
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrosion properties of steel in 1-butyl-3-methylimidazolium hydrogen sulfate ionic liquid systems for desulfurization application†
Qian Zeng,
Jinwei Zhang,
Hongye Cheng *,
Lifang Chen and
Zhiwen Qi
*,
Lifang Chen and
Zhiwen Qi
Max Planck Partner Group at the State Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: hycheng@ecust.edu.cn
First published on 17th October 2017
Abstract
Ionic liquids (IL) become more promising in industrial applications, especially in desulfurization processes. It is of great importance to investigate their corrosivity to steel and the effect of steel corrosion on desulfurization performance. In this work, the corrosion behavior of two typical metals, namely mild steel and stainless steel, in 1-butyl-3-methylimidazolium hydrogen sulfate ([BMIM]HSO4) IL systems for desulfurization application were investigated by weight loss and surface analyses. The model oil can inhibit the IL corrosivity, while the presence of water or H2O2 enhances the corrosion degree. The bonding information of [BMIM]HSO4 adsorbed on a steel surface was confirmed using ATR-FTIR. It was found that the IL film is mainly adsorbed on the metal surface by the interaction of the imidazolium group of the cation and HSO4− of the anion with the steel surface. From the ICP-MS and Raman analyses, the primary component of corrosion products is iron sulfate (ferric sulfate for mild steel and ferrous sulfate for stainless steel). Localized corrosion (pitting and/or intergranular corrosion) appears on steel surface from SEM observation, which could be a serious safety risk to the [BMIM]HSO4-based desulfurization systems for industrial application. The corrosion has a side effect on the desulfurization performance of [BMIM]HSO4.
1 Introduction
Ionic liquids (ILs), also known as room-temperature molten salts, have received extensive interest due to their unique properties and potential for diverse applications. Compared with traditional solvents, ILs can offer remarkable advantages in intrinsic ionic conductivity and having a wide electrochemical window, high thermal stability, non-flammability, and negligible vapor pressure. Since their physical and chemical properties can be adjusted by combination of different cations and anions, ILs are considered as a task-specific liquid and have potential in a number of fields such as separation, organic synthesis, catalysis, nanotechnology, electrochemistry, and so on.1–3In recent years, ILs have attracted interest in extractive desulfurization (EDS) and oxidative desulfurization (ODS) processes, as more stringent environmental regulations have been implemented all over the world to restrict the sulfur content in fuels (<10 ppm).4,5 EDS employs ILs as solvent to extract sulfur compounds from fuels,6 while in ODS, sulfur compounds are oxidized to their corresponding sulfoxides and sulfones and then extracted by ILs.7–9 Compared to EDS, ODS is more efficient since sulfoxides or sulfones are more easily extracted by ILs. Owing to the advantages of low cost, easy preparation, and high desulfurization efficiency, 1-butyl-3-methylimidazolium hydrogen sulfate ([BMIM]HSO4) IL is screened as an ideal candidate for industrial desulfurization application and has been investigated in previous studies.10–12 [BMIM]HSO4 has strong acidity due to its Brönsted acid site, which is a proton-donor and can be used as the catalyst to facilitate the oxidization of sulfur compounds in ODS.13 H2O2 is the most commonly used oxidant in ODS, because of its environmental friendliness and affordable cost.14
When applying ILs in industrial processes, they are in contact with metals and alloys in pipes and units of reactor, separation and storage tank. The corrosivity of ILs to metallic material is essential, which may cause problems of leakage, abnormal plant shutdown, and environmental pollution. Therefore, industrial application of ILs requires a better understanding of their corrosivity to metals.
The corrosion characteristics and mechanism of metallic materials in ILs are quite different from those in conventional environments.15 Uerdingen et al. reported the corrosion behavior of metals in various ILs and investigated the effect of water content on corrosion.16 It proved that stainless steel exhibited the best corrosion resistance in all ILs and the presence of water can significantly enhance the corrosivity of IL media. Molchan et al. studied the corrosion behavior of mild steel in ILs for CO2 capture application.17 The morphological changes were monitored by scanning electron microscopy (SEM), and the corrosion products including ferrites, sulfates, carbonates and oxides were detected by micro-Raman analysis. Diamanti et al. investigated the corrosion behavior of steel alloys in several imidazolium-based ILs.18 The effect of the cation and anion of ILs and the water content were studied. Since the corrosion phenomena vary with the type of ILs and water content in IL solutions,19,20 the investigation of corrosion characteristics in certain IL systems is strongly needed.
Up to now, there is no report of the corrosion behavior of steel in [BMIM]HSO4 IL for EDS and ODS applications. [BMIM]HSO4 has strong acidity, which may cause severe corrosion problems. However, because of the good corrosion inhibition of imidazolium group,21,22 the corrosion behavior of [BMIM]HSO4 should be different from other acidic environments. Moreover, the existence of water and H2O2 may facilitate the corrosion of steel in [BMIM]HSO4.23,24
In the present work, the corrosion behavior of steels including mild steel and stainless steel in [BMIM]HSO4 and [BMIM]HSO4-based desulfurization systems were investigated in details. In desulfurization experiment, model oil is always used to investigate the desulfurization mechanism,25 which is prepared by dissolving a certain amount of sulfur compounds in n-heptane or n-octane.26–28 Meanwhile, water and H2O2 are also the common components in desulfurization systems.9,29 A certain amount of water within a range (<10 wt%) has a promoted effect on desulfurization efficiency using ILs,30 while H2O2 is employed as oxidant in ODS. Therefore, the effect of the addition of model oil, water, and H2O2 in [BMIM]HSO4 on the corrosion behavior of steels were studied and a possible mechanism was proposed in this work. Finally, the side effect of corrosion on [BMIM]HSO4-based desulfurization process was investigated.
2 Experimental
2.1 Materials
Two commonly used metals, i.e. Q235 steel and 316L stainless steel (SS316L) obtained from the Biosteel Research Institute were studied. Q235 is a kind of mild steel (MS) with a ferrite-pearlite structure and with a chemical composition (in wt%) of 0.18C, 0.60Mn, 0.22Si, 0.016P, 0.02S, and Fe balance. SS316L is a kind of austenitic stainless steel, containing (in wt%) 0.018C, 1.11Mn, 0.026P, 0.001S, 0.53Si, 17.4Cr, 11.1Ni, 2.16Mo, and Fe balance. Each specimen with dimensions of 10 mm × 10 mm × 3 mm was abraded consecutively with 400, 600, 800, 1000, and 1200 grit silicon carbide abrasive paper, then washed with deionized water and degreased in acetone. The surface of each specimen is 3.2 cm2.[BMIM]HSO4 (the chemical structure shown in Fig. 1) with purity higher than 99 wt% (water content 0.7 wt%) was purchased from Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. Hydrogen peroxide (30 wt%) and n-heptane (98.5 wt%) were purchased from Aladdin Chemical Co., Ltd. Dibenzothiophene (DBT) (99 wt%) was purchased from Adamas Reagent Company Co., Ltd (Shanghai, China). The chemicals were used as received.
 | ||
| Fig. 1 Molecular structure of [BMIM]HSO4. | ||
2.2 Methods
n-Heptane is taken as model oil (MO) component with DBT as the representative sulfur compound. Model oil was prepared by adding DBT to n-heptane, with the sulfur content of 500 ppm. To study the corrosion properties of steel in pure [BMIM]HSO4 as well as in EDS and ODS processes, four IL solutions were used for immersion experiments as the following: (i) IL, (ii) IL + MO, (iii) IL + MO + H2O, and (iv) IL + MO + H2O2. The amount of IL for each solution was 60 g, while the amount of MO in the solutions (ii), (iii), and (iv) was 300 g. 6 g H2O and 6 g H2O2 were added into the solutions (iii) and (iv), respectively.In each immersion test, three specimens were fixed on the polytetrafluoroethylene holder in a conical flask, in order to suspend and immerse them in the IL media and expose their surfaces to the media as much as possible. The area of the holder being in contact with specimen is rather small that it can be neglected for the calculation of corrosion rate. In desulfurization process, the mixture is a biphasic system, because IL is not soluble in the model oil. Therefore, in immersion tests, a shaking bed is used to adequately mix up the IL phase and oil phase, making the specimen corroded just like in the real desulfurization process. The flask was sealed and fixed in the shaking bed with a frequency of 90 rpm and temperature of 45 °C. After 14 day immersion experiment, the specimens were taken from the solution for further Fourier transform infrared spectroscopy (FTIR) and Raman analyses. Then, according to the ASTM G31 norm,31 the specimens were scrubbed with a bristle brush to remove corrosion products, rinsed in deionized water to remove the IL solution, washed with acetone to clean the model oil, dried in air, and weighed accurately for the weight loss measurement and SEM analyses.31–35
After corrosion of mild steel and stainless steel for 14 days, the types and the contents of metal ions in all the four IL systems were measured by Inductively Coupled Plasma Mass Spectrometry (PerkinElmer) and spectrophotometer (Sigma-Aldrich).
