
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly chemoselective ruthenium(II)-catalyzed direct arylation of cyclic and N,N-dialkyl benzamides with aryl silanes†
Pradeep
Nareddy
,
Frank
Jordan
and
Michal
Szostak
 *
*
Department of Chemistry, Rutgers University, 73 Warren Street, Newark, NJ 07102, USA. E-mail: michal.szostak@rutgers.edu
First published on 24th February 2017
Abstract
The ruthenium(II)-catalyzed oxidative cross-coupling of C(sp2)–H bonds with organosilanes has been accomplished for the first time. This novel protocol enlists challenging cyclic and N,N-dialkyl benzamides as weakly-coordinating substrates to achieve highly regioselective C(sp2)–H arylation as a proof-of-concept, taking advantage of the attractive features of organosilanes as coupling partners. This innovative method is characterized by very high chemoselectivity, installing halide functional groups (I, Br, Cl) that are incompatible with Ru(II)-carboxylate systems employing halides as cross-coupling partners, while obviating the need for sensitive organometallic reagents and cryogenic temperatures typical to the classic directed-ortho-metallation (DoM) techniques, employing benzamides to afford bioactive structural motifs.
Introduction
The Hiyama cross-coupling reaction is recognized as one of the most powerful methods for producing carbon–carbon bonds because of the wide substrate scope and significant attributes of organosilicon reagents, including low toxicity, high-functional group tolerance, and ready availability, which offer synthetic advantages over classic organoboranes.1–3 Recent advances in the direct cross-coupling of unactivated C–H bonds with organometallics have substantially increased the utility of C–H functionalization methods.4 These versatile processes circumvent the need for prefunctionalization, while providing multiple alternative sources of carbon nucleophiles under mild, functional group tolerant oxidative conditions.5Recently, ruthenium(II) catalysts have been identified as particularly promising alternatives to other metals using common directing groups to control the C–H activation site-selectivity in broadly useful synthetic molecules.6 The economic advantages of ruthenium precursors7 and the fact that ruthenium(II) catalysts do not require the formation of ate-type complexes that necessitate substrate deprotonation8 supports the invention of new catalytic reactivity.9,10 The key advances as reported by the groups of Oi/Inoue,9a Ackermann9b–d and Dixneuf9e,f described C–H arylation using aryl halides as precursors and strongly coordinating directing groups by carboxylate assistance. Unfortunately, despite significant advances, these methods suffer major limitations, such as (i) low tolerance for synthetically valuable aryl halide functional handles,9r and (ii) low reactivity for the direct arylation of weakly coordinating groups (Fig. 1A).
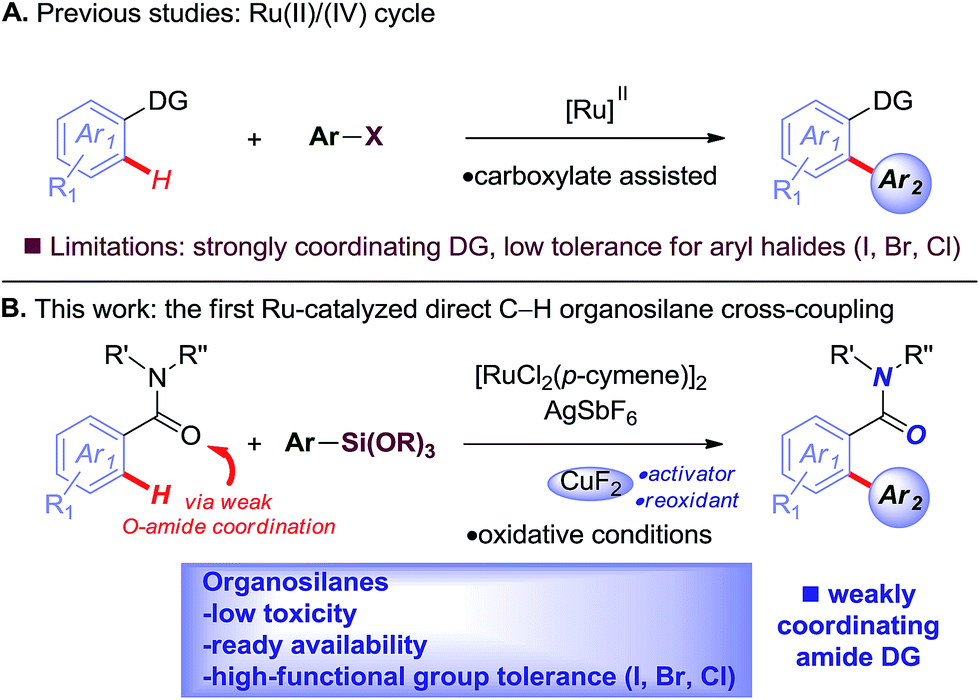 | ||
| Fig. 1 (A) Ruthenium-catalyzed arylation using aryl halides (previous studies), and (B) highly chemoselective Ru(II)-catalyzed C–H arylation using organosilanes (this study). | ||
