methanol 1:4. A fraction of crude target molecule was collected (112 mg, 88%). Crystallization from methanol–dichloromethane led to crystals of pure N-(10-chloro-9-anthracenemethyl)-1-deoxynojirimycin (6, 61 mg, 48%), [α]25D = −27.6° (c = 0.4, methanol). HRMS (MALDI) calcd for C21H22ClNO4 [M + Na]+ 410.1130, found 410.1137.
O), 8.53 (d, 2H, 3JHa,Hb = 9.1 Hz, 2 × H-a), 8.09 (d, 2H, 3JHa′,Hc = 9.2 Hz, 2 × H-a′), 7.57–7.49 (m, 4H, 2 × H-b, 2 × H-c), 4.59 (m, 2H, 2 × CCHO).
OCH3), 4.57 (m, 1H, CCHOCH3), 4.39 (m, 2H, 2 × CCHCHOCH3), 3.79 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz): δ 146.09 (HOCH3), 133.88(2C), 130.29(2C), 128.90(2C) (6 × Cquat), 126.45(2C) (2 × C-b), 125.82(2C) (2 × C-c), 125.64(2C) (2 × C-a), 125.25(2C) (2 × C-a′), 105.25 (HCHOCH3), 55.99 (CCHCHOCH3), 23.29 (OCH3). HRMS (ESP): calcd for C18H15ClONa [M + Na]+: 305.0709; found: 305.1090.
H2OH). HRMS (MALDI) calcd for C15H11ClO 242.0498, found 242.0497.
Cl). 13C NMR (CDCl3, 125 MHz): δ 130.52(3C), 128.90(3C) (6 × Cquat), 127.34(2C) (2 × C-b), 126.83(2C) (2 × C-c), 126.00(2C) (2 × C-a), 123.93(2C) (2 × C-a′), 38.99 (H2Cl). HRMS (MALDI) calcd for C15H10Cl2 260.0160, found 260.0159.
:1, then 7:3, then 1:4 and finally ethyl acetate–methanol 1:4. A fraction of crude target molecule was collected (112 mg, 88%). Crystallization from methanol–dichloromethane led to crystals of pure N-(10-chloro-9-anthracenemethyl)-1-deoxynojirimycin (6, 61 mg, 48%), [α]25D = −27.6° (c = 0.4, methanol). HRMS (MALDI) calcd for C21H22ClNO4 [M + Na]+ 410.1130, found 410.1137.
O), 8.53 (d, 2H, 3JHa,Hb = 9.1 Hz, 2 × H-a), 8.09 (d, 2H, 3JHa′,Hc = 9.2 Hz, 2 × H-a′), 7.57–7.49 (m, 4H, 2 × H-b, 2 × H-c), 4.59 (m, 2H, 2 × CCHO).
OCH3), 4.57 (m, 1H, CCHOCH3), 4.39 (m, 2H, 2 × CCHCHOCH3), 3.79 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz): δ 146.09 (HOCH3), 133.88(2C), 130.29(2C), 128.90(2C) (6 × Cquat), 126.45(2C) (2 × C-b), 125.82(2C) (2 × C-c), 125.64(2C) (2 × C-a), 125.25(2C) (2 × C-a′), 105.25 (HCHOCH3), 55.99 (CCHCHOCH3), 23.29 (OCH3). HRMS (ESP): calcd for C18H15ClONa [M + Na]+: 305.0709; found: 305.1090.
E isomer: Rf = 0.57 (PE/DCM: 3/2). 1H NMR (CDCl3, 500 MHz): δ 8.58 (d, 2H, 3JHa,Hb = 8.9 Hz, 2 × H-a), 8.30 (d, 2H, 3JHa′,Hc = 8.8 Hz, 2 × H-a′), 7.62–7.54 (m, 4H, 2 × H-b, 2 × H-c), 6.33 (d, 1H, 3J = 13.1 Hz, COCH3), 5.07 (m, 1H, CCHOCH3), 4.25 (m, 2H, 2 × CCHCHOCH3), 3.42 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz): δ 148.68 (HOCH3), 139.59(2C), 130.17(2C), 128.77(2C) (6 × Cquat), 126.35(2C) (2 × C-b), 125.93(2C) (2 × C-c), 125.61(2C) (2 × C-a), 124.85(2C) (2 × C-a′), 101.19 (HCHOCH3), 55.90 (HCHCHOCH3), 26.35 (OCH3). HRMS (ESP): calcd for C18H15ClONa [M + Na]+: 305.0709; found: 305.2644.