The acidity of different IL systems before and after corrosion of mild steel and stainless steel for 14 days was evaluated from the determination of Hammett acidity function. The ILs from different systems and the indicator (4-nitroaniline, pK(I)aq = 0.99) were dissolved in the dimethyl sulfoxide (DMSO) at a concentration of 5 × 10−5 mol L−1. The solutions were shaken vigorously and left to stand for 6 h. Then, their spectra were recorded on a P-General UV-visible spectrophotometer.
A set of 33 day immersion tests were conducted to investigate the corrosion kinetics of stainless steel in [BMIM]HSO4 IL solutions. The specimens were periodically removed from IL solutions, cleaned according to ASTM G31 norm, and then weighed accurately.31
FTIR spectra were recorded using a Perkin Elmer Spectrum 100 spectrometer in a reflection mode with an Attenuated Total Reflectance (ATR) attachment utilizing a diamond prism. ATR-FTIR spectroscopy is an acknowledged technique which has been widely used to analyze the nature of bonding for organic complexes adsorbed on the metal surface.36,37 The spectral resolution was 1 cm−1 and the wavenumber range of 650–4000 cm−1 was utilized to collect the IR spectra. The recorded spectra were compared with those of the substances selected from the Sadtler Handbook of Infrared Spectra.38 The corrosion products were detected by micro-Raman (HORIBA Jobin Yvon LabRAM HR800 system). Laser light with the wavelength of 514.5 nm was used. A long working distance lens with the magnification of 50× was applied to detect the scattered light and focus the laser spot on specimen surface. The surface morphology of the specimens before and after immersion tests were examined by SEM (Hitachi S3400N).
In order to have an intuitive understanding of the corrosion behavior of steel in IL solutions, the weight loss measurements for mild steel and stainless steel in four [BMIM]HSO4-based systems were adopted. A modified experimental plan was performed based on ASTM G31-72.31 To get high reliability and reproducibility, the processed specimens were immersed in IL solutions in triplicate, and the weight losses of three specimens were recorded.
The weight loss rates were calculated from the following equation:
| ν = (W0 − W1)/St | (1) |
With the weight loss rate, the average corrosion rate on the basis of equivalent thickness loss can be calculated as reported in the literature:39,40
| B = 8.76ν/ρ | (2) |
After 14 day immersion tests, the ILs of IL + MO and IL + MO + H2O2 systems in which stainless steel specimens were corroded were collected and processed for EDS and ODS experiments, respectively. The specimens were taken away from the IL solution after immersion period. The IL system was still a biphasic system. After settling the system for 2 h, the mixture will become two layers, namely, the IL phase and the oil phase. The IL phase along with H2O or H2O2 was the lower layer. The model oil can be easily separated from the system by decantation. Then, the IL phase was distilled in a rotary evaporation at 80 °C for 10 h to remove the residual water, hydrogen peroxide and model oil. In EDS and ODS experiments, the mass ratio of the IL to the model oil is 0.2.
3 Results and discussion
3.1 Weight loss measurement
Fig. 2 shows the changes in color of [BMIM]HSO4 IL before and after corrosion of mild steel and stainless steel specimens. The color of pristine [BMIM]HSO4 is light yellow, and after immersion tests the IL solutions become darker. The change in color varies with different corrosion systems. According to the colorimetric properties of iron ion,41,42 the darker of the color implies the higher concentration of the metallic iron and consequently the stronger corrosivity of IL solution. Therefore, by comparing the color in different IL solutions, it can be preliminarily deduced that the corrosivity of four IL solutions is in an order of IL + MO + H2O2 > IL + MO + H2O > IL > IL + MO. | ||
| Fig. 2 Appearance of (0) pristine [BMIM]HSO4 and (1) IL, (2) IL + MO, (3) IL + MO + H2O, (4) IL + MO + H2O2 after immersion with (a) Q235, (b) SS316L at 45 °C for 14 days. (For color version of this figure, the reader is referred to the online version of this article). | ||
There is an interesting phenomenon that the color of ILs appears to be yellow brown after corrosion of mild steel, while it is green in general after corrosion of stainless steel. It suggests that the primary form of the corrosion product in IL solutions is the ferric ion (Fe3+) for mild steel and the ferrous ion (Fe2+) for stainless steel, respectively. This hypothesis is confirmed by the Inductively Coupled Plasma Mass Spectrometry (ICP-MS) and spectrophotometer analyses. The type and the content of metal ions in different IL systems after corrosion of mild steel and stainless steel are listed in Table 1, which are consistent with the changes in color of different IL systems.
| System | Fe2+ (wt%) | Fe3+ (wt%) | Cr3+ (wt%) | Ni2+ (wt%) | |
|---|---|---|---|---|---|
| Mild steel | IL | 0.042 | 0.323 | — | — |
| IL + MO | 0.074 | 0.237 | — | — | |
| IL + MO + H2O | 0.108 | 0.557 | — | — | |
| IL + MO + H2O2 | 0.159 | 0.624 | — | — | |
| Stainless steel | IL | 0.021 | 0.005 | 0.0025 | 0.0012 |
| IL + MO | 0.016 | 0.003 | 0.0022 | 0.0012 | |
| IL + MO + H2O | 0.078 | 0.013 | 0.0144 | 0.0076 | |
| IL + MO + H2O2 | 0.133 | 0.028 | 0.0241 | 0.0129 | |
The difference in the type of metal ions can be ascribed to the kinetics of ferrous oxidation that lower acidity is favorable for the conversion of Fe2+ to Fe3+.43 As the mild steel experienced much more severe corrosion in IL solutions than the stainless steel, the hydrogen ion consumption should be greater and thus the acidity should be weaker in these systems. The acidity of different IL systems before and after corrosion of mild steel and stainless steel are evaluated from the determination of the Hammett acidity function (H0), calculated with the formula:44
| H0 = pK(I)aq + log([I]/[IH+]) | (3) |
The Brönsted acidity results are illustrated in Table 2. The IL systems after corrosion of mild steel indeed have a weaker acidity than those after corrosion of stainless steel. As a consequence, the oxidization rate is greatly enhanced and finally Fe2+ undergoes oxidation to Fe3+ in [BMIM]HSO4 solutions immersed with mild steel.
| Systems | Amax | [I] (%) | [IH+] (%) | H0 | |
|---|---|---|---|---|---|
| Blank | 1.305 | 100 | 0 | — | |
| Before corrosion | IL | 0.402 | 30.1 | 69.9 | 0.624 |
| IL + MO | 0.404 | 31.0 | 69.0 | 0.643 | |
| IL + MO + H2O | 0.362 | 27.7 | 72.3 | 0.574 | |
| IL + MO + H2O2 | 0.366 | 28.0 | 72.0 | 0.580 | |
| After corrosion of stainless steel | IL | 0.780 | 59.8 | 40.2 | 1.162 |
| IL + MO | 0.776 | 59.5 | 40.5 | 1.157 | |
| IL + MO + H2O | 0.751 | 57.5 | 42.5 | 1.121 | |
| IL + MO + H2O2 | 0.758 | 58.1 | 41.9 | 1.132 | |
| After corrosion of mild steel | IL | 1.046 | 80.2 | 19.8 | 1.598 |
| IL + MO | 0.997 | 76.4 | 23.6 | 1.500 | |
| IL + MO + H2O | 0.961 | 73.6 | 26.4 | 1.435 | |
| IL + MO + H2O2 | 0.969 | 74.3 | 25.7 | 1.451 | |
Fig. 3 shows the corrosion depth rates of mild steel and stainless steel specimens in different IL solutions. Primarily, it can be observed that stainless steel has a better corrosion resistance in all IL solutions than mild steel. In pure [BMIM]HSO4 IL, mild steel is attacked with a corrosion depth rate of 1.25 mm y−1, and with addition of model oil, the corrosion depth rate exhibits a slight reduction to 1.15 mm y−1. Mild steel suffers severe corrosion in aqueous IL solution with a corrosion depth rate of 1.64 mm y−1, and in the presence of H2O2, the corrosion depth rate increases up to 1.78 mm y−1. On the other hand, the stainless steel has excellent performance in water free IL solutions, i.e., in IL and IL + MO systems, where the corrosion depth rates are only 0.06 mm y−1 and 0.03 mm y−1, respectively, below the target corrosion depth rate of max 0.1 mm y−1 for the stainless steel.16 The presence of water and H2O2 significantly enhances the corrosivity of IL systems that the corrosion rate of stainless steel increases to about 0.14 mm y−1 and 0.44 mm y−1, respectively, well above the 0.1 mm y−1 target line.
