
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins: an efficient approach to chiral amines†
Wenchao
Gao
a,
Hui
Lv
 *ad,
Tonghuan
Zhang
bc,
Yuhong
Yang
b,
Lung Wa
Chung
*b,
Yun-Dong
Wu
c and
Xumu
Zhang
*ab
*ad,
Tonghuan
Zhang
bc,
Yuhong
Yang
b,
Lung Wa
Chung
*b,
Yun-Dong
Wu
c and
Xumu
Zhang
*ab
aKey Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China. E-mail: huilv@whu.edu.cn
bDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, 518055, China. E-mail: zhangxm@sustc.edu.cn; oscarchung@sustc.edu.cn
cLab of Computational Chemistry and Drug Design, Laboratory of Chemical Genomics, Peking University Shenzhen Graduate School, Shenzhen 518055, China
dEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, Wuhan, 430072, China
First published on 4th July 2017
Abstract
An efficient approach for synthesizing chiral β-amino nitroalkanes has been developed via the Ni-catalyzed asymmetric hydrogenation of challenging β-amino nitroolefins under mild conditions, affording the desired products in excellent yields and with high enantioselectivities. This protocol had good compatibility with the wide substrate scope and a range of functional groups. The synthesis of chiral β-amino nitroalkanes on a gram scale has also been achieved. In addition, the reaction mechanism was elucidated using a combined experimental and computational study, and it involved acetate-assisted heterolytic H2 cleavage followed by 1,4-hydride addition and protonation to achieve the nitroalkanes.
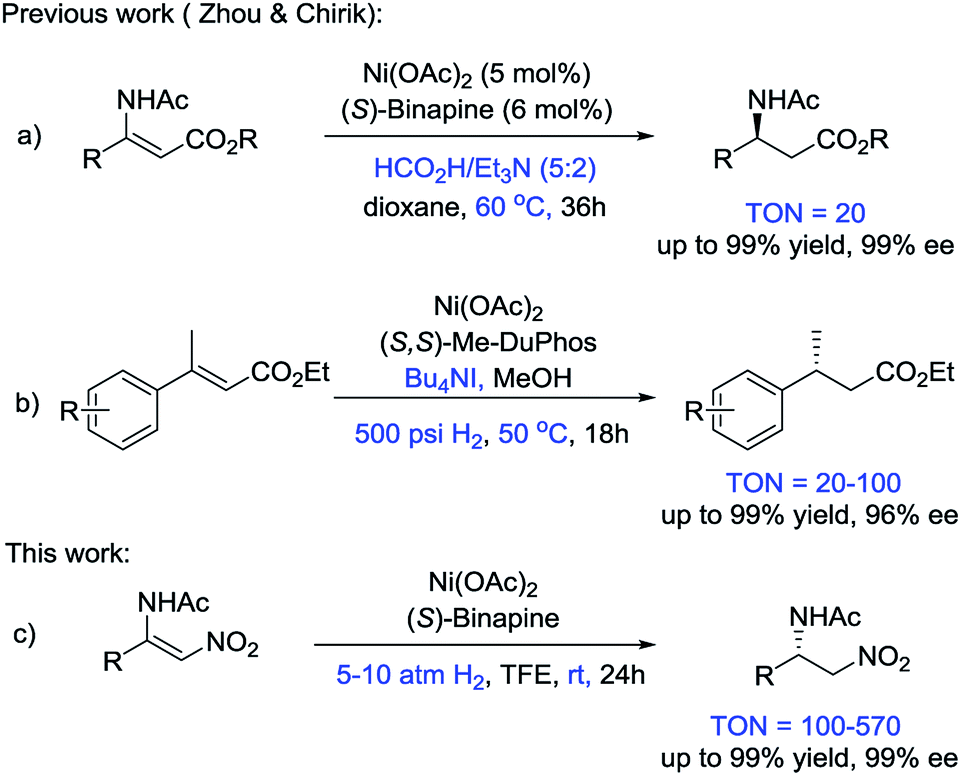
The development of new protocols for synthesizing chiral compounds in an environmentally-friendly and cost-effective manner is an important subject in both academic research and industrial applications.1 In this context, asymmetric hydrogenation, one of the most effective approaches for constructing chiral compounds, has been rapidly developed and has achieved remarkable progress. However, almost all of the catalytic systems for asymmetric hydrogenation heavily rely on noble transition metal catalysts based on Ru, Rh, Ir or Pd.1 In contrast, catalysts based on the cheap, earth-abundant first-row transition metals have potential advantages in terms of cost and sustainability. Therefore, Fe-, Co- and Ni-catalyzed asymmetric hydrogenation has attracted great attention.2
