
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting allosteric mutants that increase activity of a major antibiotic resistance enzyme†
M. J.
Latallo‡
a,
G. A.
Cortina‡
ab,
S.
Faham
a,
R. K.
Nakamoto
a and
P. M.
Kasson
 *abc
*abc
aDepartment of Molecular Physiology, University of Virginia, Box 800886, Charlottesville, VA 22908, USA. E-mail: kasson@virginia.edu
bDepartment of Biomedical Engineering, University of Virginia, USA
cScience for Life Laboratory, Department of Cell and Molecular Biology, Uppsala University, Sweden
First published on 19th July 2017
Abstract
The CTX-M family of beta lactamases mediate broad-spectrum antibiotic resistance and are present in the majority of drug-resistant Gram-negative bacterial infections worldwide. Allosteric mutations that increase catalytic rates of these drug resistance enzymes have been identified in clinical isolates but are challenging to predict prospectively. We have used molecular dynamics simulations to predict allosteric mutants increasing CTX-M9 drug resistance, experimentally testing top mutants using multiple antibiotics. Purified enzymes show an increase in catalytic rate and efficiency, while mutant crystal structures show no detectable changes from wild-type CTX-M9. We hypothesize that increased drug resistance results from changes in the conformational ensemble of an acyl intermediate in hydrolysis. Machine-learning analyses on the three top mutants identify changes to the binding-pocket conformational ensemble by which these allosteric mutations transmit their effect. These findings show how molecular simulation can predict how allosteric mutations alter active-site conformational equilibria to increase catalytic rates and thus resistance against common clinically used antibiotics.
Introduction
Antibiotic-resistant Gram-negative bacterial infections have spread rapidly in recent years, with all regions of the world reporting >50% of Enterobacteriaceae infections resistant to fluoroquinolone or cephalosporin antibiotics.3 Cephalosporin-resistant Gram-negative infections in particular are associated with a significant increase in all-cause mortality and a >30% increase in cost of care compared with their cephalosporin-sensitive counterparts.3,4 This is in part because the most common resistance mechanism is a CTX-M extended-spectrum β-lactamase, which is plasmid mediated and genetically associated with resistance to other classes of oral antibiotics.5–9 CTX-M positive infections are thus of clinical concern because they frequently require hospitalization and treatment with intravenous antibiotics.Here we are concerned with new point mutations that increase the drug resistance of CTX-M β-lactamases. Such point mutations have been observed clinically, and a number have been characterized mechanistically.10–15 However, the landscape of mutations affecting drug resistance remains incompletely characterized due to the large combinatorial space involved, and the mechanism for increased resistance by allosteric CTX-M mutants remains largely unknown. CTX-M is also an attractive system to study allosteric mutations because of the strong structural similarity across many class A β-lactamases despite large differences in sequence, substrate profiles, and catalytic rates.
Antibiotic resistance by class A β-lactamases proceeds via a two-step kinetic mechanism involving an acyl intermediate:
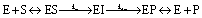
Many approaches have been taken towards computational understanding of β-lactamase function. Here, we wish to understand mutational changes that affect a chemically narrow but clinically important substrate spectrum. We therefore undertook classical molecular dynamics simulation of the acylenzyme:drug covalent complex to predict how mutations, including those distant from the active site, would affect conformational dynamics of the drug and subsequent catalytic activity in the deacylation step of hydrolysis. Our focus on the acyl intermediate thus differs from other approaches to β-lactamase function that have treated the apo and ligand-bound states.19,20 We simulated 125 different point mutants of the CTX-M9 β-lactamase, running >1000 simulations per mutant in order to improve estimation of these conformational changes. Measuring kcat changes experimentally for this many randomly selected mutants is a substantial but feasible task; the main reasons to perform molecular simulation are to enable screening for an arbitrary specified set of mutants (not just point mutants) and to facilitate detailed explanation of how allosteric mutations might affect function.
Although it would be unexpected for a single point mutation to confer substantial meropenem hydrolytic capability onto a CTX-M enzyme, we simulated CTX-M9 in complex with meropenem as a demanding test of mutations increasing drug resistance. Mutants were scored by the probability of forming hydrogen bonds believed to stabilize the deacylation transition state (Fig. S1†), and 5 high-scoring mutants as well as 4 lower-scoring ones were expressed in bacteria and tested for cefotaxime and meropenem resistance. We selected three such mutants with substantially increased resistance for further mechanistic and structural testing. All three of these mutations were at allosteric sites: T165W on a loop near the active site and S281A and L48A >20 Å away on the other side of the enzyme.
Experimental procedures
Constructs
A pET-9a plasmid containing blaCTX-M9 was the kind gift of Robert Bonomo.21 The following point mutants were constructed, were sequence-confirmed and transformed into bacterial strains as specified below: L48A; A140K; T165W; T158E; A219H; S220R; and N271D, and S281A.Molecular dynamics simulations
CTX-M9 and mutant enzymes were simulated in acylenzyme complexes with meropenem; wild-type enzyme and 3 top-scoring mutants were also simulated in complex with cefotaxime. Starting structures were constructed by least-squares fitting of a meropenem-SHV-1 acylenzyme structure22 onto the CTX-M9 apo crystal structure of CTX-M9 (PDB codes 2P74).23 Each protein was placed in an octahedral box with 2 nm minimum periodic separation and solvated with TIP3P water and 150 mM NaCl. Simulations were run using Gromacs 4.5 (ref. 24) with the AMBER99SB-ILDN force field.25 Parameters for meropenem and cefotaxime were determined using the AMBER Antechamber program using AM1-BCC partial charges.26,27 Partial charges were obtained using Ser70 covalently bound to meropenem or cefotaxime respectively. Bonded and vdW parameters were used from AMBER99SB-ILDN where available and from GAFF28 where not. Hydrogen bonds were constrained using LINCS, and short-range interactions were truncated at 1.2 nm. Long-range electrostatics were treated with Particle Mesh Ewald.29 Simulations were run at 310 K and 1 bar pressure using a velocity-rescaling thermostat30 and the Berendsen barostat.Simulations were run on two architectures: 200 simulations of CTX-M9:meropenem and 50 simulations of each of the 125 mutants tested were run on the Folding@Home platform. Additionally, 1000 simulations for each of the 125 point mutants of CTX-M9:meropenem were run using the Google Exacycle platform. Aggregate simulation averaged 5.75 microseconds per mutant for the Folding@Home simulations and 5.24 microseconds per mutant for the Exacycle simulations. Oxyanion hole scoring of mutants on these two datasets was not significantly different and is compared in Fig. S2.† Convergence analysis is provided in Fig. S3;† approximately 25 simulations averaging 57 ns each were required for a converged ranking of mutants. Three simulations of wild-type CTX-M9 in complex with cefotaxime were also run, totalling 2.5 microseconds, as well as 20 simulations of each of the CTX-M9 wild-type and the L48A, T165W, and S281A mutants of >80 ns each. Simulation snapshots were saved every 50 ps for analysis.