Strategies for the direct cross-coupling with organosilanes via C–H functionalization are difficult to achieve due to low nucleophilicity of organosilicon reagents.1,2,4 As a consequence, only a few synthetically useful methods for direct C–H Hiyama cross-coupling have been reported.11 These methods involve Pd, Rh and Ni catalysis, while employing strongly-coordinating directing groups, substrates that are electronically-biased, or contain acidic protons to form ate-complexes after deprotonation.12 Unfortunately, despite potential advantages, ruthenium-catalyzed direct arylation using organosilicon reagents remains elusive. Moreover, direct C–H arylation with organosilanes using weakly coordinating neutral directing groups remains to be reported.11
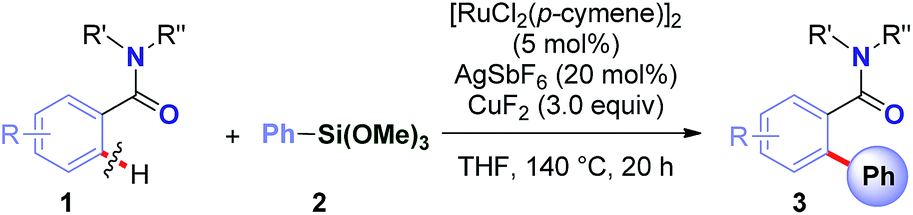
To realize the full potential of the ruthenium(II)-catalysis platform it is critical that new organometallic reagents become available as cross-coupling partners for direct C–H activation and that highly chemoselective processes tolerating a range of readily modifiable functional handles by weak neutral coordination are developed. Herein, we report the first example of ruthenium(II)-catalyzed direct cross-coupling of C(sp2)–H bonds with organosilanes (Fig. 1B). The manuscript describes the first example of transmetallation from silicon to ruthenium (including Ru(0)-catalysis)9i through C–H bond activation, and is the broadest in scope for Hiyama cross-coupling reactions using any metal for (sp2)-C–H activation reported to date.11 The reaction uses cyclic and N,N-dialkyl benzamides as weakly-coordinating substrates to achieve highly regioselective C–H arylation to give high value amide biaryls (Fig. 2).13 The feasibility of transmetallation from Si to Ru opens the door to a variety of C–H functionalization reactions by this activation manifold, enlisting a versatile, user-friendly and functional group tolerant cationic Ru(II)-catalysis platform. Notably, our protocol is characterized by a wide substrate scope tolerating an array of halide functional groups that are incompatible with Ru(II)-carboxylate-catalyzed arylations using aryl halides,6,9d while obviating the need for sensitive organometallic reagents and cryogenic temperatures typical to the classic directed-ortho-metallation (DoM) employing benzamides.14,15
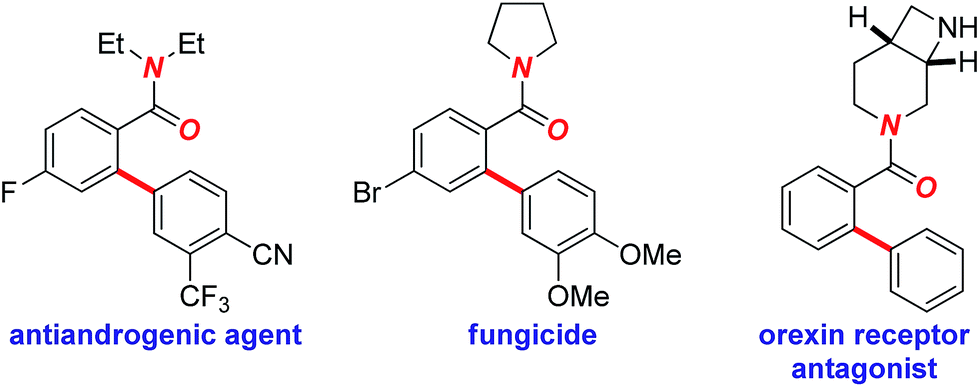 | ||
| Fig. 2 Examples of biologically-active biaryls bearing tertiary amides. | ||
Results and discussion
Recently, our group has pursued the development of a direct cross-coupling between cyclic benzamides and boronic acids via Ru(II)-catalyzed C(sp2)–H activation.16 As part of our program in functionalization of amides,17 we hypothesized that the same amide substrates could also be engaged in the markedly more challenging direct C(sp2)–H arylation using organosilanes. Over the past decade, methods for C(sp3)–H functionalization in activated N-acyl-pyrrolidine substrates have been identified.18 The ability to switch the reaction selectivity by judicious choice of the catalytic system represents an attractive strategy for organic synthesis.19We selected the reaction of pyrrolidine amide (1a) and trimethoxyphenylsilane (2a) as our model system. After extensive optimization (ESI†), we were delighted to identify a very simple reagent system consisting of [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%) and CuF2 (3.0 equiv.), affording the desired product in 87% isolated yield (>98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mono-/diarylation selectivity, >98% conversion) (Table 1, entry 17). Selected optimization results are shown in Table 1. Importantly, exclusive reaction selectivity for the arylation of aromatic C(sp2)–H bond in the presence of a weakened pyrrolidinyl C(sp3)–H bond was observed.18 Likewise, the reaction proceeded with full mono-/diarylation selectivity, which is often a complicating feature of other C–H manifolds, including Ru(II)-carboxylate catalysis.8,9