Recently, the Fe-catalyzed asymmetric hydrogenation of ketones and imines and the Co-catalyzed asymmetric hydrogenation of ketones and olefins have been reported.2–4 These methods exhibited the great potential of first-row transition metals in asymmetric hydrogenation. However, seminal studies on Ni-catalyzed asymmetric hydrogenation are rare, although heterogeneous nickel catalysts have long played a prominent role in reduction reactions. In 2008, Hamada et al. reported the Ni-catalyzed asymmetric hydrogenation of α-amino-β-ketoesters via dynamic kinetic resolution.5 Subsequently, Zhou and coworkers disclosed a series of studies on the Ni-catalyzed asymmetric transfer hydrogenation of enamides and hydrazones, and the asymmetric reductive amination of ketones (Scheme 1a).6 Recently, Chirik and coworkers developed the first example of the asymmetric hydrogenation of α,β-unsaturated esters using Ni catalysts and H2 gas (Scheme 1b).7 Given that the studies on Ni-catalyzed asymmetric hydrogenation are in their infancy, exploring a wide substrate scope, increasing the enantioselectivities of the products and improving the TON values of the catalysts are highly desirable.
 | ||
| Scheme 1 Reports on Ni-catalyzed asymmetric hydrogenation. | ||
Generally, β-acylamino nitroolefins are challenging substrates for asymmetric hydrogenation due to the weak binding affinity of the olefin with an electron-poor nitro group. There are only a few examples on the asymmetric hydrogenation of β-acylamino nitroolefins that employed precious transition metal catalysts.8 To the best of our knowledge, cheap transition metals have never been used in the asymmetric hydrogenation of β-acylamino nitroolefins. Herein, we report an efficient access route to chiral β-amino nitroalkanes via the Ni-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins under mild conditions (Scheme 1c).
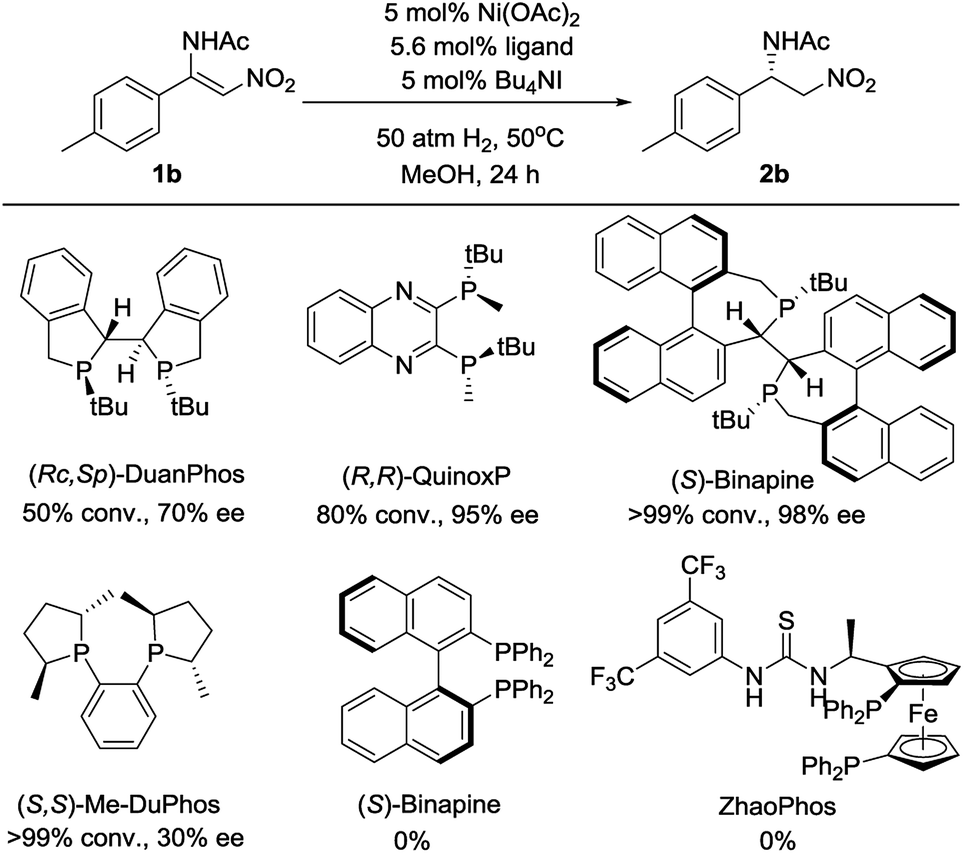
Initially, (Z)-N-(2-nitro-1-(p-tolyl)vinyl)acetamide 1b was chosen as a model substrate for optimizing the reaction conditions. Some chiral diphosphine ligands were evaluated. When the reaction was carried out in the presence of 5 mol% Ni(OAc)2 and 5.6 mol% ligand under 50 atm of H2 at 50 °C in MeOH for 24 h, using Bu4NI as the additive, all of the P-chiral diphosphine ligands could catalyze the reaction with different conversions and enantioselectivities (Fig. 1). (S)-Binapine was found to give the best results (>99% conversion, 98% ee). Axial chiral and planar chiral diphosphine ligands had no activity for this reaction. The solvent screening experiments indicated that CF3CH2OH was the best choice (Table 1, entries 1–6). Further investigation showed that the Bu4NI additive had no effect on this reaction (Table 1, entries 