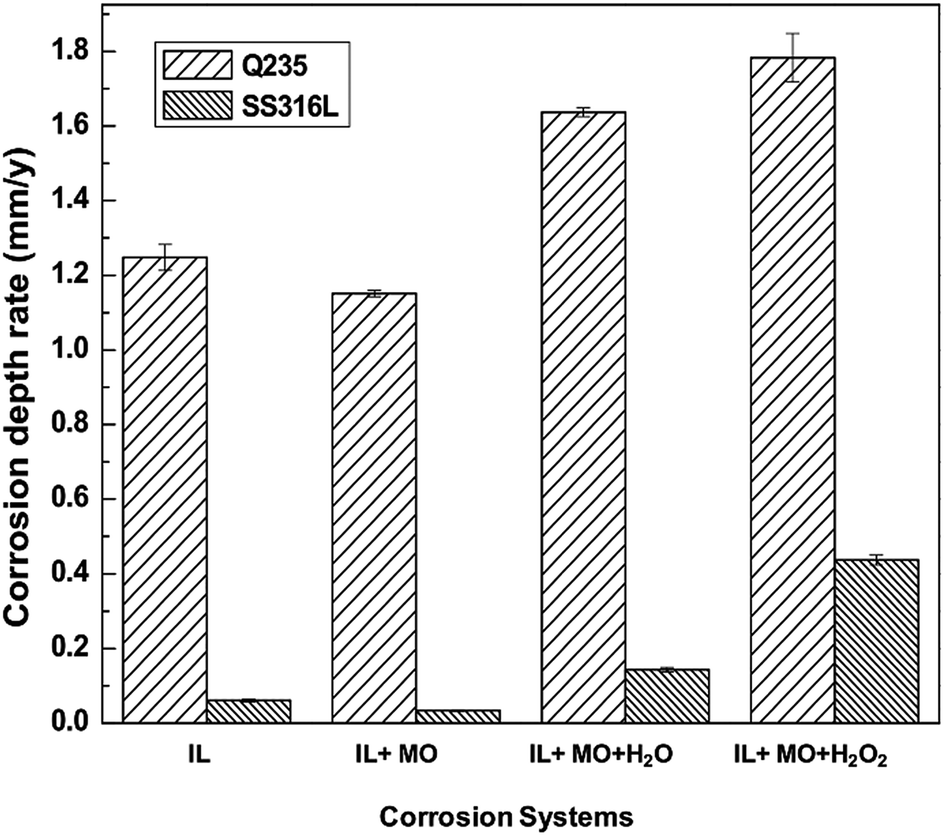 | ||
| Fig. 3 Corrosion rate of steel specimens immersed in different [BMIM]HSO4 systems at 45 °C for 14 days. | ||
The addition of 10% of water can significantly increase the corrosivity of [BMIM]HSO4. However, the corrosion depth rates of mild steel and stainless steel in MO + H2O systems are only 0.12 mm y−1 and 0.006 mm y−1, respectively, which are far less than that in IL + MO + H2O systems. Uerdingen et al. reported that with the presence of 10% water, carbon steel was severely corroded in IL with methyl sulfate anion (2.5 mm y−1).16 This was explained by the formation of sulfuric acid via hydrolysis of the methyl sulfate. In this work, the addition of 10% of water leads to a stronger acidity in IL + MO + H2O system than nonaqueous IL systems (see Table 2), which increases the corrosion depth rate of steel. Similarly, hydrogen peroxide is not corrosive to the steel as well that the corrosion depth rates of mild steel and stainless steel in MO + H2O2 systems are only 0.007 mm y−1 and 0.003 mm y−1, respectively.
For [BMIM]HSO4, the imidazolium cation plays as an inhibitor for steel while the anion of HSO4− is corrosive to the steel. It was reported that ILs with imidazolium cation have been employed as inhibitors for steel in H2SO4 solution.45 For example, the addition of 1-octyl-3-methylimidazolium bromide and 1-allyl-3-octylimidazolium bromide ionic liquids in 0.5 M H2SO4 solutions can significantly decrease the corrosion rate. The corrosion depth rate of 316 stainless steel in 98% H2SO4 is close to 0.13 mm y−1,46 which is higher than the corrosion depth rate of stainless steel in pure [BMIM]HSO4 system, indicating the corrosion inhibition property of imidazolium cation of [BMIM]HSO4. On the other hand, the corrosion property of ionic liquid with the anion of HSO4− is related to cation structure. The imidazolium cation is a better corrosion inhibitor than quaternary ammonium moiety.16 The corrosion depth rate of 316L stainless steel in 1-methylimidazole hydrogensulfate ([HMIM]HSO4) was 0.011 mm y−1, similar to the corrosion depth rate in [BMIM]HSO4 (0.06 mm y−1). But the corrosion depth rate of stainless steel in N-triethylammonium sulfate ([Et3NH]HSO4) was 1.3 mm y−1, which is more corrosive than imidazolium-based hydrogensulfate ILs.47
The weight loss of SS316L specimens in different IL solutions are shown as a function of time in Fig. 4. A continuous increase in weight loss at an approximately linear rate take places for SS316L in each IL systems. The weight loss rate constants and correlation coefficients are shown in Table S1 (see ESI†). The correlation coefficients (R2) of regression analyses approach to 1, indicating that the corrosion kinetics is linear as the function of time for the weight loss of SS316L in four IL solutions.
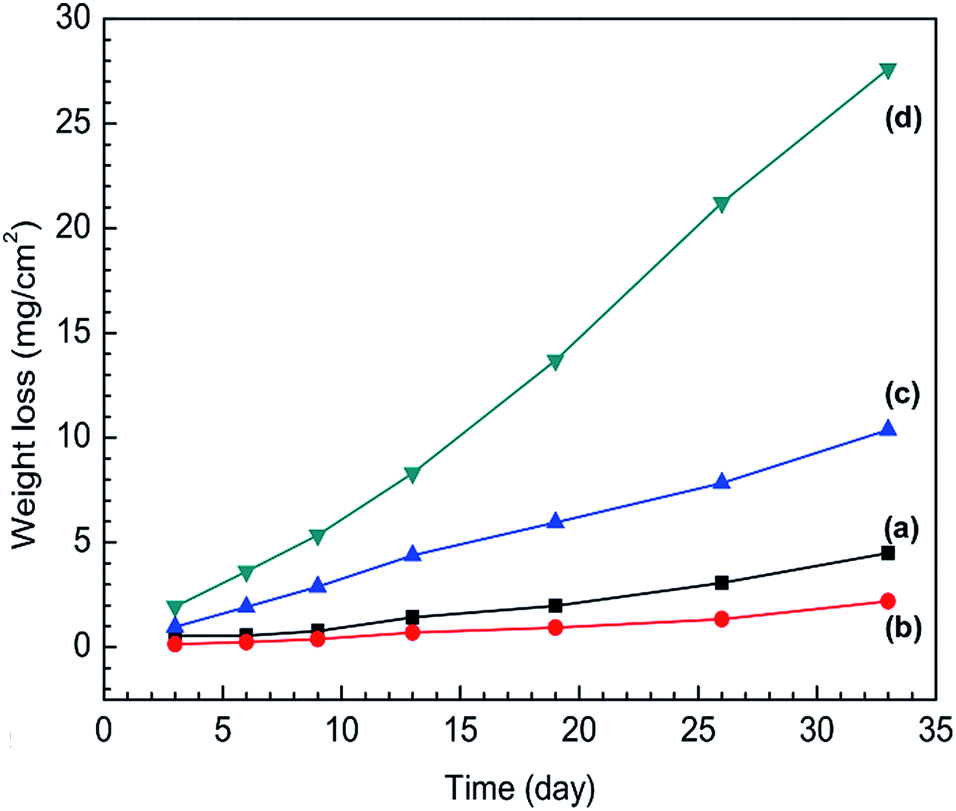 | ||
| Fig. 4 Weight loss evolution of stainless steel specimens in (a) IL, (b) IL + MO, (c) IL + MO + H2O, (d) IL + MO + H2O2 as a function of time. | ||
To explain the experimental results, the corrosion mechanism of steel in various [BMIM]HSO4-based desulfurization systems is proposed. In acid media, metallic materials could be corroded via two distinct reactions, namely, hydrogen evolution corrosion and oxygen reduction corrosion.