:2 mono-/diarylation selectivity, >98% conversion) (Table 1, entry 17). Selected optimization results are shown in Table 1. Importantly, exclusive reaction selectivity for the arylation of aromatic C(sp2)–H bond in the presence of a weakened pyrrolidinyl C(sp3)–H bond was observed.18 Likewise, the reaction proceeded with full mono-/diarylation selectivity, which is often a complicating feature of other C–H manifolds, including Ru(II)-carboxylate catalysis.8,9
|
|
||
|---|---|---|
| Entry | Conditions | Yieldb (%) |
| a Conditions: amide (R′R′′ = pyrrolidine, 1.0 equiv.), [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%), PhSi(OMe)3 (2.0 equiv.), CuF2 (3.0 equiv.), DCE, 140 °C (0.20 M), 20 h. b Determined by 1H NMR and/or GC. c Isolated yield. See ESI for full details. | ||
| 1 | 2a (1.2 equiv.), CuF2 (1.0 equiv.), DCE | <5 |
| 2 | 2a (1.2 equiv.), CuF2 (2.0 equiv.), DCE | 30 |
| 3 | 2a (1.2 equiv.), CuF2 (3.0 equiv.), DCE | 41 |
| 4 | 2a (1.5 equiv.), CuF2 (3.0 equiv.), DCE | 49 |
| 5 | 2a (1.5 equiv.), CuF2 (3.5 equiv.), DCE | 70 |
| 6 | 2a (2.0 equiv.), CuF2 (4.0 equiv.), DCE | 87 |
| 7 | 2a (2.0 equiv.), CuF2 (3.0 equiv.), DCE | 93 |
| 8 | 2a (2.5 equiv.), CuF2 (2.5 equiv.), DCE | 68 |
| 9 | 2a (2.5 equiv.), CuF2 (3.0 equiv.), DCE | 83 |
| 10 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), DCE | 90 |
| 11 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), dioxane | 60 |
| 12 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), CH3CN | 22 |
| 13 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), THF | 90 |
| 14 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), toluene | <5 |
| 15 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), DMF | 42 |
| 16 | 2a (2.5 equiv.), CuF2 (3.5 equiv.), DMAc | 10 |
| 17 | 2a (2.0 equiv.), CuF2 (3.0 equiv.), THF | >98 (87)c |
| 18 | 2a (2.5 equiv.), CuF2 (5.0 equiv.), DCE | 89 |

The central challenge in optimization experiments was to identify a catalytic system that would activate organosilane1,2 and reoxidize ruthenium catalyst to complete the cycle.4 Initial screenings using KF, CsF, AgF, TBAF, CuF2 in the presence of various established Ru(0) re-oxidants (Ag2O, Ag2CO3, Cu(OAc)2, Cu(OTf)2)6,9 led to low or no formation of the desired cross-coupling product 3a (see ESI†), presumably due to incompatibility of the activation/oxidation events. We hypothesized that identification of a single activator/oxidant would facilitate the catalyst turnover. The use of CuF2 delivered the desired product with excellent selectivity. The developed process represents (i) the first example of a Ru(II)-catalyzed direct C–H arylation using organosilanes, and (ii) the first example of a direct C–H Hiyama cross-coupling using neutral weakly-coordinating directing groups.6,11