7 and 8). When the catalyst loading was reduced from 5 mol% to 1 mol%, the reaction completed smoothly with similar results (Table 1, entry 9). Decreasing the hydrogen pressure to 5 atm did not affect the reaction. The excellent enantioselectivity was maintained when further decreasing the hydrogen pressure to 1 atm, but the yield dramatically decreased (Table 1, entries 10–14).
 | ||
| Fig. 1 The performance of chiral phosphines in the asymmetric hydrogenation of 1b. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X (mol%) | Solvent | H2 (atm) | Temp. (°C) | Conv.b (%) | eec (%) |
a Conditions: Ni(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(S)-binapine:Bu4NI = 1:1.1:1, and 1b (0.1 mmol) in 1 ml of solvent.
b The conversion was determined using 1H NMR and HPLC analysis.
c The ee values were determined using HPLC analysis with a chiral stationary phase.
d Without using Bu4NI as an additive. :(S)-binapine:Bu4NI = 1:1.1:1, and 1b (0.1 mmol) in 1 ml of solvent.
b The conversion was determined using 1H NMR and HPLC analysis.
c The ee values were determined using HPLC analysis with a chiral stationary phase.
d Without using Bu4NI as an additive.
|
||||||
| 1 | 5 | MeOH | 50 | 50 | >99 | 98 |
| 2 | 5 | EtOH | 50 | 50 | 38 | 90 |
| 3 | 5 | iPrOH | 50 | 50 | 40 | 92 |
| 4 | 5 | DCM | 50 | 50 | 26 | 89 |
| 5 | 5 | THF | 50 | 50 | Trace | 60 |
| 6 | 5 | Toluene | 50 | 50 | 12 | 78 |
| 7 | 5 | TFE | 50 | 50 | >99 | >99 |
| 8d | 5 | TFE | 50 | 50 | >99 | >99 |
| 9d | 1 | TFE | 50 | 50 | >99 | >99 |
| 10d | 1 | TFE | 50 | 40 | >99 | >99 |
| 11d | 1 | TFE | 50 | rt | >99 | >99 |
| 12d | 1 | TFE | 10 | rt | >99 | >99 |
| 13d | 1 | TFE | 5 | rt | >99 | >99 |
| 14d | 1 | TFE | 1 | rt | 72 | >99 |
Under the optimized reaction conditions, the substrate scope was examined. As shown in Scheme 2, various electron-rich or electron-poor aromatic group substituted β-acylamino nitroolefins could be hydrogenated smoothly to afford the corresponding β-amino nitroalkanes in high yields and with excellent enantioselectivities. The position of the substituents on the benzene ring had no influence on the reactivity and enantioselectivity. In addition, a heteroaromatic group substituted β-acylamino nitroolefin was also well-tolerated, albeit with a slight decrease in the yield and enantioselectivity. It is worth noting that alkyl substituted β-acylamino nitroolefins, which are challenging substrates for Rh or Ir catalysts, could also be hydrogenated in high yields and with excellent enantioselectivities.