Simulations were scored by the probability of forming hydrogen bonds that would stabilize an oxyanion in the deacylation transition state (the “oxyanion hole”). Distances between the carbonyl oxygen on the acylated β-lactam antibiotic and each of two hydrogen bond donors (backbone amide hydrogens on residues 237 and 70) were measured, using 3 Å as a distance cutoff for hydrogen bond formation. All point mutant simulations were scored by the fraction of simulation snapshots satisfying both these hydrogen bonding criteria. Simulations were performed to create two independent data sets; scoring of these data sets was highly concordant, particularly for top mutants (Fig. S2†). Furthermore, simulations of cefotaxime acylenzymes ranked the three top experimental mutations in the same order as the meropenem acylenzyme simulations (Table S1†). CTX-M9 wild-type simulation snapshots never satisfied both hydrogen bonding criteria simultaneously in complex with meropenem, whereas separate simulations with cefotaxime satisfied the criteria in >90% of snapshots. Separate simulations of the carbapenemase KPC-2 in complex with meropenem also frequently satisfied the hydrogen bonding criteria, leading us to conclude that these criteria may be a good predictor of deacylation activity.
Positional mutual information was calculated for simulations of CTX-M9 and the L48A, T165W, and S281A mutants in complex with meropenem and with cefotaxime in a manner similar to that described previously31 except that here each simulation snapshot was rigid-body aligned to the CTX-M9 crystal structure using binding pocket atoms, where the binding pocket was defined as all residues having at least one non-hydrogen atom within 1 nm of the drug in >90% of wild-type simulation snapshots. The aligned distance of each binding-pocket atom to its reference position in the crystal structure was measured, and the probability density function was estimated by binning distances across all snapshots of a given mutant using a 1.5 Å bin width (top-10 rankings were identical for bin widths from 1.1 to 3.5 Å). Mutual information was calculated between the position of each binding-pocket atom and the corresponding protein sequence being simulated. The top 10 atoms were selected using this criterion and used to train a decision tree classifier using a Gini impurity criterion and “best split” strategy. Classification accuracy was tested using 10-fold cross-validation on the training set and separately on a randomly selected test set consisting of 20% of the original data set.
Drug resistance assays
Phenotypic testing was performed using MG1655 Omp C-/F- E. coli (gift of Linus Sandegren) transformed with each blaCTX-M9 mutant plasmid as indicated. Antibiotic susceptibility was tested using the Kirby–Bauer disc diffusion method.32,33 Discs containing the following amounts of antibiotics were purchased from BD Medical (Franklin Lakes, New Jersey): meropenem (10 μg), cefotaxime (30 μg), ceftriaxone (30 μg), cefepime (30 μg), cefuroxime (30 μg), ceftazidime (30 μg), and fold-change measurements were calculated using wild-type CTX-M9 tested with each batch as an internal control. Bacteria were grown to an OD600 of 0.1 in liquid broth and then evenly spread on Miller–Huntington agar plates, and the size of the inhibition zone was measured after 14–16 h of incubation at 37 °C. Resistance was assessed as fold change of inhibitory concentration, calculated as the square diameter of clearance of wild-type CTX-M9 over square of the diameter of the mutant. Four samples were tested for each mutant–drug combination. Bacterial growth rates were determined by optical density (OD600) measurements every 30 minutes at 37 °C with continuous shaking at 220 rpm in LB liquid broth.Additional methods
Details of enzyme purification, measurement of hydrolysis kinetics, thermostability assays, simulation preparation, and crystallization, X-ray diffraction, and refinement are given in the ESI.†Results
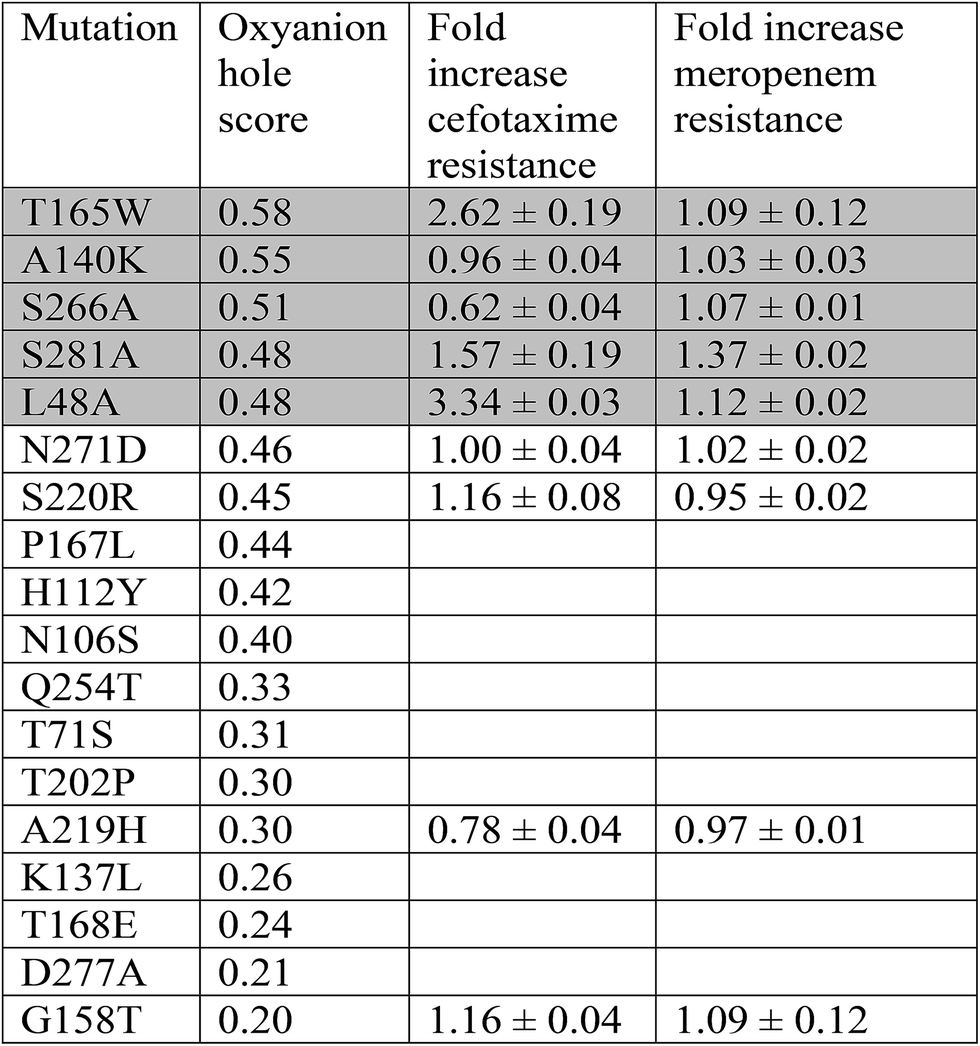