With the optimized conditions in hand, the scope of this new Ru(II)-catalyzed direct arylation was explored (Table 2). The cross-coupling of electronically-diverse amide substrates, including electron-neutral (3a–b), electron-rich (3c) and electron-deficient (3d) substrates proceeded uneventfully. Importantly, our protocol exhibits an excellent chemoselectivity profile, accommodating an assembly of functional groups poised for further manipulation, such as strategically-positioned fluorides (para, ortho, 3e–f), chlorides (3g–h), bromide (3i), iodide (3j), nitro (3k) and ester groups (3l). Moreover, polyarenes (3m, >98:2 regioselectivity) and heterocycles are well-tolerated (3n–o). Notably, the remarkable functional group tolerance for halides (I, Br, Cl) affords handles for palladium-catalyzed cross-coupling post-functionalization. The results provide the first example of an aryl iodide tolerated in Hiyama C–H cross-coupling,11 clearly a testament to the high chemoselectivity profile of the cationic oxidative Ru(II) catalysis platform.6,9


The scope of this direct arylation with respect to organosilane component was evaluated (Table 3). Triethoxyphenylsilane was similarly effective as trimethoxyphenylsilane (Table 3, entry 1 vs. entry 2).12a The optimized conditions were found to be general using various arylsilanes, including electronically-diverse aryltrimethoxysilanes containing methyl (3p), methoxy (3q), trifluoromethyl (3r) groups on the ring. Moreover, the reaction tolerates sensitive halide functional handles such as fluoro (3s), chloro (3t) and bromo (3u), furnishing arylation products in good yields. Arylation with a meta-substituted organosilane proceeded uneventfully (3v). At present, ortho-substituted organosilanes are not compatible with the reaction conditions. It is particularly noteworthy that electrophilic chloro and bromo substituents are well-accommodated. The results provide the first example of a direct C(sp2)–H Hiyama cross-coupling tolerating a bromo-substituent on the silane,11 demonstrating the practicality of our Ru(II)-catalyzed protocol. The reaction scope using weakly-coordinating directing groups compares favourably with methods employing boronic acids by strong coordination,9q which is unusual for C–H functionalization processes involving organosilanes.11











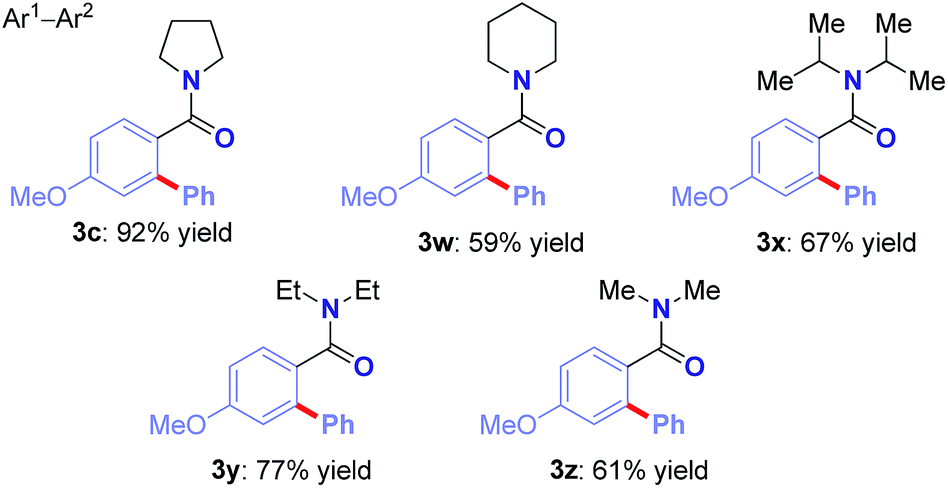
We were pleased to find that the reaction could be extended to a host of cyclic and alicyclic N,N-disubstituted benzamides to afford biaryl motifs with synthetically-valuable piperidinyl (3v), sterically-demanding N,N-(i-Pr)2 (3w), and simple, unhindered N,N-(Et)2 (3x), N,N-(Me)2 (3y) substitution (Table 4).13–15 It is worth noting that arylation of dimethylbenzamide under these conditions also proceeds in 60% yield (not shown). Taken together, the results demonstrate high levels of selectivity across sterically- and electronically-diverse amide substrates, thus opening the door for the development of future techniques for direct Ru(II)-catalyzed C–H Hiyama cross-coupling to afford N-containing substrates.13 The observed selectivity has the potential to supersede the classic DoM and Ru(II)/Ru(IV) techniques.6,14
Arylation selectivity in meta-substituted amides was evaluated (Table 5). The reaction proceeds with exquisite arylation regioselectivity using an electron-withdrawing CF3 (3aa) and a neutral Me group (3ab), while the use of a strongly electron-donating MeO substituent resulted in mixture of regioisomers (3ac). In all examples, only monoarylated products were formed (>98:2 selectivity).