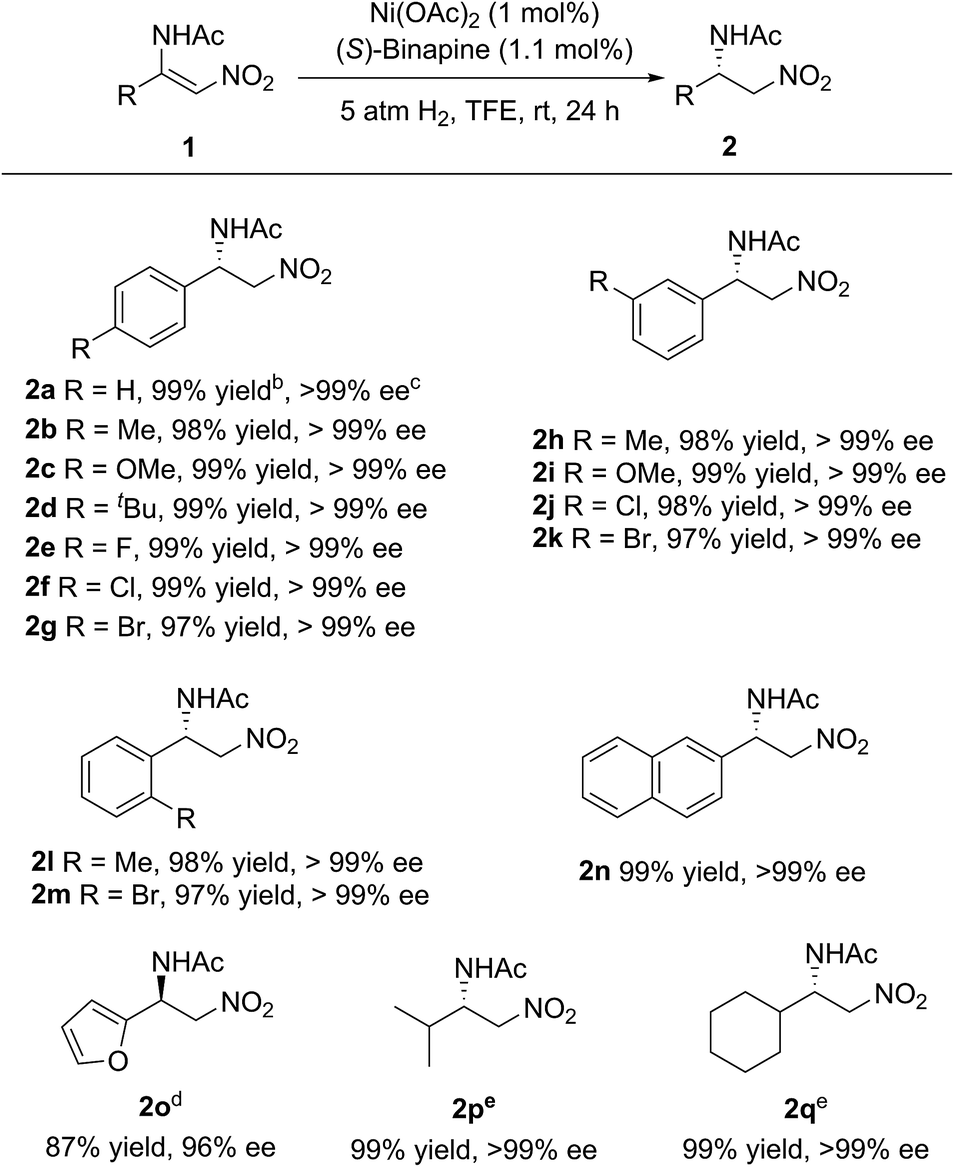 | ||
| Scheme 2 Substrate scope. a The general reaction conditions: the ratio 1 (0.1 mmol):Ni(OAc)2:(S)-binapine = 100:1:1.1, 1 ml of TFE as the solvent under 5 atm of H2 at rt for 24 h. b Isolated yield. c Determined using HPLC analysis with a chiral stationary phase. d 6 mol% catalyst, 50 °C, 50 atm of H2. e 10 atm of H2. | ||
To explore the potential application of this methodology, the synthesis of the chiral β-acylamino nitroalkane on a gram scale was examined. The reaction was carried out under 5 atm of hydrogen pressure at room temperature in the presence of 1 mol% Ni(OAc)2/(S)-binapine complex, affording the desired compound 2a in 99% yield and with 99% ee. When the catalyst loading was decreased to 0.1 mol%, we achieved 2a in 57% yield and with 99% ee (Scheme 3).