We simulated 125 point mutants of the CTX-M9 β-lactamase in complex with meropenem, using residues from the structurally similar but highly drug-resistant enzyme KPC-2 as a source of mutational diversity. Mutants were scored by probability of forming two key hydrogen bonds that stabilize the deacylation transition state (Fig. S1; all scores listed in Table S1†). Nine of ten top-scoring mutants were identical whether hydrogen-bond length or both length and angle were used as scoring criteria (Table S2†). Five high-scoring mutants and several comparators were expressed in E. coli and tested for increased resistance to cefotaxime and meropenem compared to wild-type CTX-M9. The three top-scoring mutants identified by this procedure were then purified and characterized via crystallography, thermal stability, and steady-state kinetics to understand the mechanism of allosteric modulation of drug resistance by these mutants.Prediction and testing of mutants
Point mutants of CTX-M9 at every site differing from KPC-2 were generated computationally and simulated in an acylenzyme complex with meropenem using classical molecular dynamics. The probability of forming hydrogen bonds that stabilize the nascent oxyanion in the deacylation transition state (Fig. S1†) was calculated over these simulations, averaging 5.75 microseconds in total for each of the 125 mutants (minimum of 5.3 μs per mutant), and used to score the mutants. A set of 5 high-scoring mutants and an arbitrary selection of mid-scoring mutants were expressed in E. coli and tested for cefotaxime and meropenem resistance using disc diffusion assays. Results of these tests are shown in Fig. 1 and Table 1. As discussed below, strongly increased hydrogen bond probability is hypothesized to be a predictor of increased resistance but not necessarily in a linear fashion. Three allosteric mutants, L48A, T165W, and S281A, scored particularly well in these tests and were selected for further characterization. Since two of these were alanine mutants, we compared fold-increase in cefotaxime resistance to a set of 13 allosteric CTX-M9 alanine mutants that we tested under identical conditions and reported previously;31 this comparator group showed a 0.38- to 1.05-fold gain in cefotaxime resistance, so the 1.5- to 3.3-fold gain seen in our three mutants is substantially greater than expected due to chance. | ||
| Fig. 1 CTX-M9 mutants increasing β-lactam hydrolysis. The seven top-scoring point mutants from molecular dynamics simulations and two moderate-scoring mutants were transformed into E. coli, and drug resistance was assayed using disc diffusion tests at 37 °C. These top mutations, rendered in magenta on the CTX-M9 structure (1YLJ2), are located primarily outside the drug-binding pocket (catalytic serine in orange). Of the five top-scoring mutants from simulation, three (in spheres) showed a substantial increase in cefotaxime resistance and a moderate increase in meropenem resistance. | ||
|
Broad drug-resistance and bacterial growth rates of high-scoring mutants
The three top-scoring mutants via experimental gain in cefotaxime resistance were tested against a range of cephalosporin drugs via disc-diffusion assays and showed increased resistance against all of them compared to wild-type CTX-M9 (Fig. 2). None of these enzymes retarded growth of transformed bacteria compared to wild-type CTX-M9, thus arguing against a fitness penalty for these mutations in the absence of drug (Fig. S4†). Thermostability assays on purified enzymes showed a 1.5 °C melting temperature stabilization in both the apo and meropenem-acyl forms of CTX-M9 T165W and a mild destabilization (0.5–1.5 °C) of the CTX-M9 L48A and S281A mutants (Table 2 and Fig. S6†). Although these shifts were relatively small in magnitude, they were precisely reproducible across three independent experiments per enzyme and are thus considered notable. Based on these data, T165W in particular does not display the stability-function tradeoff that has been previously found in some β-lactamase mutants.2,34,35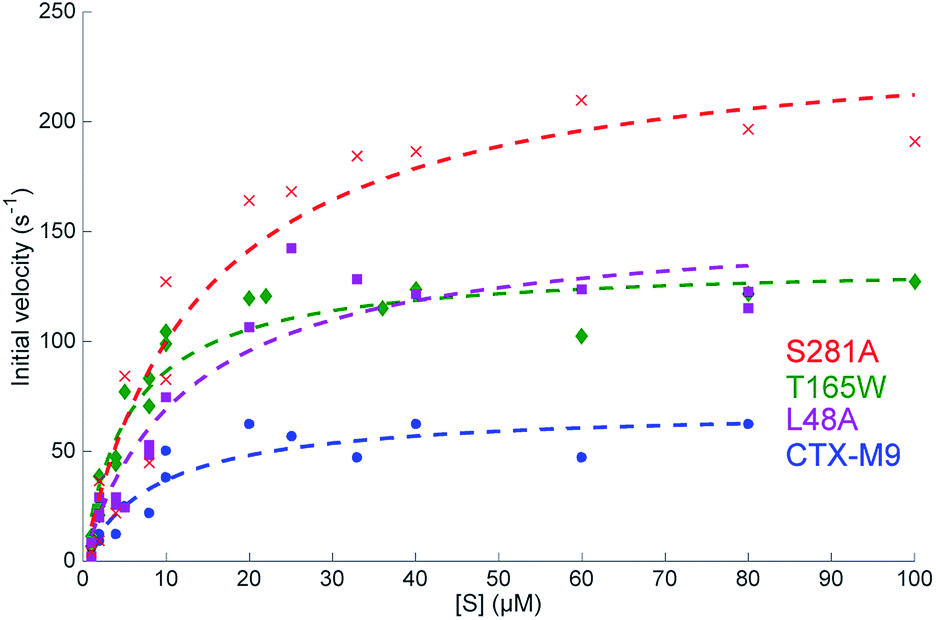 | ||
| Fig. 2 Steady-state reaction kinetics show an increased hydrolysis rate of top mutants. Initial velocities for nitrocefin hydrolysis at 28 °C are plotted as a function of substrate concentration with Michaelis–Menten fits overlaid. Experiments were performed at 0.2 nM enzyme, and at least two biological replicas per enzyme were used for fitting. Fit parameters are listed in Table 3 and show a substantial increase in kcat as well as kcat/KM for all three mutants. | ||
| CTX-M9 | L48A | T165W | S281A | |
|---|---|---|---|---|
| Apo enzyme | 48.0 ± 0.5 °C | 47.5 ± 0.5 °C | 49.5 ± 0.5 °C | 47.0 ± 0.5 °C |
| Acylenzyme | 45.0 ± 0.5 °C | 43.5 ± 0.5 °C | 46.5 ± 0.5 °C | 43.5 ± 0.5 °C |
Steady-state enzyme kinetics