As [BMIM]HSO4 IL is a proton-conducting nonaqueous electrolyte in reaction,48 the cathodic and anodic reactions are written in the same form for the nonaqueous and aqueous IL solutions. The hydrogen evolution corrosive process can be expressed as:
Anodic reaction (iron dissolution):
| Fe → Fe2+ + 2e− | (4) |
For stainless steel, in addition to the dissolution of Fe, the anodic dissolution of Cr and Ni can also occur via the following reactions:49
| Cr → Cr3+ + 3e− | (5) |
| Ni → Ni2+ + 2e− | (6) |
The mechanism is in accordance with the ICP results (Table 1).
Cathodic reaction (reduction of hydrogen ion):
| 2H+ + 2e− → H2 | (7) |
The presence of water can decrease the cation–anion interaction and increase the number of H-bond between anion and water,50 which could lead to an increase of the number of H+ in the systems. This hypothesis has been demonstrated by the acidity measurements that the IL + MO + H2O system has a higher acidity than the nonaqueous IL systems, as illustrated in Table 2. As a result, the corrosion depth rate is enhanced and the steel suffers more severe attack in the aqueous IL systems (IL + MO + H2O and IL + MO + H2O2 systems) than in the nonaqueous systems (IL and IL + MO systems). In the nonaqueous systems, the corrosion depth rate is inhibited with the addition of MO, due to the DBT in MO working as an inhibitor via adsorption on steel surface. The mechanism of DBT adsorption on the steel surface is similar to the imidazolium ring of [BMIM]HSO4 that the interaction takes place through heteroatoms and aromatic rings onto the active site of steel surface.21
At the same time, the steel can be corroded through oxygen reduction corrosion as well.51
Cathodic reaction:
| O2 + 4e− + 4H+ → 2H2O | (8) |
However, this cathodic reaction is limited by the concentration of O2 in IL systems.52 This cathodic reaction unlikely happens in IL and IL + MO systems. In the presence of H2O2, the cathodic reaction can also take place via the following equation:
Cathodic reaction:
| H2O2 + 2e− + 2H+ → 2H2O | (9) |
As H2O2 is a more powerful oxidant, and the concentration of H2O2 is relatively higher in IL + MO + H2O2 system, which results in a faster cathodic reaction.
In addition, H2O2 can be easily decomposed into O2 and H2O, enhancing the concentration of O2 in the IL system:
| 2H2O2 → O2 + 2H2O | (10) |
As a result, the steel suffers the most severe corrosion in the IL systems with the addition of H2O2.
3.2 ATR-FTIR analysis
The bonding information of pristine [BMIM]HSO4 IL as well as the surface films on the steel specimens after immersion in different IL solutions were investigated by ATR-FTIR analysis, as illustrated in Fig. 5. The spectra of the surface films of the stainless steel immersed in corresponding IL solutions are almost the same as those of mild steel, suggesting that the adsorption of IL molecules on the stainless steel surface is similar to that on the mild steel, as illustrated in Fig. S1 (see ESI†).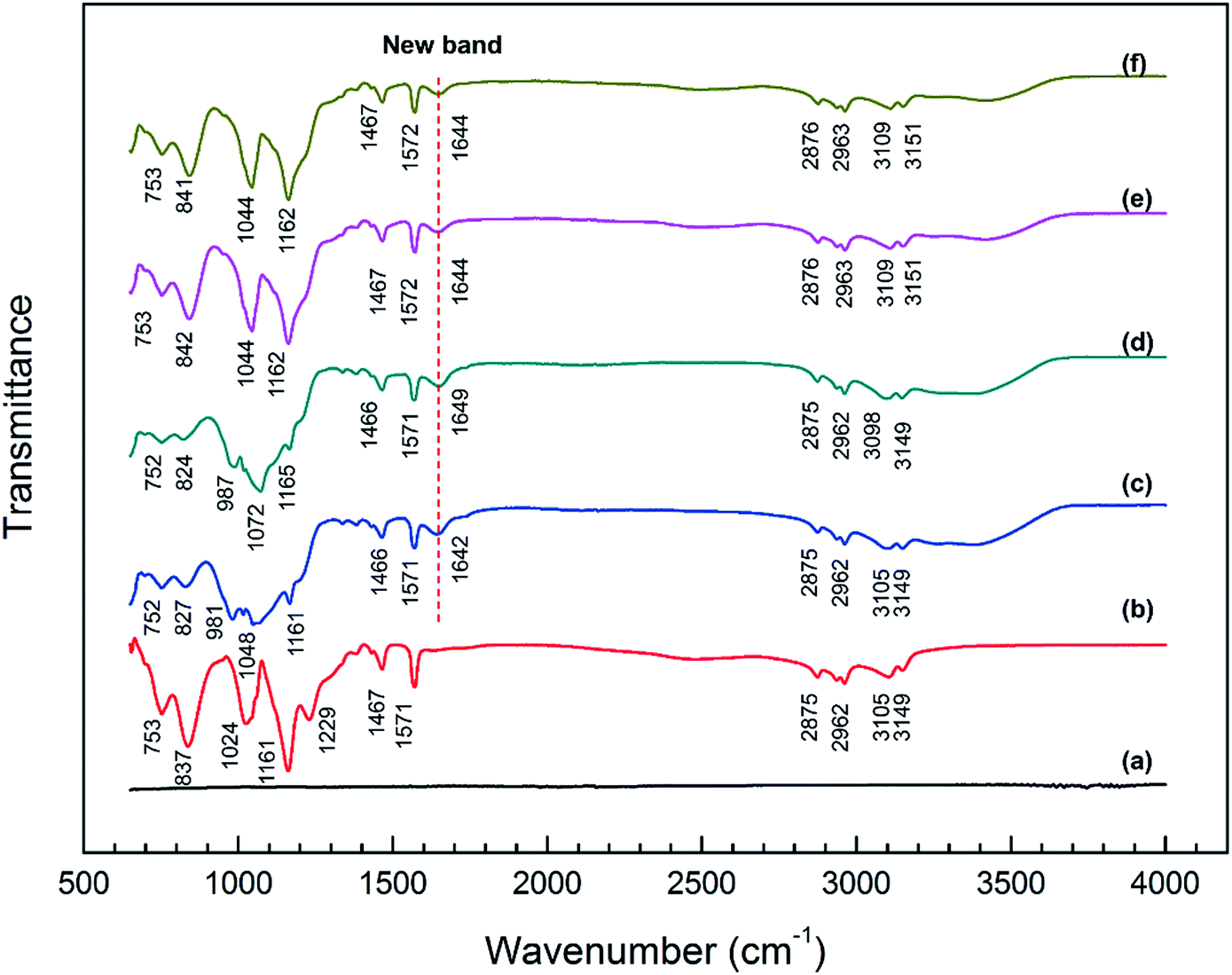 | ||
| Fig. 5 IR spectra in a range of 650–4000 cm−1 of (a) mild steel, (b) pristine [BMIM]HSO4 and surface films of mild steel specimens after immersion in (c) IL, (d) IL + MO, (e) IL + MO + H2O and (f) IL + MO + H2O2 at 45 °C for 14 days. | ||
No obvious band is observed from the spectrum of mild steel (see Fig. 5a). The spectrum of pristine [BMIM]HSO4 IL is shown in Fig. 5b. The bands at 3149 and 3105 cm−1 are attributed to the C–H stretching vibration in imidazolium ring. The bands at 2962 and 2875 cm−1 are assigned to the aliphatic C–H asymmetric and symmetric stretching vibration of alkyl chain. The bands at 1467 and 1571 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching vibrations of imidazolium ring, respectively. The peaks at 1024 and 1229 cm−1 are assigned to the S–O and S–OH stretching vibrations of HSO4−.53 The band at 1161 cm−1 indicate the stretching vibration of C–N. The band at 837 cm−1 is due to C–H in-plane bending vibration of imidazolium ring and the band at 753 cm−1 can be assign to the C–H out-of-plane bending vibration of imidazolium ring.38,54
C and CN stretching vibrations of imidazolium ring, respectively. The peaks at 1024 and 1229 cm−1 are assigned to the S–O and S–OH stretching vibrations of HSO4−.53 The band at 1161 cm−1 indicate the stretching vibration of C–N. The band at 837 cm−1 is due to C–H in-plane bending vibration of imidazolium ring and the band at 753 cm−1 can be assign to the C–H out-of-plane bending vibration of imidazolium ring.38,54
By comparing the spectra in Fig. 5, it is found that the majority of the bands of pristine IL are observed in the spectra of the surface film on steel specimens immersed in different IL solutions, which confirms the presence of [BMIM]HSO4 IL in the surface films. However, it is remarkable that there are some distinctive differences in spectra between the surface film and the pristine IL:
(i) In the region of 1640–1650 cm−1, a new band is observed all over the four steel surfaces compared with the pristine [BMIM]HSO4 IL. The new band can be ascribed to the formation of the surface chemical bond, i.e., Fe–N bond in the interaction of the IL with the steel surface.55,56
(ii) In the spectrum of steel surface immersed in pure IL (Fig. 5c), the bands appeared at 1024, 1161, and 1229 cm−1 in the spectra of pristine [BMIM]HSO4 are shifted to a broad band with multiple peaks at 981, 1048, and 1161 cm−1, which shows a possible interaction of the imidazolium groups of cation and HSO4− of anion with the steel surface. The quantum chemical property of [BMIM]HSO4 showed that the highest occupied molecular orbital (HOMO) distributed in the anion while the lowest unoccupied molecular orbital (LUMO) distributed in the imidazolium group of cation.57 Hence, [BMIM]HSO4 can easily interact with the steel surface by donating the electron from HSO4− to steel surface and accepting the electron from steel surface to the imidazolium ring. Meanwhile, the shift of peaks of C–N bond to lower number has been demonstrated in previous research,54 when the imidazolium or analogue structure has an interaction with the metal surface. The spectrum of specimen immersed in IL + MO solution (Fig. 5e) is highly similar to that in Fig. 5c, implying a similar interaction of the IL molecules and the steel surface in IL + MO solution.