A series of Hammett and Taft studies was conducted to shed light on the mechanism of this unique Ru(II)-catalyzed Hiyama C–H cross-coupling (Table 6, see ESI†). Moreover, deuterium incorporation studies revealed reversibility of the C–H activation step in both the amide substrate (34% H/D exchange) (Scheme 1A) and the ortho-arylated product (>98% H/D exchange) (Scheme 1B), providing evidence for facile cyclometallation. Overall, these mechanistic findings strongly support reversible C–H functionalization20 with coordination to amide oxygen to facilitate transmetallation.21
| Entry | Structure | Hammett ρ | Hammett ρ* |
|---|---|---|---|
| a Entries 1, 2 and 4: values determined in the reaction with phenyltrimethoxysilane (2a). Entry 3: values determined in the reaction with phenyl(pyrrolidin-1-yl)methanone (1a). See ESI for full details. | |||
| 1 |

|
−0.93 | −0.60 |
| 2 |

|
+1.28 | +0.78 |
| 3 |
|
+0.98 | +0.61 |
| Entry | Structure | Charton ν | Taft ES |
|---|---|---|---|
| 4 |

|
+1.93 | +0.98 |
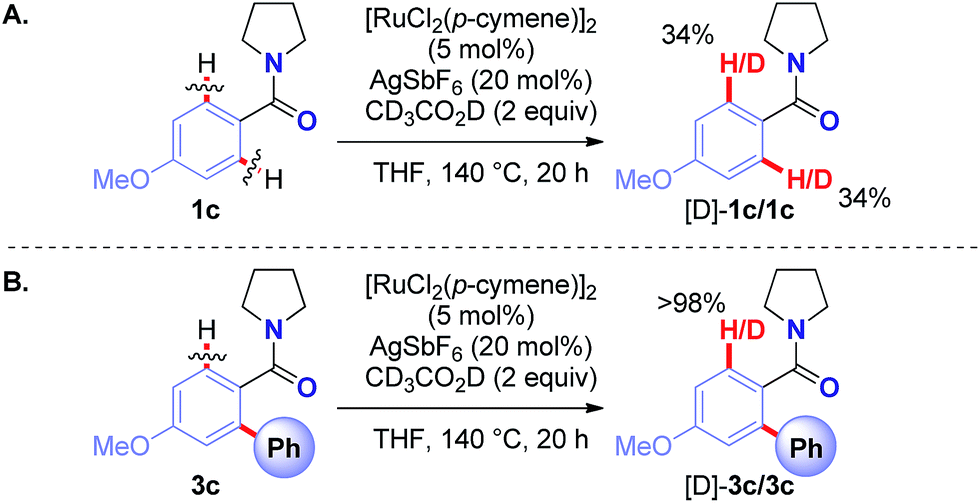 | ||
| Scheme 1 Deuterium incorporation studies. | ||
A mechanism for this C–H arylation could involve a rate-limiting transmetallation and/or reductive elimination preceded by C–H cleavage equilibrium as suggested by the H/D exchange and the observed electronic effects on the amide substrates and arylsilanes (Scheme 2). The mechanism could involve a cationic Ru as suggested by the Hammett plot with substituents para to amides and the use of AgSbF6 for cleaving C–H bond by an intermolecular fluoride base. The rate limiting transition state may involve a F-bridged [ArRu-F-SiAr]− anion for transmetallation. Studies to elucidate the mechanism are ongoing.