Catalytic parameters of all three top-scoring mutants were determined by measuring the hydrolysis of the cephalosporin antibiotic nitrocefin by purified enzymes and fitting initial velocities according to Michaelis–Menten kinetics. At 28 °C, the S281A mutant displayed the highest nitrocefin kcat, followed by T165W and L48A, with all showing a >two-fold increased kcat over the wild-type enzyme and a >33% increase in catalytic efficiency (Fig. 3 and Table 3). Nitrocefin undergoes a shift in visible absorbance upon hydrolysis; these measurements under steady-state conditions yield an aggregate kcat for hydrolysis. These results confirm the phenotypic drug-resistance assays and show that the increased resistance derives from either an increase in kac or kdac but are insufficient alone to specify which rate constants are altered. Pre-steady-state kinetics of nitrocefin hydrolysis by CTX-M9 suggest that acylation is not rate-limiting (Fig. S7†). This is consistent with the finding that β-lactam drugs such as carbapenems that CTX-M9 is unable to hydrolyze are nonetheless readily acylated, although testing nitrocefin was necessary because other β-lactamases have demonstrated different rate-limiting steps on different classes of substrates. Additionally, E. coli expression levels (Fig. S8†) show that the increase in drug resistance cannot be explained by increased expression of high-scoring CTX-M9 mutants, while preserved or increased KM argue against increased binding of ligand, suggesting that kcat changes are primarily responsible for the increased resistance.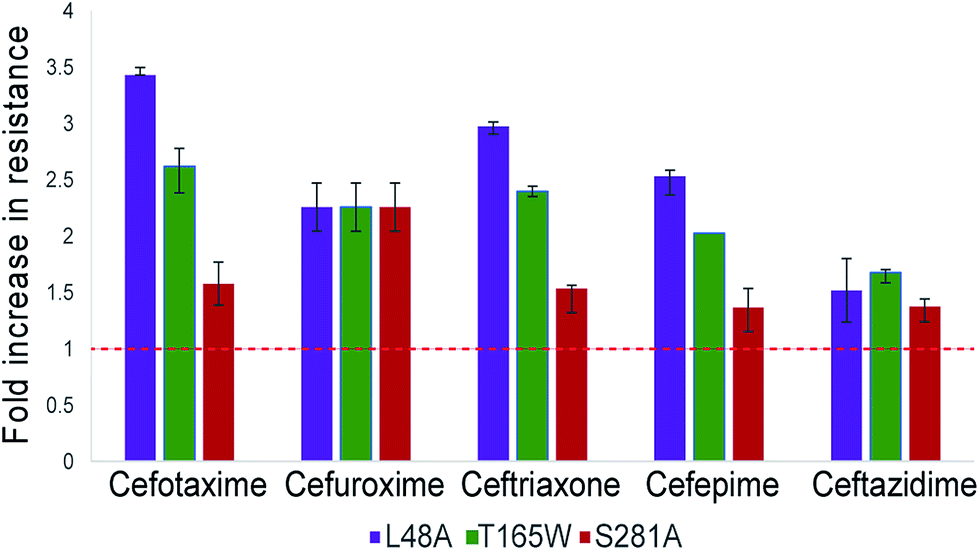 | ||
| Fig. 3 Top-scoring mutants increase drug resistance broadly against cephalosporin antibiotics. Drug resistance to five cephalosporins was measured via Kirby–Bauer disc assays for three top-scoring mutants from initial testing and compared to CTX-M9 wild type. As described in the methods, the fold-increase in inhibitory drug concentration was calculated from disc diffusion assays; values plotted are the median of four independent assays. Wild-type CTX-M9 does not confer resistance to ceftazidime in culture according to clinical lab criteria, although the clinical efficacy of this drug for such infections has been debated.58 Error bars show inter-quartile ranges. Chemical structures for the drugs used are given in Fig. S5.† | ||
| Enzyme | K M | k cat | k cat/KM |
|---|---|---|---|
| CTX-M9 | 8.9 μM (8.8–10.6 μM) | 70 s−1 (69–78 s−1) | 7.8 μM−1 s−1 (7.4–7.8 μM−1 s−1) |
| L48A | 12 μM (11.5–13.9 μM) | 156 s−1 (151–165 s−1) | 12 μM−1 s−1 (11.7–13.3 μM−1 s−1) |
| T165W | 5.7 μM (5.4–6.0 μM) | 136 s−1 (134–140 s−1) | 23 μM−1 s−1 (22.8–24.9 μM−1 s−1) |
| S281A | 14 μM (12.2–15.8 μM) | 243 s−1 (236–253 s−1) | 17 μM−1 s−1 (15.7–19.4 μM−1 s−1) |
Structures of CTX-M9 mutants
To rule out a structural change in the low-free-energy conformations of these mutants, we crystallized all three mutants and performed X-ray diffraction studies. The S281A mutant only diffracted to 6 Å and was not refined, but the structures of CTX-M9 L48A and T165W were solved to 1.73 and 1.8 Å resolution and were indistinguishable from the wild-type enzyme with RMSD of 0.3 Å (Fig. 5; crystallographic statistics in Table S3†). Structures have been deposited as PDB codes 5KMT and 5KMU. Ligand soaks of these crystals with cefoxitin as previously reported2 yielded low occupancy, but based on the close similarity of catalytic side chain structure in the apo and acylenzyme forms of CTX-M9 (Fig. S10†), we expect the low-free-energy conformation to be similar in any trapped acylenzyme state. We expect this because a structural difference associated with a more catalytically active mutant would manifest as an acylenzyme intermediate more closely resembling the deacylation transition state. Since these two structures are already quite similar for wild-type CTX-M9, we conclude such differences would be quite small, and indeed our simulations of the acylenzyme complexes also do not show large structural differences.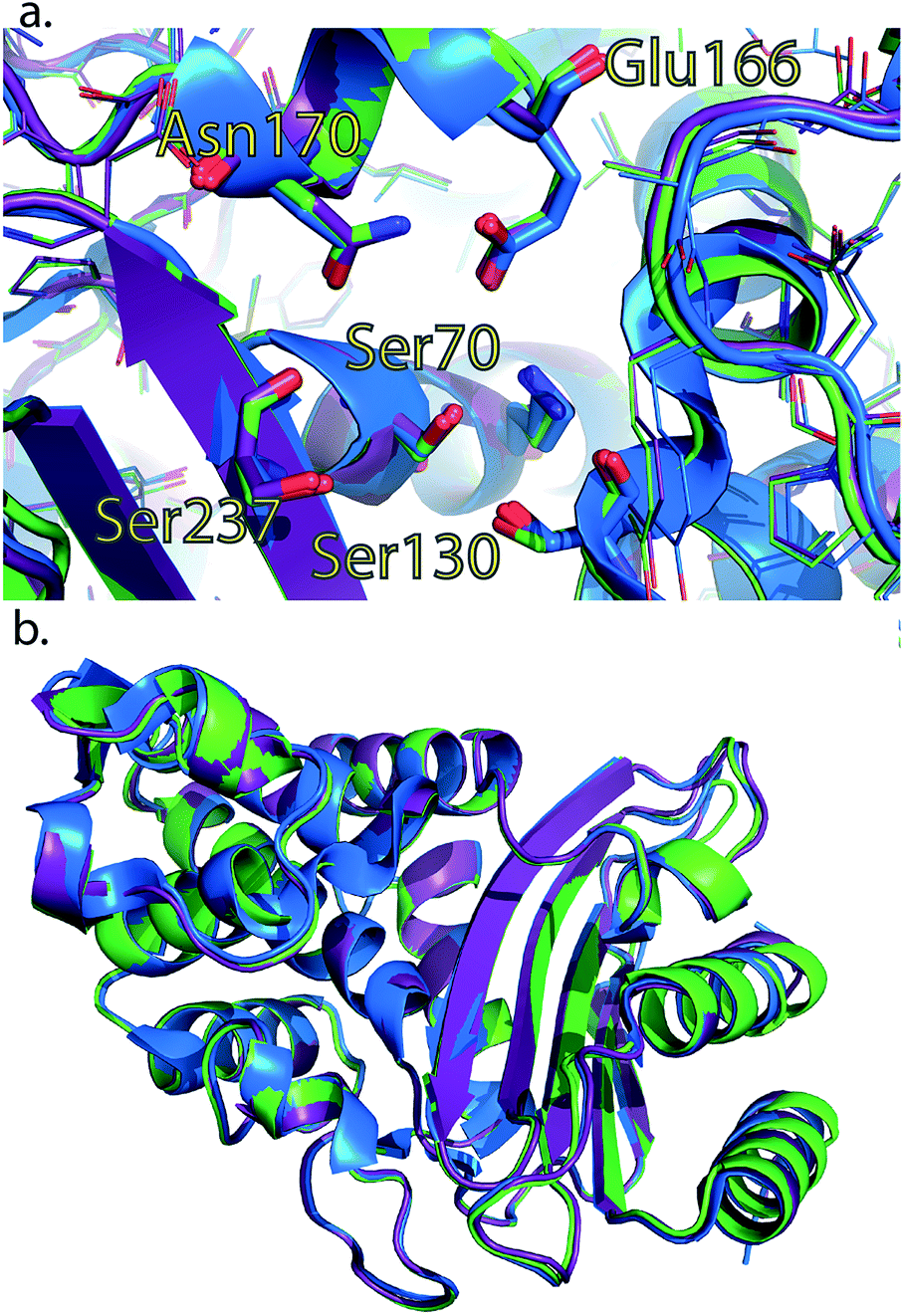 | ||
| Fig. 4 Crystal structures of L48A and T165W mutants show no substantial changes from wild-type apo enzyme. Wild-type CTX-M9 (blue) is rendered overlaid with the L48A (purple) and T165W (green) mutants. Crystal structures of these two mutants show no substantial change in the apo form in the active site region (panel a) or globally (panel b). All-atom RMSD values are 0.3 Å respectively between each of the mutants and wild-type CTX-M9. An analogous view to panel a showing catalytic side chains in previously determined crystal structures throughout the catalytic cycle is shown in Fig. S8.† | ||