(iii) In the spectrum of specimen immersed in IL + MO + H2O solution (Fig. 5e), the bands of characteristic stretching vibration of S–O and C–N shifted to 1162 and 1044 cm−1, which is different from Fig. 5c, revealing that the presence of water has an effect on the interaction between IL and steel. This phenomenon could be ascribed to the charge screening of water,50 which leads to an increase of hydrogen ions and a competitive adsorption against imidazolium ring. The spectrum of specimen immersed in IL + MO + H2O2 solution (Fig. 5f) is almost the same as that in Fig. 5e.
There are no obvious changes in bands at 2800–3000 cm−1 in the spectra of steel surface, indicating that the aliphatic hydrocarbon chain in the IL molecule does not participate to the adsorption process.
From ATR-FTIR analysis, the IL film adsorbed on the metal surface is mainly by the interaction of imidazolium group of cation and HSO4− of anion with the metal surface. The interaction of imidazolium group with the metal surface can form a barrier between the steel and the corrosive environment and then inhibit the corrosion. The corrosion behavior of steel in different IL systems could be partly attributed to the interaction of the imidazolium group and the steel surface.
3.3 Raman analysis
To investigate the corrosion products of steel corroded by [BMIM]HSO4 and [BMIM]HSO4-based desulfurization systems, Raman spectrometer was applied to identify the compounds on the steel surface after immersion tests.Fig. 6 shows the Raman spectra of corrosion products formed on mild steel surfaces after immersion tests. In these spectra, a strong and sharp peak at about 1018 cm−1 can be observed all over the mild steel surfaces, which is ascribed to the symmetric stretching vibrations of the sulfate ion in FeSO4 or Fe2(SO4)3. The formation of FeSO4 or Fe2(SO4)3 can be described as:58
| Fe2+ + SO42− → FeSO4 | (11) |
| 2Fe3+ + 3SO42− → Fe2(SO4)3 | (12) |
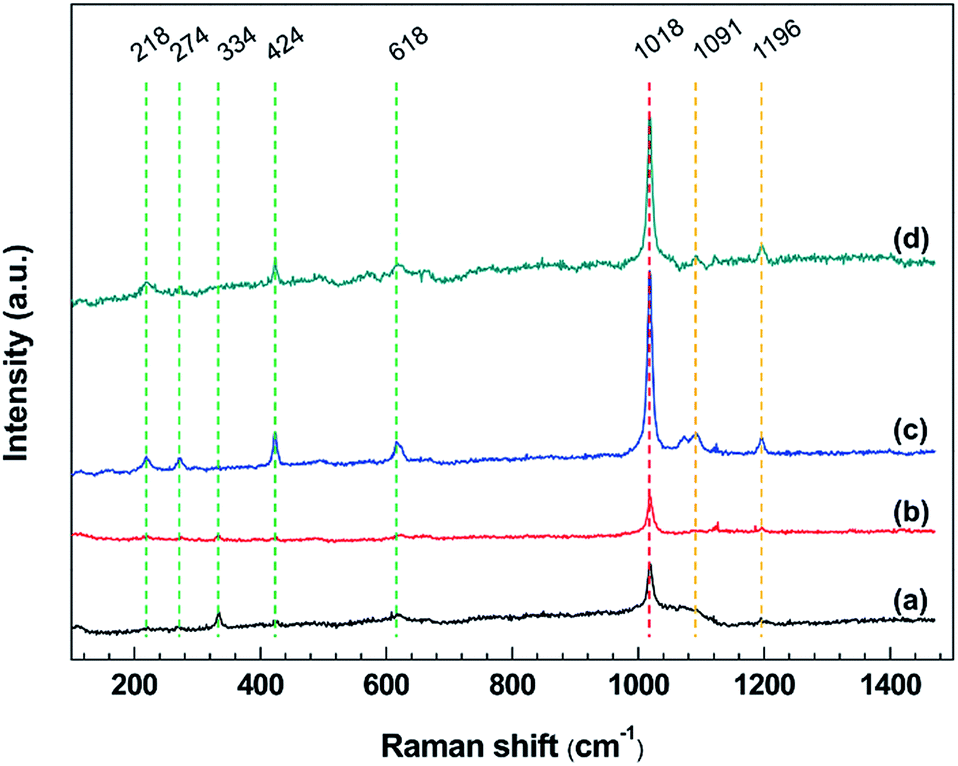 | ||
| Fig. 6 Raman spectra in a range of 100–1500 cm−1 of mild steel surfaces after exposure to (a) IL, (b) IL + MO, (c) IL + MO + H2O, (d) IL + MO + H2O2 at 45 °C for 14 days. | ||
The peaks at about 218, 274, 334, and 618 cm−1 are attributed to the hematite (α-Fe2O3). In Fig. 6c and d, a sharp peak appearing at 424 cm−1 suggests the formation of the FeOOH in the [BMIM]HSO4 solution containing water.59 This is because Fe can react with H2O and O2 to form FeOOH via the following reaction:
| 4Fe + 3O2 + 2H2O → 4FeOOH | (13) |
There are some weak bands at about 1091 and 1196 cm−1, which are related to the vibration of the imidazolium ring that adsorbed on the steel surface,60 agreeing well with ATR-FTIR analysis. The intensity of Raman band at the same position varies with the specimen, due to the difference in the percentage of elemental composition on specimen surface.61 The Raman bands of some corrosion products (Fe2O3 and FeOOH) are slightly different from those reported in the literature,17,59 possibly because the specific corrosion circumstance in this work.