 | ||
| Scheme 2 Synthesis of benzylic biaryl amines. | ||
The present method provides rapid access to valuable biaryl amides.15 Moreover, the strategy could be employed to quickly access benzylic biaryl amines.22 For example, reduction of 3a with LiAlH4 smoothly gave the biologically-relevant pyrrolidine in 93% yield (Scheme 3). Biological activity of such benzylic biaryl amines is well-established.22b
 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have described the first example of ruthenium(II)-catalyzed cross-coupling of C(sp2)–H bonds with organosilanes. The reaction occurs with excellent arylation selectivity using versatile, user-friendly, and commercially available ruthenium(II) catalyst system. Low toxicity of arylsilanes, the functional group tolerance for aryl halides and the formation of high value biaryl products by weak coordination make this protocol very attractive. We envision that the new Ru(II)/CuF2 catalyst system in which CuF2 acts as a dual silane activator and Ru re-oxidant will find broad use in organic synthesis. The Ru(II)-based strategy could significantly expand the use of organosilicon compounds in C–H functionalization.Acknowledgements
Financial support was provided by Rutgers University. The 500 MHz spectrometer was supported by the NSF-MRI grant (CHE-1229030). We thank SEED Grant (Rutgers University) to the Center for Sustainable Synthesis for partial support.Notes and references
-
(a)
A. de Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley, New York, 2014 Search PubMed
; (b) G. Molander, J. P. Wolfe and M. Larhed, Science of Synthesis: Cross-Coupling and Heck-Type Reactions, Thieme, Stuttgart, 2013 Search PubMed
- Reviews:
(a) T. Hiyama and Y. Hatanaka, Pure Appl. Chem., 1994, 66, 1471 CrossRef CAS
- Reports on mutagenicity of boronic acids:
(a) M. M. Hansen, R. A. Jolly and R. J. Linder, Org. Process Res. Dev., 2015, 19, 1507 CrossRef CAS
- A comprehensive review on C–H arylation with organometallics: R. Giri, S. Thapa and A. Kafle, Adv. Synth. Catal., 2014, 356, 1395 CrossRef CAS
- Select reviews on C–H activation:
(a)
J. Q. Yu, Science of Synthesis: Catalytic Transformations via C–H Activation, Thieme, Stuttgart, 2015 Search PubMed
- Reviews on Ru(II) catalysis:
(a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed
- In January 2017, the average prices of Rh, Ir, Pd and Ru were 795, 690, 761, and 42 US$/oz, https://taxfreegold.co.uk, accessed 10/1/2017.
- K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed
- Select examples of Ru-catalyzed arylation:
(a) S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579 CrossRef CAS PubMed
-
(a) M. K. Singh, H. K. Akula, S. Satishkumar, L. Stahl and M. K. Lakshman, ACS Catal., 2016, 6, 1921 CrossRef CAS PubMed
- Pd: acetanilides:
(a) S. Yang, B. Li, X. Wan and Z. J. Shi, J. Am. Chem. Soc., 2007, 129, 6066 CrossRef CAS PubMed
- Direct C–H Hiyama cross-coupling using electronically-activated or acidic substrates: Ni:
(a) H. Hachiya, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2010, 49, 2202 CrossRef CAS PubMed
-
(a) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed
-
(a) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS
-
A. J. Burke and C. S. Marques, Modern Arylation Methods, Wiley-VCH, Weinheim, 2015 Search PubMed
- P. Nareddy, F. Jordan, S. E. Brenner-Moyer and M. Szostak, ACS Catal., 2016, 6, 4755 CrossRef CAS
-
(a) G. Meng and M. Szostak, Org. Lett., 2015, 17, 4364 CrossRef CAS PubMed
-
(a) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed
-
(a) J. Mahatthananchai, A. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef CAS PubMed
-
(a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed
- D. V. Partyka, Chem. Rev., 2011, 111, 1529 CrossRef CAS PubMed
-
(a) Functional group transformations of aromatic amides are well-established: T. Bailey, P. R. Blakemore, V. Cesare, J. K. Cha and G. Cook, Science of Synthesis, Thieme, 2014, vol. 21 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c7sc00156h |
| This journal is © The Royal Society of Chemistry 2017 |