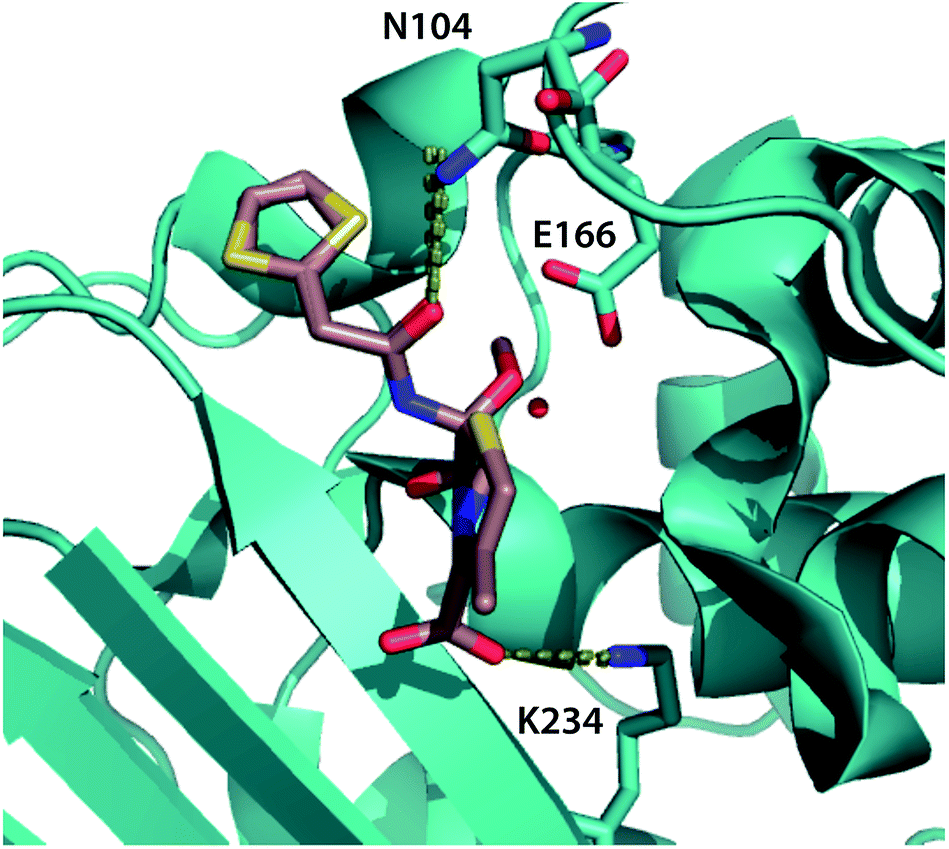 | ||
| Fig. 5 Top binding-pocket residues predicted to transmit allosteric mutations. The top three binding-pocket residues that change position with allosteric mutations affecting cephalosporin resistance are rendered in cyan sticks, visualized using the crystal structure of CTX-M9 in the acyl intermediate form with cefoxitin1 (rendered in tan). Dotted lines show key interactions between the ligand and N104 and K234; E166 is believed to interact via a catalytic water molecule. Top-scoring atoms by positional mutual information are tabulated in Table S3,† and decision trees are rendered in Fig. S9.† | ||
Simulations yield a hypothesis for allosteric effects
Based on our experimental data, we predict that the three mutant enzymes display differences in the population distribution of the acylenzyme conformational ensemble rather than large shifts in the lowest-free-energy structure. To localize potential differences and predict how distant mutations may affect enzyme–drug interactions, we calculated root-mean-square fluctuation (RMSF) for all non-hydrogen atoms in simulations of each mutant acylenzyme with meropenem and compared them to the wild-type acylenzyme simulations. Residues showing the highest percent magnitude difference from wild-type are shown and visualized on the enzyme structure in Fig. S10.† For CTX-M9 L48A and T165W, we observed a substantial increase in conformational flexibility of the loop in the region 103–105 that has been shown important in substrate binding and hydrolysis for a number of class A β-lactamases.36–39 No such increase was observed in S281A. All three mutants showed an increase in flexibility of N170, an active-site residue thought to be involved in positioning the catalytic water for hydrolysis.40 In addition, R276, a residue at the edge of the binding pocket that can interact with the free carboxylate of meropenem or cefotaxime,18,41 increased in flexibility, as did the 221–225 helix in the S281A simulations, although the significance of this last region for enzyme–drug interactions is unclear. There was also an increase in the flexibility of other omega loop residues in the T165W and L48A simulations with meropenem. These findings from simulation lead us to speculate that some combination of V103–Y105 loop interactions and N170 interactions with the drug might be related to the increased drug resistance of the mutant enzymes.We also used simulations to directly predict how our top-scoring allosteric mutations S281A, L48A, and T165W alter the conformational ensemble of the substrate-binding pocket to effect a change in drug resistance. We employed a machine-learning technique called max-mutual information feature selection42 on each of the meropenem and cefotaxime acylenzyme datasets as follows. In each snapshot from molecular dynamics simulations of wild-type and top-scoring mutant acyl-enzymes, we calculated the mutual information, a nonlinear statistical metric of relatedness, between the position of each binding-pocket atom and sequence of the enzyme. Averaged over the entire mutant dataset, this yields an information-theoretic score for which atoms in the binding pocket are most positionally responsive to the mutations under study. For the meropenem acylenzyme dataset, top-scoring atoms by this criterion are from residues 234, 166, and 104 (Table S4† and Fig. 4). As a further validation, we trained a decision tree machine-learning classifier. Using only the top 10 binding-pocket atoms, we predict the mutation that corresponds to a given binding-pocket conformation with 95.8% accuracy via 10-fold cross-validation (and 99.4% accuracy using the full binding pocket; Fig. S11†). Simulations of these mutations in acylenzyme complex with cefotaxime identify these residues as well as residue 105 as top-scoring (Table S4 and Fig. S11†). We thus predict that the S281A, L48A, and T165W mutant enzymes alter the acylenzyme conformational ensemble and thus the catalytic rate by changing the positions of residues 104, 166, and 234 in this ensemble, although not in the apo enzyme crystal structures (displacement histograms in Fig. S12†). Of these residues, glutamate 166 is involved in coordinating catalytic water molecules in hydrolysis,1 asparagine 104 has been implicated in substrate positioning in the acyl-intermediate form of other β-lactamases,12,43 and lysine 234 is highly conserved and thought to interact with the cephalosporin C3 carboxylate moiety.40