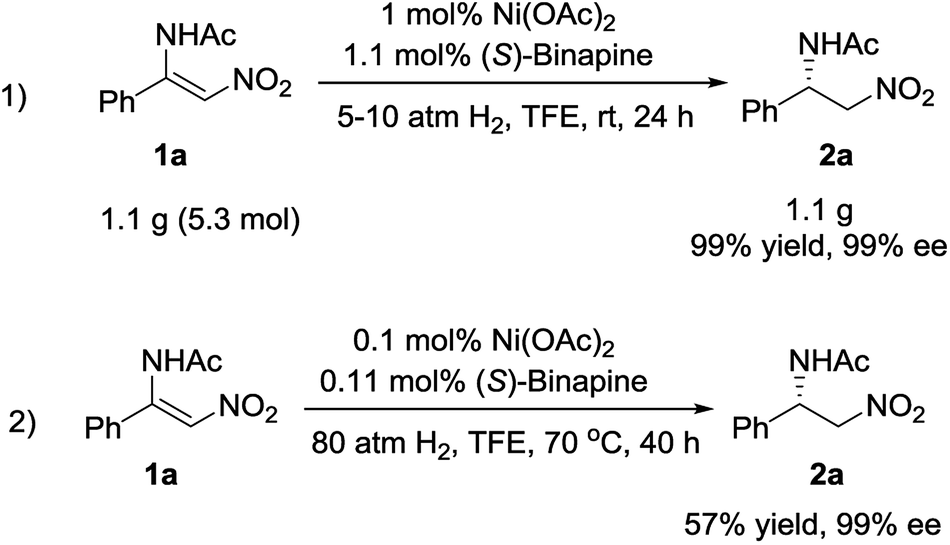 | ||
| Scheme 3 Gram-scale reaction and S/C evaluation. | ||
To decipher the possible reaction mechanism for the Ni-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins, a series of isotopic labeling studies were conducted. Firstly, when 1a was hydrogenated with 10 atm of D2 in TFE solution, the deuterium atom was solely added at the β position (Scheme 4a). When the experiment was repeated with H2 and CD3OD, the deuterium atoms were incorporated at the α position (Scheme 4b). Performing the hydrogenation reaction under 30 atm of D2 in CD3OD solution gave the expected compound with the deuterium atoms at both the α and β positions (Scheme 4c). Finally, when 2a was dissolved in CD3OD solution and stirred, the deuterium atoms were found to be incorporated at the α position, showing the H/D scrambling of the product (Scheme 4d).9
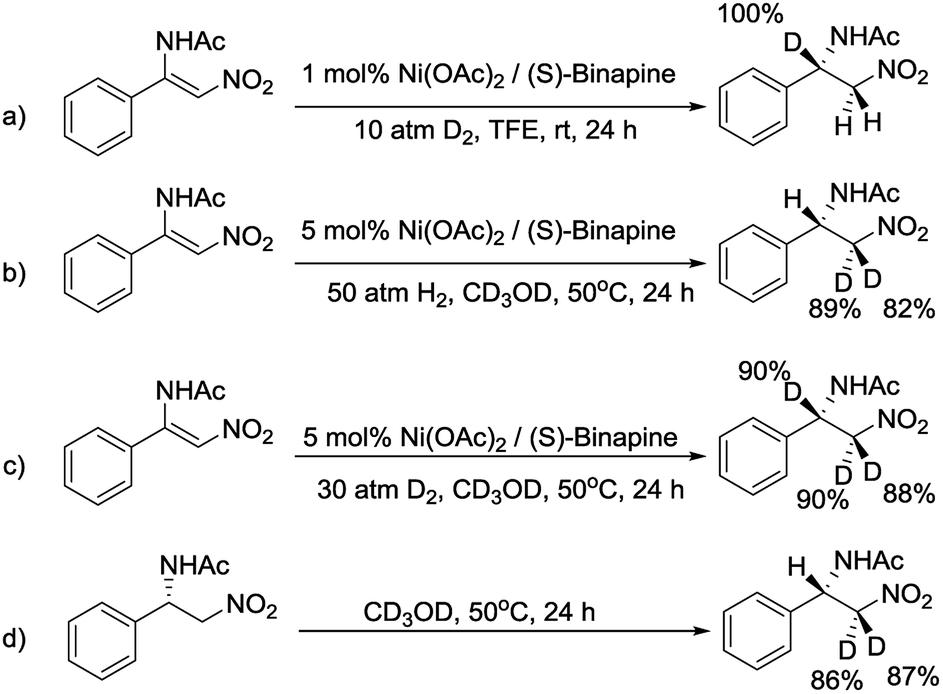 | ||
| Scheme 4 Deuterium labeling studies for the hydrogenation of 1a. | ||
To gain further insight into the reaction mechanism, DFT calculations using M06-L-D3 and B3LYP-D3 methods have been performed (Scheme 5).10,11 Our favored computed catalytic cycle starts with the acetate-assisted heterolytic cleavage of H2 to give a Ni(II)–H intermediate (III) with a barrier of ∼23.3–24.1 kcal mol−1 in solution. Then, ligand exchange with the nitroolefin substrate takes place, followed by the regio-determining 1,4-addition of the hydride to the β position of the nitroolefin to preferentially form a Ni(II) intermediate VIS1viaTSIIS1. Such a major pathway requires a lower barrier than the minor pathway by 4.0 kcal mol−1, and it forms the (S)-product. Subsequently, an AcOH molecule can re-coordinate to the Ni metal and undergo protonation to afford the desired (S)-product 2aviaTSIIIS1. These computational results are qualitatively consistent with the observed enantioselectivity and isotope labeling. 