The Raman spectra of stainless steel surface corroded by IL solutions are given in Fig. 7. In these spectra, the bands appearing at about 615 and 1018 cm−1 are similar to the spectra for mild steel, which could be assigned to α-Fe2O3 and iron sulfate, respectively. However, these bands are broader and less distinct, indicating that the structure of compounds formed on the stainless steel is amorphous rather than crystalline.62 In addition, the bands located at about 840 cm−1 in the stainless steel samples are not present in the mild steel samples, which are corresponding to the CrIII oxide or CrVI oxide.63,64 In the IL, IL + MO and IL + MO + H2O systems, the formation of CrIII oxide with a low solubility in IL solutions provides the primary corrosion resistance. Therefore, the corrosion rates of stainless steel in these systems are significantly lower than those of mild steel. In the presence of H2O2, the IL system becomes excessively oxidizing, where the dissolution of passive film occurs, specially by the oxidation of CrIII (as insoluble Cr2O3) to CrVI (as soluble Cr2O72−). This leads to a fast and accelerating transpassive corrosion, in the form of intergranular corrosion,65,66 which is confirmed by the morphology results (see Fig. 8). The bands at 615 and 840 cm−1 are weak, which could be attributed to the amorphous structure of the compounds formed on the surface and the low content of the corrosion products.61,62
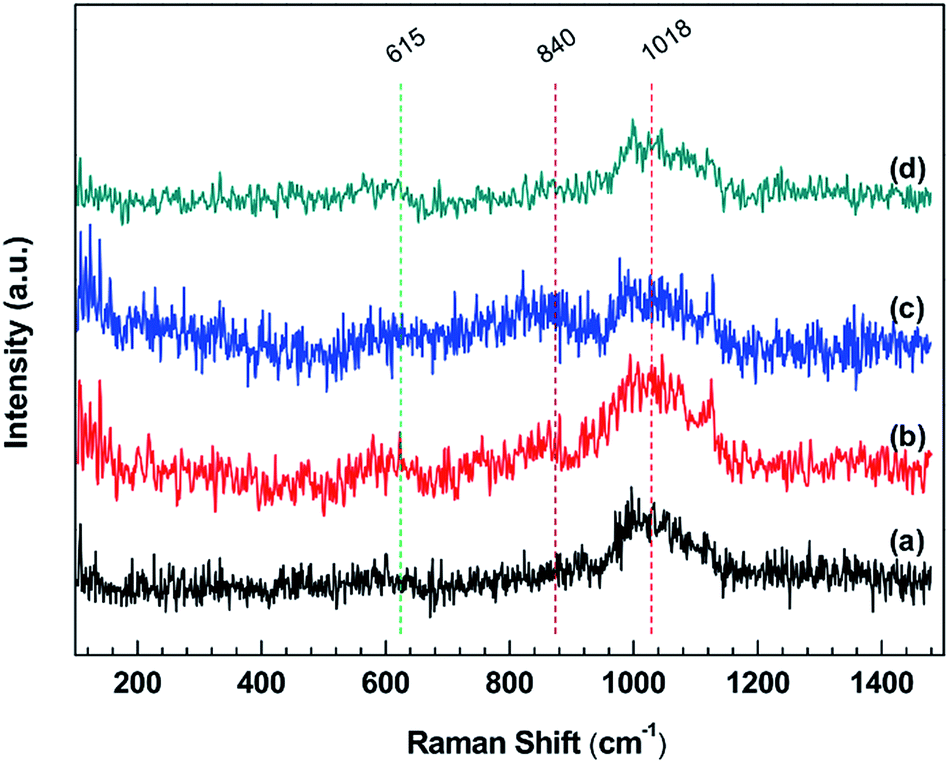 | ||
| Fig. 7 Raman spectra in a range of 100–1500 cm−1 of stainless steel surfaces after exposure to (a) IL, (b) IL + MO, (c) IL + MO + H2O, (d) IL + MO + H2O2 at 45 °C for 14 days. | ||
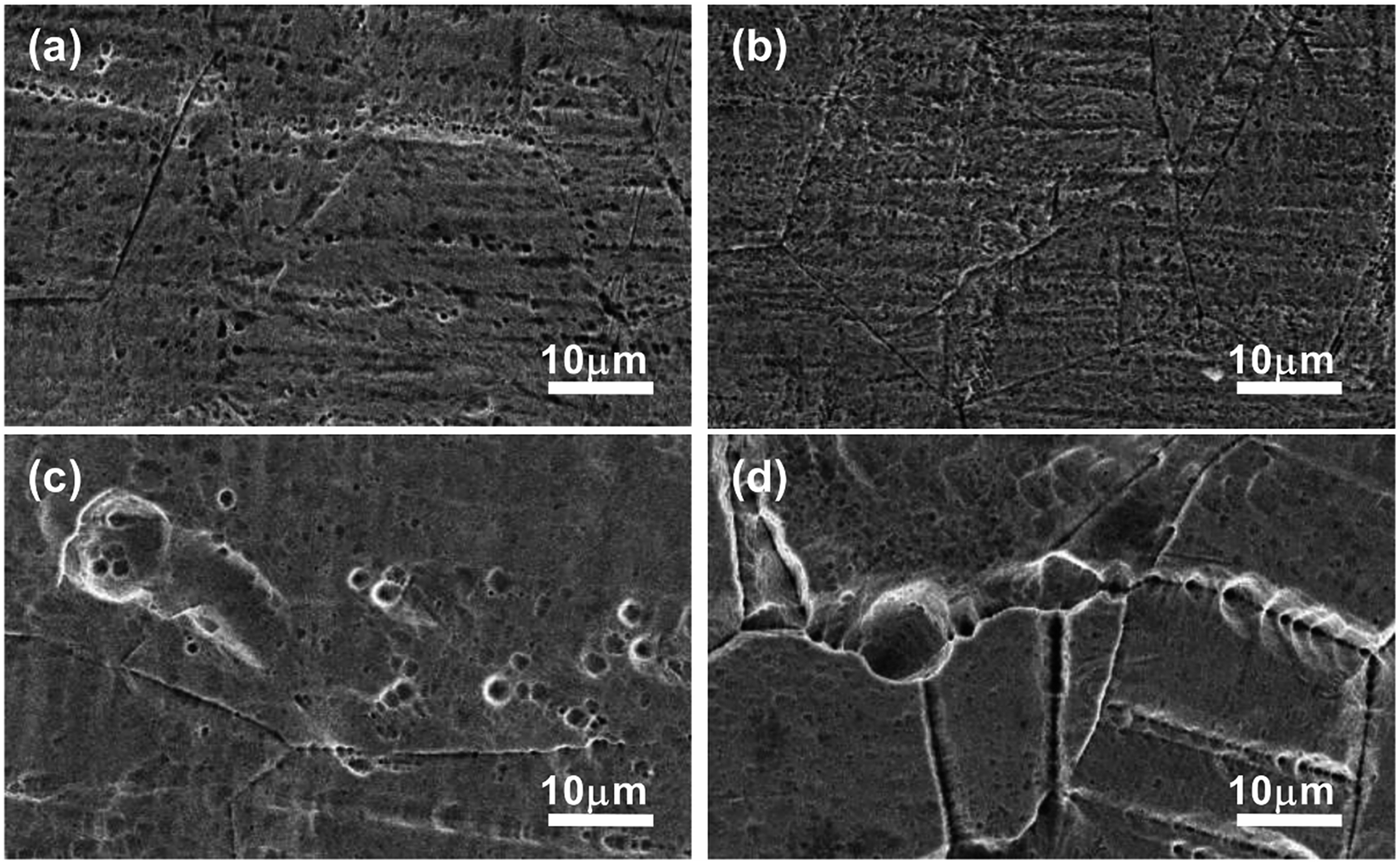 | ||
| Fig. 8 SEM of the surfaces of stainless steel after immersion in (a) IL, (b) IL + MO, (c) IL + MO + H2O, (d) IL + MO + H2O2 at 45 °C for 14 days. | ||
After the cleaning procedures, the surface of the specimens were analyzed by Raman spectrometer. The bands observed over the cleaned surfaces are extremely weak and almost invisible, as illustrated in Fig. S2 (see ESI†). It can be considered that most of the corrosion products are removed from the specimen surfaces after the cleaning procedures.
3.4 SEM observation
The morphological changes of steel surfaces before and after immersion in IL solutions are monitored by SEM. The micrographs of the steel specimens without corrosion are shown in Fig. S3 (see ESI†).Fig. 8 shows the SEM of stainless steel surfaces after immersion in IL solutions. As illustrated in Fig. 8a, the pits with size of 1–2 μm can be clearly seen on the rough surface of the stainless steel specimen immersed in pure IL. For the stainless steel immersed in IL + MO system (Fig. 8b), the stainless steel surface is less damaged as the size of pits is not exceeding 1 μm and the original scratches can still be observed over the surface. In the IL solution with containing water (Fig. 8c), the stainless steel specimen is strongly corroded. Both the width and depth of the pits are increased. When adding H2O2 in the IL solution, the specimen suffers the most severe damage. The pits on the stainless surface are visible with a maximum width of nearly 10 μm and a deep depth (Fig. 8d). The cross-section micrographs of stainless steel surfaces after immersion in IL + MO + H2O and IL + MO + H2O2 systems are shown in Fig. 9. In addition to the pitting corrosion, there also appears the intergranular corrosion over the surface, which is attributed to the polarization of the stainless steel specimen in its transpassive domain in H2O2.40
 | ||
| Fig. 9 SEM of the cross-section of stainless steel after immersion in (a) IL + MO + H2O, (b) IL + MO + H2O2 at 45 °C for 14 days. | ||
Compared to stainless steel, mild steel exposed to the same IL solution suffers more severe corrosion by localized attack (see Fig. 10). The craters formed on mild steel surface are significantly greater and deeper than those on stainless steel, and the distribution and the shape of these craters are irregular.
 | ||
| Fig. 10 SEM of the surfaces of mild steel after immersion in (a) IL, (b) IL + MO, (c) IL + MO + H2O, (d) IL + MO + H2O2 at 45 °C for 14 days. | ||
From SEM observation, it can be seen that the severity of localized attack on stainless steel and mild steel is consistent in an increasing order of IL + MO < IL < IL + MO + H2O < IL + MO + H2O2, which is in good agreement with the corrosion degree determined by weight loss measurement. The localized attack (pitting and/or intergranular corrosion) revealed by SEM could be a serious safety risk to the [BMIM]HSO4-based desulfurization systems for the industrial application.