Although all these active-site residues and the allosteric sites 48 and 165 are ≥ 99% conserved within the CTX-M family, approximately 25% of CTX-M sequences contain the S281A mutation. Using a multiple sequence alignment of 500 beta lactamases most closely related to CTX-M9, residues 281 and 105 showed a strong sequence co-variation, quantified as phylogenetically corrected sequence mutual information44 (Table S5†). We also analyzed residue–residue dynamic relationships via positional mutual information (as opposed to residue–mutant relationships). Four simulation datasets were examined per mutation site: wild-type meropenem acylenzyme, wild-type cefotaxime acylenzyme, mutant meropenem acylenzyme, and mutant cefotaxime acylenzyme. Residue K234 ranked in the top 5 binding-pocket residues linked to residue 48 in 3 of 4 datasets (Table S6†). E166 was most closely linked to residue 165 in all simulation datasets (although this can be explained by proximity), and 104 scored in the top 5 once. K234 again scored in the top 5 binding-pocket residues linked to residue 281 for 3 of 4 datasets. No other binding-pocket residue was similarly enriched in linkage from more than one allosteric site.
Discussion
Allosteric mutants that increase the activity of an already competent β-lactamase represent a challenging prediction target, and successful prediction often yields moderate rate enhancement in these cases, in contrast to the larger gains when successfully engineering new substrate activity. Our results demonstrate that simulations of acylenzyme conformational dynamics can prospectively identify new allosteric mutations and that the mechanism of such mutations is consistent with a change to the acylenzyme intermediate or the deacylation transition state. Clearly, simulations of these conformational dynamics—the classical dynamics in the acylenzyme state—neglect much of the hydrolysis process and thus cannot by themselves be expected to capture mutations that affect ligand binding, the acylation transition state, or the reactive chemistry directly. Those steps have been the subject of other simulation studies,45–53 and it is hoped that a combined multimodal approach may yield a more comprehensive understanding of mutations affecting CTX β-lactamase function. Our results are striking in that they show how acylenzyme conformational dynamics can identify allosteric mutations that do not substantially alter minimum-free-energy structures (of the apo enzyme and, we speculate, other intermediates) yet increase catalytic rates and resistance to clinically used antibiotics several-fold. As might be expected, the allosteric mutations thus identified do not lie in the thermostable or rigid core of the molecule—in our tests, mutations there are more likely decrease rather than enhance function (Table S7†). The oxyanion hole stabilization metric used to score mutants is believed necessary but not sufficient for hydrolysis and is likely noisy due to simulation sampling. It thus is not expected to be a linear predictor of kcat but nonetheless demonstrates good predictive ability in identifying mutants with increased resistance (3/5 top-scoring mutants have increased resistance).If we classify the conformation ensemble of the acylenzyme state as an equilibrium between microstates that would stabilize an oxyanion (OXY) and microstates that would not (NON), our simulation dataset samples the OXY → NON conversion rate much better than the NON → OXY conversion rate. We ascribe the predictiveness of oxyanion hole scores computed using meropenem acylenzyme simulations for cephalosporin resistance to a correlation between OXY → NON conversion rates across mutants for these drugs. Additionally, since meropenem acylenzyme simulations display somewhat faster OXY → NON rates, these simulations provide statistically better-converged estimators of the oxyanion hole scores. We also believe that undersampling of the slow NON → OXY conversion rate also likely explains why our simulation dataset is not predictive of meropenem resistance.