O), 8.53 (d, 2H, 3JHa,Hb = 9.2 Hz, 2 × H-a), 8.16 (d, 2H, 3JHa′,Hc = 9.2 Hz, 2 × H-a′), 7.57–7.50 (m, 4H, 2 × H-b, 2 × H-c), 3.88 (m, 2H, 2 × CCH2CHO), 2.89 (m, 2H, 2 × CH2CCHO).
R f = 0.50 (DCM/MeOH: 7/1). 1H NMR (DMSO-d6, 500 MHz): δ 8.47 (t, 4H, 3JHa,Hb = 3JHa′,Hc = 8.8 Hz, 2 × H-a, 2 × H-a′), 7.74 (m, 2H, 2 × H-b), 7.65 (m, 2H, 2 × H-c), 4.70 (m, 2H, C3–OH, C4–OH), 4.67 (d, 1H, 3JH2,OH = 4.5 Hz, C2–OH), 4.36 (t, 1H, 3JH6,OH = 5.1 Hz, C6–OH), 3.85 (m, 1H, H-6), 3.67–3.49 (m, 3H, H-6, 2 × H-3′), 3.33–3.30 (m, 1H, H-2), 3.14–3.05 (m, 2H, H-1′, H-4), 2.98–2.85 (m, 2H, H-3, H-1), 2.71 (m, 1H, H-1′), 2.01–1.72 (m, 4H, H-5, H-1, 2 × H-2′). 13C NMR (DMSO-d6, 125 MHz): δ 129.51(3C), 127.94(3C) (6 × Cquat), 127.20(2C) (2 × C-b), 126.14(2C) (2 × C-c), 125.17(2C) (2 × C-a), 124.66(2C) (2 × C-a′), 79.17 (C-3), 71.02 (C-4), 69.50 (C-2), 66.87 (C-5), 59.58 (C-6), 57.17 (C-1), 52.06 (C-1′), 26.90 (C-2′), 25.44 (C-3′). HRMS (ESP): calcd for C23H27ClNO4 [M + H]+: 416.1629; found: 416.1632.
3)3), 0.87 (br, 18H, 2 × C(C3)3), 0.85 (s, 9H, C(C3)3), 0.13, 0.12, 0.10, 0.08, 0.07, 0.06 (6s, 18H, 6 × SiC3), 0.05 (s, 6H, 2 × SiC3). 13C NMR (CDCl3, 125 MHz): δ 171.28 (NOCH), 77.92 (C-2), 74.87 (C-3), 73.83 (C-4), 58.32 (C-5), 63.86 (C-6), 25.93(3C), 25.85(6C), 25.80(3C) (12 × C(H3)), 18.32, 18.22, 18.01, 17.96 (4 × (CH3)3), −3.72, −4.12, −4.53, −4.74, −4.86, −5.06, −5.25, −5.29 (8 × SiH3). HRMS (ESP): calcd for C30H68NO5Si4 [M + H]+: 635.2160; found: 635.3584.
3)3), 0.35, 0.25, 0.11, 0.05, −0.09, −0.21, −0.45, −0.72 (8s, 24H, 8 × SiC3). 13C NMR (CDCl3, 125 MHz): δ 170.48 (NOCH), 132.03(2C), 128.91(2C), 128.25(2C) (6 × Cquat), 126.93(2C) (2 × C-b), 126.67(2C) (2 × C-c), 125.82(2C) (2 × C-a), 125.21(2C) (2 × C-a′), 81.58 (C-3), 76.25 (C-2), 70.14 (C-4), 64.58 (C-5), 61.86 (C-6), 42.59 (C-1′), 26.46(3C), 26.04(3C), 25.77(3C), 25.72(3C) (12 × C(H3)), 18.90, 18.01, 17.96, 17.93 (4 × (CH3)3), −3.17, −3.90, −4.17, −4.49, −4.54, −4.69, −5.55, −6.40 (8 × Si(H3)). HRMS (ESP): calcd for C45H77ClNO5Si4 [M + H]+: 858.4561; found: 858.4529.