3.5 Effect of corrosion on desulfurization efficiency
To investigate whether the corrosion processes have effect on the desulfurization efficiency of IL, the oxidative-extraction of DBT experiments using [BMIM]HSO4 IL before and after immersion were carried out at 45 °C under different O/S molar ratios for 2 h.Fig. 11 displays the oxidative-extraction desulfurization efficiency of [BMIM]HSO4 IL before and after corrosion. When the pristine IL is used, the oxidative desulfurization efficiency increases at first and then decreases with increasing of H2O2. The removal of DBT increases sharply from 7% in the absence of H2O2 to more than 70% at the O/S ratio getting to 5, and then increases slowly to a plateau of 90% when the O/S ratio is 20. The sulfur removal drops as the dosage of H2O2 further increasing, which is possibly because the polarity of IL (the main driving force of desulfurization) is diluted with the high content of water. However, the desulfurization efficiency of [BMIM]HSO4 IL after corrosion is different to that of pristine IL. As the O/S ratio increases from 0 to 5, the removal of DBT from the model oil increases from 7% to 47%, and then increases to 57% with the O/S ratio reaching to 20. The removal of sulfur has no evident change with further increase of H2O2.
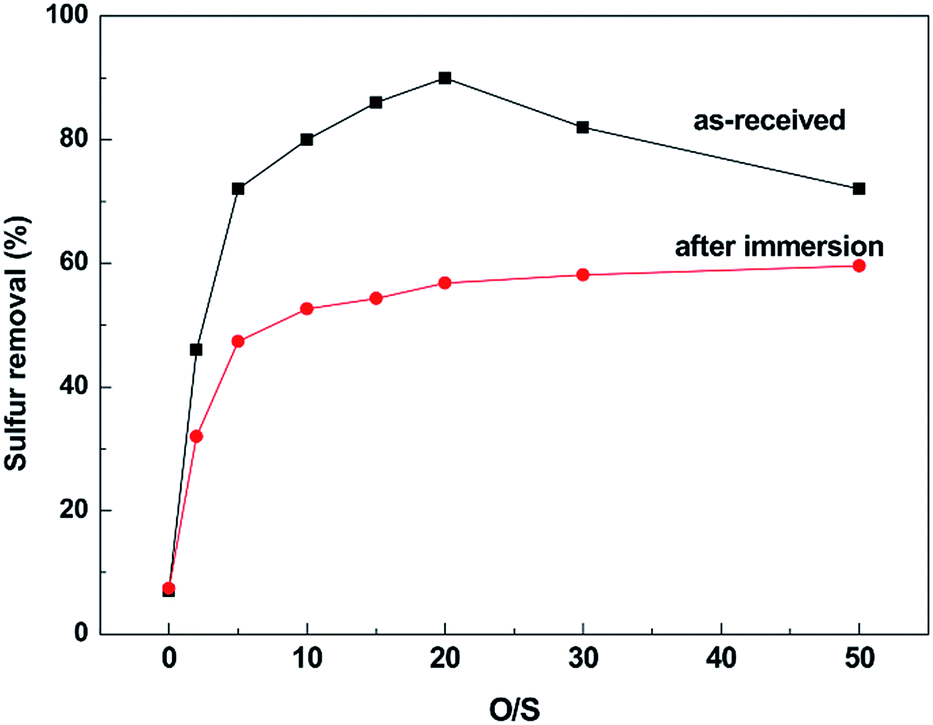 | ||
| Fig. 11 Desulfurization efficiency of [BMIM]HSO4 before and after corrosion of stainless steel at 45 °C for 14 days. | ||
It is clear that the corrosion of steel in [BMIM]HSO4 IL has changed the desulfurization capacity. The mechanism of IL extraction desulfurization of aromatic sulfur compounds such as DBT is mainly attributed to the π–π interactions between the imidazolium group and aromatic ring.5,67 In the oxidation process, DBT is oxidized to DBTO2, which has much higher polarity than DBT8,68 and can be more easily inserted into the IL network.9 Thus, the oxidation of DBT can greatly enhance the desulfurization efficiency. In the oxidation process, acid is necessary for H2O2 decomposition, and the decomposition rate increases as the pH decreases.69 [BMIM]HSO4 can provide the hydrogen ion in oxidation process, which is benefit to the oxidization of DBT to DBTO2 in the IL phase. However, as stated above, the corrosion of steel has consumed considerable quantity of H+, leading to a weaker acidity, which will reduce the catalytic activity of [BMIM]HSO4 that the desulfurization efficiency is worse than pristine IL. Furthermore, the corrosion products transferred into the IL phase during the immersion tests, which also influences the removal of sulfur compounds that the metallic ion such as Fe3+ can form π complexes with the aromatic sulfur compounds.4,7
In summary, the weakened acidity and the presence of corrosion products (iron ion) could be the two main reasons for the differences of desulfurization efficiency between ILs before and after corrosion.
4 Conclusions
The corrosion behavior of steel (mild steel and stainless steel) in [BMIM]HSO4-based desulfurization systems was investigated by weight loss measurements, ATR-FTIR, Raman and SEM from different aspects. The corrosion degree determined by all these methods are in good agreement, which is in an order of IL + MO < IL < IL + MO + H2O < IL + MO + H2O2. The addition of model oil can inhibit the corrosivity of IL media to some extent, while the presence of water or H2O2 enhances the corrosivity of IL solution.From the combination of the changes in IL color and the ICP-MS and spectrophotometer and Raman analyses, iron sulfate proved to be the primary component of corrosion products, which are ferric ion (Fe3+) for the mild steel and ferrous ion (Fe2+) for the stainless steel, respectively. From ATR-FTIR analysis, the adsorption of IL on the steel surface is mainly via the interaction of the imidazolium group of cation and HSO4− of anion with the metal surface, and the interaction is different between the water-free solutions and the aqueous solutions. SEM examination reveals that the steel suffers localized corrosion (pitting and/or intergranular corrosion) in IL solutions. The side effect of corrosion on the IL desulfurization efficiency is clearly suggested from the desulfurization experiments using pristine IL and IL after immersion tests. The differences in desulfurization efficiency are probably caused by the change of acidity and the existence of metallic ion in IL.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (21406063, 21776074 and U1462123), Fundamental Research Funds for the Central Universities (222201514005).References
- J. F. Wishart, Energy Environ. Sci., 2009, 2, 956–961 CAS.
- G. W. Meindersma, A. J. Podt and A. B. de Haan, Fuel Process. Technol., 2005, 87, 59–70 CrossRef.
- Z. X. Lyu, T. Zhou, L. F. Chen, Y. M. Ye, K. Sundmacher and Z. W. Qi, Chem. Eng. Sci., 2014, 113, 45–53 CrossRef CAS.
- P. S. Kulkarni and C. A. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- O. V. Oliveira, A. S. Paluch and L. T. Costa, Fuel, 2016, 175, 225–231 CrossRef CAS.
- Z. Song, T. Zhou, Z. W. Qi and K. Sundmacher, ACS Sustainable Chem. Eng., 2017, 5, 3382–3389 CrossRef CAS.
- J. M. Campos-Martin, M. d. C. Capel-Sanchez, P. Perez-Presas and J. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879–890 CrossRef CAS.
- D. Zhao, J. Wang and E. Zhou, Green Chem., 2007, 9, 1219–1222 RSC.
- H. Lü, S. Wang, C. Deng, W. Ren and B. Guo, J. Hazard. Mater., 2014, 279, 220–225 CrossRef PubMed.
- W. Zhang, K. Xu, Q. Zhang, D. Liu, S. Wu, F. Verpoort and X. M. Song, Ind. Eng. Chem. Res., 2010, 49, 11760–11763 CrossRef CAS.
- Z. Song, T. Zhou, J. Zhang, H. Cheng, L. Chen and Z. Qi, Chem. Eng. Sci., 2015, 129, 69–77 CrossRef CAS.
- Z. Song, J. Zhang, Q. Zeng, H. Cheng, L. Chen and Z. Qi, Fluid Phase Equilib., 2016, 425, 244–251 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- R. Abro, A. A. Abdeltawab, S. S. Al-Deyab, G. R. Yu, A. B. Qazi, S. R. Gao and X. C. Chen, RSC Adv., 2014, 4, 35302–35317 RSC.
- R. Anantharaj and T. Banerjee, AIChE J., 2011, 57, 749–764 CrossRef CAS.
- M. Uerdingen, C. Treber, M. Balser, G. Schmitt and C. Werner, Green Chem., 2005, 7, 321–325 RSC.
- I. Molchan, G. Thompson, P. Skeldon, R. Lindsay, J. Walton, E. Kouvelos, G. E. Romanos, P. Falaras, A. Kontos and M. Arfanis, RSC Adv., 2015, 5, 35181–35194 RSC.
- M. V. Diamanti, U. V. Velardi, A. Brenna, A. Mele, M. P. Pedeferri and M. Ormellese, Electrochim. Acta, 2016, 192, 414–421 CrossRef CAS.