This modulation of activity without substantially changing dominant conformation raises the question of whether our high-scoring mutations alter the free energy of the deacylation transition state and thus control the rate-limiting step for β-lactam hydrolysis by CTX-M9. Classical molecular dynamics calculations that do not explicitly treat reactivity will not capture this directly. However, the scoring method we use here to assess hydrogen bonds that would stabilize an oxyanion in the deacylation transition state does succeed in predicting (either mechanistically or serendipitously) a set of mutants that increase catalytic activity. We therefore hypothesize that the predicted mutations either decrease the free energy of the deacylation transition state ensemble, likely via oxyanion stabilization, or alter the free energy of the acyl intermediate conformational ensemble with an overall effect of reducing the ΔG‡ for deacylation. The kinetic data support an increase in kcat and, although they do not specifically prove an increase in kdac, are consistent with this hypothesis.
Point mutations that increases resistance to clinically used drugs at no apparent fitness cost—identical growth rates and for at least one mutant improved thermal stability—raise the question of why these mutants have not fixed in the bacterial population. We hypothesize that these indicate that the primary selection factor for CTX-M9 β-lactamase fitness in the wild must not be cephalosporin hydrolysis (or likely any clinically used β-lactam) but some non-pharmacological toxin. This would not be surprising given that β-lactamases likely arose as defences against microbial toxins. However, even circumstantial evidence that these and not pharmacological therapy dominate selection raises interesting implications for the further evolution of drug resistance. Such evidence implies that selection results from the interplay of microbial chemo-ecology and human intervention rather than a scenario where diversity was generated by microbial interactions and current selection is primarily driven by human factors.54–56
Conclusions
Allosteric mutations that enhance kcat raise an important mechanistic question of how the allosteric change is manifested in the binding pocket. General theories of allostery include analyses of spatial transmission paths or modulation of protein conformational ensembles.57 Here, we show allosteric mutations that enhance kcat without substantially altering the active-site conformation in apo enzyme crystal structures. Machine-learning analyses can query how the conformational ensemble of the binding pocket is altered by these mutations in simulations and thus predict how distant changes may manifest in the binding pocket. Our results suggest a particular subset of residues involved in substrate positioning or coordinating catalytic water change in their conformational distribution with allosteric mutations that increase kcat. These findings will help guide future analyses of CTX-M9 substrate specificity and provide a generalizable method for identifying specific binding-pocket residues with altered conformational ensembles in allosteric mutants.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Folding@Home donors for computing time and T. Fenn for many helpful discussions. We thank Z. Derewenda, D. Konerding, K. Skorupka, C. Stroupe, J. Wagner, and J. Zimmer for kind advice and use of equipment or reagents and J. Morgan for assistance with initial crystallographic refinement. Plasmids and cell lines were kindly provided by R. Bonomo, Z. Derewenda, and L. Sandegren. This work was funded by a grant from The Hartwell Foundation to P. M. K. and a Google Exacycle Award to P. M. K. Additional simulation resources were provided by the Swedish National Infrastructure for Computing at High Performance Computing Center North. G. A. C was supported by the National Institutes of Health (5T32AI007046). Crystallographic data are deposited as PDB codes 5KMT and 5KMU. Simulation data are available from the Dryad Digital Repository: DOI: 10.5061/dryad.ck3dh.Notes and references
- Y. Chen, B. Shoichet and R. Bonnet, J. Am. Chem. Soc., 2005, 127, 5423–5434 CrossRef CAS PubMed.
- Y. Chen, J. Delmas, J. Sirot, B. Shoichet and R. Bonnet, J. Mol. Biol., 2005, 348, 349–362 CrossRef CAS PubMed.
- W. H. Organization, Antimicrobial resistance: global report on surveillance, World Health Organization, 2014 Search PubMed.
- P. D. Mauldin, C. D. Salgado, I. S. Hansen, D. T. Durup and J. A. Bosso, Antimicrob. Agents Chemother., 2010, 54, 109–115 CrossRef CAS PubMed.
- G. A. Jacoby, K. E. Walsh, D. M. Mills, V. J. Walker, H. Oh, A. Robicsek and D. C. Hooper, Antimicrob. Agents Chemother., 2006, 50, 1178–1182 CrossRef CAS PubMed.
- J. R. Johnson, B. Johnston, C. Clabots, M. A. Kuskowski and M. Castanheira, Clin. Infect. Dis., 2010, 51, 286–294 CrossRef PubMed.
- Y. Matsumura, M. Yamamoto, M. Nagao, Y. Ito, S. Takakura, S. Ichiyama and G. Kyoto-Shiga, Clinical Microbiology Study, Antimicrob. Agents Chemother., 2013, 57, 4736–4742 CrossRef CAS PubMed.
- T. M. Coque, F. Baquero and R. Canton, Eurosurveillance, 2008, 13(48), 19051 Search PubMed.
- D. M. Livermore, R. Canton, M. Gniadkowski, P. Nordmann, G. M. Rossolini, G. Arlet, J. Ayala, T. M. Coque, I. Kern-Zdanowicz, F. Luzzaro, L. Poirel and N. Woodford, J. Antimicrob. Chemother., 2007, 59, 165–174 CrossRef CAS PubMed.
- C. Aumeran, C. Chanal, R. Labia, D. Sirot, J. Sirot and R. Bonnet, Antimicrob. Agents Chemother., 2003, 47, 2958–2961 CrossRef CAS PubMed.
- M. Cartelle, M. del Mar Tomas, F. Molina, R. Moure, R. Villanueva and G. Bou, Antimicrob. Agents Chemother., 2004, 48, 2308–2313 CrossRef CAS PubMed.
- F. J. Perez-Llarena, F. Kerff, O. Abian, S. Mallo, M. C. Fernandez, M. Galleni, J. Sancho and G. Bou, Antimicrob. Agents Chemother., 2011, 55, 4361–4368 CrossRef CAS PubMed.
- A. M. Hujer, K. M. Hujer and R. A. Bonomo, Biochim. Biophys. Acta, 2001, 1547, 37–50 CrossRef CAS.