2a could undergo H exchange at the α position with TFE (or AcOH) to give the compound 2a′. This reaction mechanism for the Ni catalyst is different to that for the Rh-dihydride catalysts, in which alcohol solvents play a critical role in the catalytic system.12
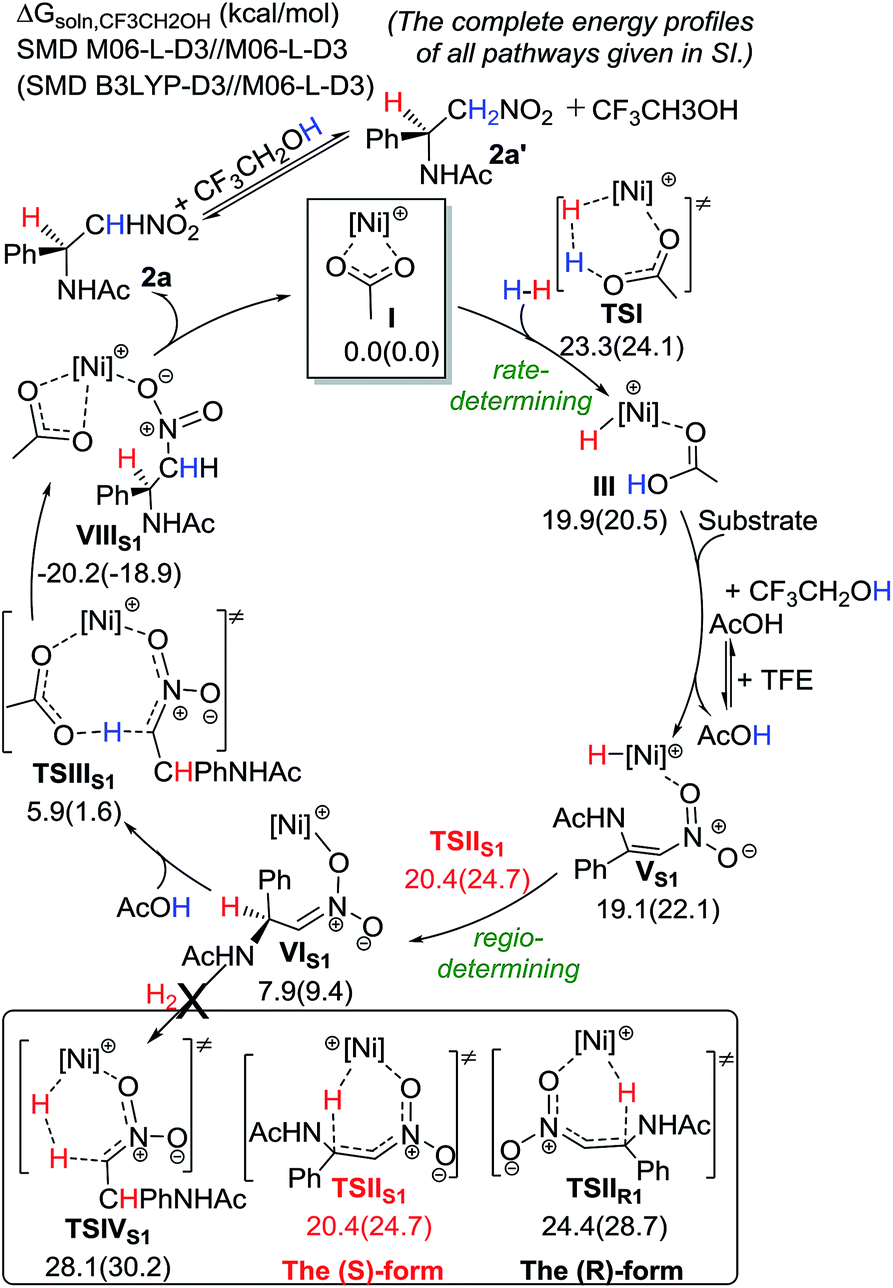 | ||
| Scheme 5 Computed energetics of the proposed catalytic cycle for the Ni-catalyzed hydrogenation of 1a with the (S)-binapine ligand. | ||
Conclusions
In conclusion, the Ni-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins using H2 as the reductant has been achieved, affording chiral β-amino nitroalkanes in high yields and with excellent enantioselectivities. Notably, this catalytic system was carried out under mild conditions and higher turnover numbers were achieved. Compared to noble metal catalysts, such as Rh species, the Ni catalyst is more attractive in the synthesis of chiral β-amino nitroalkanes. Moreover, deuterium labeling and computational studies were performed to reveal a possible mechanism for the Ni-catalyzed asymmetric hydrogenation. A further investigation on Ni-catalyzed asymmetric hydrogenation is ongoing in our laboratory.Notes and references
- (a) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS; (b) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (c) Q.-H. Fan, Y.-M. Li and A. S.-C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS PubMed; (d) W.-J. Tang and X.-M. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (e) I. D. Gridnev and T. Imamoto, Acc. Chem. Res., 2004, 37, 633 CrossRef CAS PubMed; (f) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed; (g) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357 CrossRef CAS PubMed; (h) H. Shimizu, I. Nagasski, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed; (i) W. Zhang, Y. Chi and X.-M. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed; (j) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (k) Y. Liu, Z. Wang and K.-L. Ding, Acta Chim. Sin., 2012, 70, 1464 CrossRef CAS; (l) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427 CrossRef CAS; (m) B.-G. Zhao, Z.-B. Han and K.-L. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 ( Angew. Chem. , 2013 , 125 , 4844 ) CrossRef CAS PubMed; (n) Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894 CrossRef CAS PubMed; (o) Z.-F. Zhang, N. A. Butt and W.-B. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS PubMed.
- (a) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (b) Y. Y. Li, S. L. Yu, W. Y. Shen and J. X. Gao, Acc. Chem. Res., 2015, 48, 2587 CrossRef CAS PubMed; (c) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS PubMed; (d) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687 CrossRef CAS PubMed.
- (a) S. L. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121 CrossRef CAS PubMed; (b) J. F. Sonnenberg, N. Coombs, P. A. Dube and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 5893 CrossRef CAS PubMed; (c) W. W. Zou, A. J. Lough, Y. F. Young and R. H. Morris, Science, 2013, 342, 1080 CrossRef PubMed; (d) P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367 CrossRef CAS PubMed; (e) Y. Y. Li, S. L. Yu, X. F. Wu, J. L. Xiao, W. Y. Shen, Z. R. Dong and J. X. Gao, J. Am. Chem. Soc., 2014, 136, 4031 CrossRef CAS PubMed.