- Y.-C. Wang, T.-C. Lee, J.-Y. Lin, J.-K. Chang and C.-M. Tseng, Corros. Sci., 2014, 78, 81–88 CrossRef CAS.
- I. Perissi, U. Bardi, S. Caporali and A. Lavacchi, Corros. Sci., 2006, 48, 2349–2362 CrossRef CAS.
- M. Mashuga, L. Olasunkanmi, A. Adekunle, S. Yesudass, M. Kabanda and E. Ebenso, Materials, 2015, 8, 3607–3632 CrossRef.
- E. S. Sherif, H. Abdo and S. Abedin, Materials, 2015, 8, 3883–3895 CrossRef PubMed.
- R. J. Wilbraham, C. Boxall, D. T. Goddard, R. J. Taylor and S. E. Woodbury, J. Nucl. Mater., 2015, 464, 86–96 CrossRef CAS.
- T. Zhou, L. Chen, Y. M. Ye, L. F. Chen, Z. W. Qi, H. Freund and K. Sundmacher, Ind. Eng. Chem. Res., 2012, 51, 6256–6264 CrossRef CAS.
- L. Alonso, A. Arce, M. Francisco, O. Rodriguez and A. Soto, AIChE J., 2007, 53, 3108–3115 CrossRef CAS.
- X. Chen, S. Yuan, A. A. Abdeltawab, S. S. Al-Deyab, J. Zhang, L. Yu and G. Yu, Sep. Purif. Technol., 2014, 133, 187–193 CrossRef CAS.
- R. Abro, A. Abdeltawab, S. Aldeyab, G. Yu, A. Qazi, S. Gao and X. Chen, RSC Adv., 2014, 4, 35302–35317 RSC.
- J. L. Wang, D. S. Zhao, E. P. Zhou and Z. Dong, J. Fuel Chem. Technol., 2007, 35, 293–296 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2015, 304, 313–319 CrossRef PubMed.
- Z. Song, D. Yu, Q. Zeng, J. J. Zhang, H. Y. Cheng, L. F. Chen and Z. W. Qi, Chin. J. Chem. Eng., 2017, 25, 159–165 CrossRef CAS.
- A. Standard, American Society for Testing and Materials G31-72, 2004.
- T. Konstantinova, A. Spirieva and T. Petkova, Dyes Pigm., 2000, 45, 125–129 CrossRef CAS.
- J. J. Bell, T. E. Sargeant and J. A. Watson, J. Biol. Chem., 1976, 251, 1745–1758 CAS.
- A. P. Hanza, R. Naderi, E. Kowsari and M. Sayebani, Corros. Sci., 2016, 107, 96–106 CrossRef.
- R. P. Morco, A. Y. Musa, M. Momeni and J. Wren, Corros. Sci., 2016, 102, 1–15 CrossRef CAS.
- X. Li, S. Deng, H. Fu and G. Mu, Corros. Sci., 2009, 51, 620–634 CrossRef CAS.
- A. Yousefi, S. Javadian, N. Dalir, J. Kakemam and J. Akbari, RSC Adv., 2015, 5, 11697–11713 RSC.
- C. Eaborn, The Sadtler Handbook of Infrared Spectra, ed. W.W. Simons, Heyden and Son, Ltd, London, 1978 Search PubMed.
- Z. Shi, M. Liu and A. Atrens, Corros. Sci., 2010, 52, 579–588 CrossRef CAS.
- B. Gwinner, M. Auroy, F. Balbaud-Célérier, P. Fauvet, N. Larabi-Gruet, P. Laghoutaris and R. Robin, Corros. Sci., 2016, 107, 60–75 CrossRef CAS.
- R. M. Cornell, U. Schwertmann, R. Cornell and U. Schwertmann, Mineral. Mag., 2003, 34, 740–741 Search PubMed.
- C. Fan, X. Huang, L. Han, Z. Lu, Z. Wang and Y. Yi, Sens. Actuators, B, 2015, 224, 592–599 CrossRef.
- A. M. Jones, P. J. Griffin and T. D. Waite, Geochim. Cosmochim. Acta, 2015, 160, 117–131 CrossRef CAS.
- Z. Duan, Y. Gu, J. Zhang, L. Zhu and Y. Deng, J. Mol. Catal. A: Chem., 2006, 250, 163–168 CrossRef CAS.
- X. W. Zheng, S. T. Zhang, W. P. Li, M. Gong and L. L. Yin, Corros. Sci., 2015, 95, 168–179 CrossRef CAS.
- D. K. Louie, Handbook of Sulphuric Acid Manufacturing, DKL Engineering, Inc., Thornhill, Ontario, Canada, 2005 Search PubMed.
- D. J. Tao, X. M. Lu, J. F. Lu, K. Huang, Z. Zhou and Y. T. Wu, Chem. Eng. J., 2011, 171, 1333–1339 CrossRef CAS.
- Z. Du, Z. Li, S. Guo, J. Zhang, L. Zhu and Y. Deng, J. Phys. Chem. B, 2005, 109, 19542–19546 CrossRef CAS PubMed.
- A. Pardo, M. C. Merino, A. E. Coy, F. Viejo, R. Arrabal and E. Matykina, Corros. Sci., 2008, 50, 780–794 CrossRef CAS.
- T. C. Schutt, G. A. Hegde, V. S. Bharadwaj, A. J. Johns and C. M. Maupin, J. Phys. Chem. B, 2017, 121, 843–853 CrossRef CAS PubMed.
- A. Davydov, K. V. Rybalka, L. A. Beketaeva, G. R. Engelhardt, P. Jayaweera and D. D. Macdonald, Corros. Sci., 2005, 47, 195–215 CrossRef CAS.
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Cheminform, 2007, 39, 1208–1216 Search PubMed.
- Y. Chaker, H. Ilikti, M. Debdab, T. Moumene, E. Belarbi, A. Wadouachi, O. Abbas, B. Khelifa and S. Bresson, J. Mol. Struct., 2016, 1113, 182–190 CrossRef CAS.
- L. Feng, H. Yang and F. Wang, Electrochim. Acta, 2011, 58, 427–436 CrossRef CAS.
- M. A. Larrubia, G. Ramis and G. Busca, Appl. Catal., B, 2001, 30, 101–110 CrossRef CAS.
- G. Ramis and M. A. Larrubia, J. Mol. Catal. A: Chem., 2004, 215, 161–167 CrossRef CAS.
- J. Zhang, H. Cheng, L. Chen and Z. Qi, CIESC Journal, 2017 DOI:10.11949/j.issn.0438-1157.20170232.
- Y. S. Choi, S. Nesic and D. Young, Environ. Sci. Technol., 2010, 44, 9233–9238 CrossRef CAS PubMed.
- D. Larroumet, D. Greenfield, R. Akid and J. Yarwood, J. Raman Spectrosc., 2007, 38, 1577–1585 CrossRef CAS.
- M. Mączka, N. L. Marinho Costa, A. Gągor, W. Paraguassu, A. Sieradzki and J. Hanuza, Phys. Chem. Chem. Phys., 2016, 18, 13993–14000 RSC.
- M. Ortiz-Morales, C. Frausto-Reyes, J. Soto-Bernal, S. Acosta-Ortiz, R. Gonzalez-Mota and I. Rosales-Candelas, Spectrochim. Acta, Part A, 2014, 128, 681–685 CrossRef CAS PubMed.
- C. Joseph, P. Bourson and M. Fontana, J. Raman Spectrosc., 2012, 43, 1146–1150 CrossRef CAS.
- J. E. Maslar, W. S. Hurst, W. J. Bowers Jr, J. H. Hendricks, M. I. Aquino and I. Levin, Appl. Surf. Sci., 2001, 180, 102–118 CrossRef CAS.
- Y. Hedberg, J. Hedberg, Y. Liu and I. O. Wallinder, BioMetals, 2011, 24, 1099–1114 CrossRef CAS PubMed.
- J. Horvath and H. H. Uhlig, J. Electrochem. Soc., 1968, 115, 791–795 CrossRef CAS.
- P. Fauvet, F. Balbaud and R. Robin, J. Nucl. Mater., 2008, 375, 52–64 CrossRef CAS.
- H. Cheng, J. Zhang and Z. Qi, Mol. Simul., 2017, 1–8, DOI:10.1080/08927022.2017.1337273.
- E. Ito and J. R. Van Veen, Catal. Today, 2006, 116, 446–460 CrossRef CAS.
- G. Yu, S. Lu, H. Chen and Z. Zhu, Energy Fuels, 2005, 19, 447–452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09137k |
| This journal is © The Royal Society of Chemistry 2017 |