- T. Sun, C. R. Bethel, R. A. Bonomo and J. R. Knox, Biochemistry, 2004, 43, 14111–14117 CrossRef CAS PubMed.
- M. A. Totir, P. S. Padayatti, M. S. Helfand, M. P. Carey, R. A. Bonomo, P. R. Carey and F. van den Akker, Biochemistry, 2006, 45, 11895–11904 CrossRef CAS PubMed.
- H. Christensen, M. T. Martin and S. G. Waley, Biochem. J., 1990, 266, 853–861 CAS.
- I. Saves, O. Burlet-Schiltz, L. Maveyraud, J. P. Samama, J. C. Prome and J. M. Masson, Biochemistry, 1995, 34, 11660–11667 CrossRef CAS PubMed.
- C. J. Adamski, A. M. Cardenas, N. G. Brown, L. B. Horton, B. Sankaran, B. V. Prasad, H. F. Gilbert and T. Palzkill, Biochemistry, 2015, 54, 447–457 CrossRef CAS PubMed.
- T. S. Zou, V. A. Risso, J. A. Gavira, J. M. Sanchez-Ruiz and S. B. Ozkan, Mol. Biol. Evol., 2015, 32, 132–143 CrossRef CAS PubMed.
- K. M. Hart, C. M. W. Ho, S. Dutta, M. L. Gross and G. R. Bowman, Nat. Commun., 2016, 7, 12965 CrossRef CAS PubMed.
- C. R. Bethel, M. Taracila, T. Shyr, J. M. Thomson, A. M. Distler, K. M. Hujer, A. M. Hujer, A. Endimiani, K. Papp-Wallace, R. Bonnet and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 3465–3475 CrossRef CAS PubMed.
- M. Nukaga, C. R. Bethel, J. M. Thomson, A. M. Hujer, A. Distler, V. E. Anderson, J. R. Knox and R. A. Bonomo, J. Am. Chem. Soc., 2008, 130, 12656–12662 CrossRef CAS PubMed.
- Y. Chen, R. Bonnet and B. K. Shoichet, J. Am. Chem. Soc., 2007, 129, 5378–5380 CrossRef CAS PubMed.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- G. A. Cortina and P. M. Kasson, Bioinformatics, 2016, 32, 3420–3427 CAS.
- J. Hudzicki, Am. Soc. Microbiol., 2009, http://www.asmscience.org/content/education/protocol/protocol.3189 Search PubMed.
- A. W. Bauer, W. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493–496 CAS.
- X. Wang, G. Minasov and B. K. Shoichet, J. Mol. Biol., 2002, 320, 85–95 CrossRef CAS PubMed.
- V. L. Thomas, A. C. McReynolds and B. K. Shoichet, J. Mol. Biol., 2010, 396, 47–59 CrossRef CAS PubMed.
- K. M. Papp-Wallace, M. Taracila, C. J. Wallace, K. M. Hujer, C. R. Bethel, J. M. Hornick and R. A. Bonomo, Protein Sci., 2010, 19, 1714–1727 CrossRef CAS PubMed.
- N. Doucet, P. Y. De Wals and J. N. Pelletier, J. Biol. Chem., 2004, 279, 46295–46303 CrossRef CAS PubMed.
- W. A. Escobar, J. Miller and A. L. Fink, Biochem. J., 1994, 303(2), 555–558 CrossRef CAS PubMed.
- A. Petit, L. Maveyraud, F. Lenfant, J. P. Samama, R. Labia and J. M. Masson, Biochem. J., 1995, 305(1), 33–40 CrossRef CAS PubMed.
- J. Delmas, Y. Chen, F. Prati, F. Robin, B. K. Shoichet and R. Bonnet, J. Mol. Biol., 2008, 375, 192–201 CrossRef CAS PubMed.
- F. J. Perez-Llarena, M. Cartelle, S. Mallo, A. Beceiro, A. Perez, R. Villanueva, A. Romero, R. Bonnet and G. Bou, J. Antimicrob. Chemother., 2008, 61, 792–797 CrossRef CAS PubMed.
- H. Peng, F. Long and C. Ding, IEEE Trans. Pattern Anal. Mach. Intell., 2005, 27, 1226–1238 CrossRef PubMed.
- T. Shimamura, A. Ibuka, S. Fushinobu, T. Wakagi, M. Ishiguro, Y. Ishii and H. Matsuzawa, J. Biol. Chem., 2002, 277, 46601–46608 CrossRef CAS PubMed.
- S. D. Dunn, L. M. Wahl and G. B. Gloor, Bioinformatics, 2008, 24, 333–340 CrossRef CAS PubMed.
- R. C. Wade, R. R. Gabdoulline, S. K. Ludemann and V. Lounnas, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5942–5949 CrossRef CAS.
- D. Suarez, N. Diaz and K. M. Merz Jr, J. Comput. Chem., 2002, 23, 1587–1600 CrossRef CAS PubMed.
- E. I. Chudyk, M. A. Limb, C. Jones, J. Spencer, M. W. van der Kamp and A. J. Mulholland, Chem. Commun., 2014, 50, 14736–14739 RSC.
- F. Fonseca, E. I. Chudyk, M. W. van der Kamp, A. Correia, A. J. Mulholland and J. Spencer, J. Am. Chem. Soc., 2012, 134, 18275–18285 CrossRef CAS PubMed.
- J. C. Hermann, C. Hensen, L. Ridder, A. J. Mulholland and H. D. Holtje, J. Am. Chem. Soc., 2005, 127, 4454–4465 CrossRef CAS PubMed.
- N. J. Bernstein and R. F. Pratt, Biochemistry, 1999, 38, 10499–10510 CrossRef CAS PubMed.
- J. Lamotte-Brasseur, V. Lounnas, X. Raquet and R. C. Wade, Protein Sci., 1999, 8, 404–409 CrossRef CAS PubMed.
- F. Bos and J. Pleiss, Biophys. J., 2009, 97, 2550–2558 CrossRef PubMed.
- P. Bisignano, S. Doerr, M. J. Harvey, A. D. Favia, A. Cavalli and G. De Fabritiis, J. Chem. Inf. Model., 2014, 54, 362–366 CrossRef CAS PubMed.
- R. I. Aminov and R. I. Mackie, FEMS Microbiol. Lett., 2007, 271, 147–161 CrossRef CAS PubMed.
- J. L. Martinez, Science, 2008, 321, 365–367 CrossRef CAS PubMed.
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- H. N. Motlagh, J. O. Wrabl, J. Li and V. J. Hilser, Nature, 2014, 508, 331–339 CrossRef CAS PubMed.
- R. Bonnet, Antimicrob. Agents Chemother., 2004, 48(1), 1–14 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02676e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2017 |