- (a) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561 CrossRef CAS PubMed; (b) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076 CrossRef CAS PubMed; (c) J. H. Chen, C. H. Chen, C. L. Ji and Z. Lu, Org. Lett., 2016, 18, 1594 CrossRef CAS PubMed; (d) M. R. Friedfeld, M. Shevlin, G. W. Margulieux, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3314 CrossRef CAS PubMed.
- Y. Hamada, Y. Koseki, T. Fujii, T. Maeda, T. Hibino and K. Makino, Chem. Commun., 2008, 6206 RSC.
- (a) P. Yang, H. Y. Xu and J. R. Zhou, Angew. Chem., Int. Ed., 2014, 53, 12210 CrossRef CAS PubMed; (b) S. Y. Guo, P. Yang and J. R. Zhou, Chem. Commun., 2015, 51, 12115 RSC; (c) H. Y. Xu, P. Yang, P. Chuanprasit, H. Hirao and J. R. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5112 CrossRef CAS PubMed; (d) P. Yang, L. H. Lim, P. Chuanprasit, H. Hirao and J. R. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083 CrossRef CAS PubMed; (e) S. Y. Guo and J. R. Zhou, Org. Lett., 2016, 18, 5344 CrossRef CAS PubMed.
- M. Shevlin, M. R. Fried, H. Sheng, N. A. Pierson, J. M. Hoyt, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562 CrossRef CAS PubMed.
- (a) M. Zhou, D. J. Dong, B. L. Zhu, H. L. Geng, Y. Wang and X. M. Zhang, Org. Lett., 2013, 15, 5524 CrossRef CAS PubMed; (b) Q. Z. Yan, M. Liu, D. Y. Kong, G. F. Zi and G. H. Hou, Chem. Commun., 2014, 50, 12870 RSC; (c) P. Li, M. Zhou, Q. Y. Zhao, W. L. Wu, X. Q. Hu, X. Q. Dong and X. M. Zhang, Org. Lett., 2016, 18, 40 CrossRef CAS PubMed; (d) A. Ferraro, L. Bernardi and M. Fochi, Adv. Synth. Catal., 2016, 358, 1561 CrossRef CAS.
- Because CF3CD2OD was too expensive, we chose CD3OD as the solvent for conducting the isotopic labeling studies, and the reaction conditions were a little different to the optimized reaction conditions.
- (a) See computational details in the ESI.† Our calculated pathways in Scheme 6 are qualitatively supported using M06-L-D3 and B3LYP-D3 methods and are lower in energy than the other pathways (Tables S1–S4†). Recent computational studies on hydrogenation; (b) K. H. Hopmann, Organometallics, 2013, 32, 6388 CrossRef CAS; (c) G. Ganguly, T. Malakar and A. Paul, ACS Catal., 2015, 5, 2754 CrossRef CAS; (d) J. Yu, J. Long, Y. Yang, W. Wu, P. Xue, L. W. Chung, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 690 CrossRef CAS PubMed; (e) X. Ma and M. Lei, J. Org. Chem., 2017, 82, 2703 CrossRef CAS PubMed , and ref. 6c and d.
- Previous reports on the mechanism of asymmetric hydrogenation: (a) M. T. Ashby and J. Haipern, J. Am. Chem. Soc., 1991, 113, 589 CrossRef CAS; (b) T. J. Korstanje, J. I. Van der Vlugt, C. J. Elsevier and B. De Bruin, Science, 2015, 350, 298 CrossRef CAS PubMed.
- (a) A. S. C. Chan, J. J. Pluth and J. Halpern, J. Am. Chem. Soc., 1980, 102, 5952 CrossRef CAS; (b) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (c) R. Giernoth, H. Heinrich, N. J. Adams, R. J. Deeth, J. Bargon and J. M. Brown, J. Am. Chem. Soc., 2000, 122, 12381 CrossRef CAS; (d) M. Kitamura, M. Tsukamoto, Y. Bessho, M. Yoshimura, U. Kobs, M. Widhalm and R. Noyori, J. Am. Chem. Soc., 2002, 124, 6649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02669b |
| This journal is © The Royal Society of Chemistry 2017 |