OCH), 131.21(2C), 128.62(2C), 127.82(2C) (6 × Cquat), 127.34(2C) (2 × C-b), 126.83(2C) (2 × C-c), 124.89(2C) (2 × C-a), 124.85(2C) (2 × C-a′), 74.12 (C-3), 70.19 (C-2), 70.14 (C-4), 61.79 (C-5), 59.66 (C-6), 39.14 (C-1′). HRMS (ESP): calcd for C21H21ClNO5 [M + H]+: 402.1108; found: 402.1109.
Determination of the pKa values of iminosugars 6, 12 and 15 by fluorescence titration
Iminosugars 6, 12 and 15 were each dissolved in DMSO to give 8 μmol L−1 stock solutions. 0.05 mL was added to 0.45 mL of phosphate buffers with different pH values (2.2, 3.0, 4.0, 4.4, 5.0, 5.2, 5.4, 5.6, 6.0, 6.4, 6.6, 6.8, 7.0, 7.4, 7.8, 8.0). Fluorescence spectra of each solution were recorded with excitation at 358 nm.Procedure for fluorescence measurements
The inhibitors 6, 12, 15 and 19 were dissolved at 0.012 mg mL−1 in DMSO and diluted 4 times with DMSO to create stock solutions of approximately 8.0 μM. Normally the fluorescence was measured for the following types of solution:(A) Blank (0.45 mL phosphate buffer and 0.05 mL DMSO).
(B) Inhibitor (0.45 mL phosphate buffer and 0.05 mL inhibitor stock solution, [I] = 0.8 μM).
(C) Enzyme (0.1–0.2 mL enzyme stock solution in phosphate buffer, 0.25–0.35 mL phosphate buffer and 0.05 mL DMSO. [E] = 5.8–72 μM).
(D) Enzyme and inhibitor (0.1–0.2 mL enzyme stock solution in phosphate buffer, 0.25–0.35 mL phosphate buffer and 0.05 mL inhibitor stock solution. [I] = 0.8 μM, [E] = 5.8–72 μM).
Fluorescence spectra were taken for samples A–D excited at 358 nm on a Perkin Elmer LS50 instrument. The subtracted spectra (B − A), (D − C) and (D − C − B + A) gave the net effect of the inhibitor fluorescence when bound.
Procedure for measuring glycosidase inhibition
These experiments were performed at 37 °C in aqueous phosphate buffer (0.1 M) containing 10% DMSO to make sure the inhibitor was completely dissolved. The substrate was either 4-nitrophenyl α- or β-D-glucopyranoside, depending on whether the enzyme was α- or β-glucosidase. In a platereader, 5–8 reactions with varying substrate concentrations (1–20 mM) were simultaneously started by the addition of enzyme (0.3 nM), and the formation of 4-nitrophenol followed. The absorbance was measured at 400 nm for 10–20 minutes. This procedure was performed with and without the inhibitor at a concentration close to the expected Ki. From these data, Km could be determined with and without inhibition, and from those values Ki was calculated (using Ki = [I]/(Km′/Km − 1)).Analysis of inhibition–pH data
The previously published method was used.18 1/Ki has the following dependence on the H concentration:
The EHI and EI binding curves shown in Fig. 4 and 5 were made in a spreadsheet by entering the relevant acid constants (5: 10−5.1, β-glucosidase: 10−4.4 and 10−6.7) and reasonable values for the constants Ki(1–4).
Conflicts of interest
The authors of this manuscript have no conflicts of interest.Acknowledgements
We thank Christian Schwalbe & Birgitta Kegel for technical assistance and the Danish National Research council (FNU) for financial support.Notes and references
-
(a) A. E. Stuetz and T. M. Wrodnigg, Carbohydr. Chem., 2013, 39, 120–149 CAS
; (b) Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond, ed. A. E. Stüzt, John Wiley & sons, 1999 Search PubMed
-
(a) R. Nash, Chem. World, 2004, 1, 42–44 CAS
-
(a) H.-J. Wu, C.-W. Ho, T.-P. Ko, S. D. Popat, C.-H. Lin and A. H.-J. Wang, Angew. Chem., Int. Ed., 2010, 49, 337–340 CrossRef CAS PubMed
-
(a) S. Sugimoto, H. Nakajima, K. Kosaka and H. Hosoi, Nutr. Metab., 2015, 12, 1–7 CrossRef PubMed
- M. P. Dale, H. E. Ensley, K. Kern, K. A. R. Sastry and L. D. Byers, Biochemistry, 1985, 24, 3530–3539 CrossRef CAS PubMed
-
(a) T. Kajimoto, K. K.-C. Liu, R. L. Pederson, Z. Zhong, Y. Ichikawa, J. A. Porco Jr and C.-H. Wong, J. Am. Chem. Soc., 1991, 113, 6187–6196 CrossRef CAS
-
G. Legler, in Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond, John Wiley & sons, 1999, pp. 31–67 Search PubMed
- G. Legler, Adv. Carbohydr. Chem. Biochem., 1990, 48, 319–383 CrossRef CAS PubMed
-
(a) H. Hettkamp, G. Legler and E. Bause, Eur. J. Biochem., 1984, 142, 85–90 CrossRef CAS PubMed
-
(a) W. G. Overend, C. W. Rees and J. S. Sequeira, J. Chem. Soc., 1962, 3429 RSC
-
S. G. Withers, M. Namchuk and R. Mosi, in Imino sugars as Glycosidase Inhibitors, Nojirimycin and beyond, John Wiley & sons, 1999, pp. 188–206 Search PubMed
- J. Wicki, S. J. Williams and S. G. Withers, J. Am. Chem. Soc., 2007, 129, 4530–4531 CrossRef CAS PubMed
- T. M. Gloster, P. Meloncelli, R. V. Stick, D. Zechel, A. Vasella and G. J. Davies, J. Am. Chem. Soc., 2007, 129, 2345–2354 CrossRef CAS PubMed
- T. M. Gloster and G. J. Davies, Org. Biomol. Chem., 2010, 8, 305–320 CAS
-
(a) S. U. Hansen, I. W. Plesner and M. Bols, ChemBioChem, 2000, 1, 177–180 CrossRef CAS PubMed
- A. Varrot, C. A. Tarling, J. M. MacDonald, R. V. Stick, D. L. Zechel, S. G. Withers and G. J. Davies, J. Am. Chem. Soc., 2003, 125, 7496–7497 CrossRef CAS PubMed
-
(a) A. P. de Silva and R. A. D. D. Rupasinghe, Chem. Commun., 1985, 1669–1670 RSC
- Ó. López, F.-L. Qing, C. M. Pedersen and M. Bols, Bioorg. Med. Chem., 2013, 21, 4755–4761 CrossRef PubMed
- E. Lindbäck, B. W. Laursen, J. C. N. Poulsen, K. Kilså, C. M. Pedersen and M. Bols, Org. Biomol. Chem., 2015, 13, 6562–6566 Search PubMed
- H. S. Overkleft, J. van Wiltenburg and U. K. Pandit, Tetrahedron, 1994, 34, 4215–4224 CrossRef
- C. M. Pedersen and M. Bols, Org. Biomol. Chem., 2017, 15, 1164–1173 CAS
- Ó. López and M. Bols, ChemBioChem, 2007, 8, 657–661 CrossRef PubMed
-
(a) B. Li and V. Renganathan, Appl. Environ. Microbiol., 1998, 64, 2748–2754 CAS
- B. V. McCleary, T. S. Gibson, H. Sheehan, A. Casey, L. Horgan and J. O’Flaherty, Carbohydr. Res., 1989, 185, 147–162 CrossRef CAS
- For a recent paper where protonation of aniline occurs in a protease see: J. Schiebel, R. Gaspari, A. Sandner, K. Ngo, H.-D. Gerber, A. Cavalli, A. Ostermann, A. Heine and G. Klebe, Angew. Chem., Int. Ed., 2017, 56, 4887–4890 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of new compounds, experimental details and Fig. S1–S5 (19 pages). See DOI: 10.1039/c7sc01540b |
| This journal is © The Royal Society of Chemistry 2017 |