
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unrivalled diversity: the many roles and reactions of bacterial cytochromes P450 in secondary metabolism†
Anja
Greule
ab,
Jeanette E.
Stok
 c,
James J.
De Voss
*c and
Max J.
Cryle
*abd
c,
James J.
De Voss
*c and
Max J.
Cryle
*abd
aThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Clayton, Victoria 3800, Australia. E-mail: max.cryle@monash.edu
bEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
cSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane 4072, Australia. E-mail: j.devoss@uq.edu.au
dDepartment of Biomolecular Mechanisms, Max Planck Institute for Medical Research, Jahnstrasse 29, 69120 Heidelberg, Germany. E-mail: max.cryle@mpimf-heidelberg.mpg.de
First published on 18th April 2018
Abstract
Covering: 2000 up to 2018
The cytochromes P450 (P450s) are a superfamily of heme-containing monooxygenases that perform diverse catalytic roles in many species, including bacteria. The P450 superfamily is widely known for the hydroxylation of unactivated C–H bonds, but the diversity of reactions that P450s can perform vastly exceeds this undoubtedly impressive chemical transformation. Within bacteria, P450s play important roles in many biosynthetic and biodegradative processes that span a wide range of secondary metabolite pathways and present diverse chemical transformations. In this review, we aim to provide an overview of the range of chemical transformations that P450 enzymes can catalyse within bacterial secondary metabolism, with the intention to provide an important resource to aid in understanding of the potential roles of P450 enzymes within newly identified bacterial biosynthetic pathways.
 Anja Greule | AG studied Biology with focus on Microbiology/Biotechnology at the University of Tübingen and obtained her Ph.D. degree in Pharmaceutical Biology and Biotechnology from the University of Freiburg, Germany in 2016. Her doctoral research investigated the studies of different Actinomycetes strains for their ability to produce various secondary metabolites. Since January 2017, she has been working as a postdoctoral fellow in the Cryle group, where her interests lie in P450 monooxygenases and other enzymes involved in the biosynthesis of the glycopeptide antibiotics. |
 Jeanette E. Stok | JES completed her Ph.D. under the supervision of James De Voss at the University of Queensland. She then had the pleasure of working with carboxylesterases and epoxide hydrolases with Prof. Bruce Hammock at the University of California, Davis; and isopenicillin N synthase with Prof. Sir Jack Baldwin at the University of Oxford. Since then she has returned to the De Voss laboratory to work with cytochromes P450 from a variety of organisms, including bacteria and plants. |
 James J. De Voss | JDV received a B.Sc (Hons) from the University of Queensland and undertook his Ph.D. on Vitamin B12 as a Royal Commission for the Exhibition of 1851 scholar at Cambridge under the supervision of Prof. Sir Alan R. Battersby. After working at Johns Hopkins on the DNA cleaving enediyne calicheamicin he was introduced to the cytochromes P450 by Paul Ortiz de Montellano at UCSF. Since returning to the University of Queensland, where he is now a Professor, he has continued his interest in the interface of chemistry and biology and currently works on P450 mechanism and the phytochemistry of herbal medicines. |
 Max J. Cryle | MJC obtained his Ph.D. in chemistry from the University of Queensland in 2006. He then moved to the Max Planck Institute for Medical Research in Heidelberg Germany as an HFSP Cross-Disciplinary Fellow and later as an Emmy Noether group leader funded by the DFG. Since 2016 he is an EMBL Australia group leader based within the Biomedicine Discovery Institute at Monash University, where his team focusses on understanding antibiotic biosynthesis (with particular interest in non-ribosomal peptide synthesis) as well as developing new antibiotics. Since 2018 he is funded as a Career Development Fellow by the National Health and Medical Research Council. |
1. Introduction to P450s
The cytochromes P450 (P450s) are a superfamily of heme-containing monooxygenases that play diverse catalytic roles in nature, both in terms of the substrates that they accept and the array of reactions that they catalyse.1 P450 enzymes are easily identified by bioinformatic, structural or spectroscopic approaches due to the remarkable conservation of structure and catalytic mechanism that is found across the P450 superfamily, in spite of the low sequence identity that can exist between P450s.2,3 In order to attempt the classification of P450s based on sequence conservation, the concept of families and subfamilies within P450s has been implemented, which leads to the official CYP nomenclature (families and subfamilies must share 40% and 55% sequence identity respectively).4 Whilst relatively successful in higher eukaryotes, this approach has limited utility in bacterial systems, where substrate selectivity and catalytic function can vary widely within P450 families – classic examples of P450 families that encompass members displaying highly diverse functions include the CYP105, CYP107 and CYP109 families.2,3,5,6 Thus, care must be taken in assigning P450 functions in bacteria based purely on sequence conservation data, unless the level of conservation is very high (>55–65%).2,3 Bacterial P450s are almost exclusively cytosolic proteins, which is a further significant difference to P450s in eukaryotic systems; none the less, bacterial P450s with high pI values can still associate with membranes even if there is no specific membrane anchoring element.7 The structure of P450s is remarkably conserved, containing mostly α-helical secondary structure (standard helices are labelled A–L) supplemented by one β-sheet region (Fig. 1).7 The core of a P450 is formed by a four-helix bundle comprising the D, E, I and L-helices, with the prosthetic heme moiety at the centre of the P450 active site sandwiched between the I- and L-helices.7 The I-helix, which runs across the top face of the heme moiety contains the typically conserved acid/alcohol pair usually important for the P450 active cycle (vide infra),7 whilst the loop just prior to the start of the L-helix contains the cysteine residue that provides the crucial proximal axial thiolate ligand for the heme iron of the P450.7 The proximal face of a P450 is also the site where interactions with redox partner proteins occur, with these processes largely appearing to be driven by electrostatic interactions.8 The active site of P450s comprises regions that surround the distal face of the heme moiety, and include the so-called BC loop region between the B- and C-helices, the C-terminal region of the F-helix, the N-terminal portion of the G-helix, the centre of the I-helix, the β-strand following the K-helix and the C-terminal loop that impinges of the P450 active site (such regions are also referred to as substrate recognition sites, SRSs 1–6).9 Variability within these regions leads to the ability of P450s to accept a wide range of different substrates (even those that are protein bound), whilst the retention of the general structure surrounding the active site heme group and axial cysteine ligand support the remarkable consistency of the P450 catalytic mechanism.7,10,11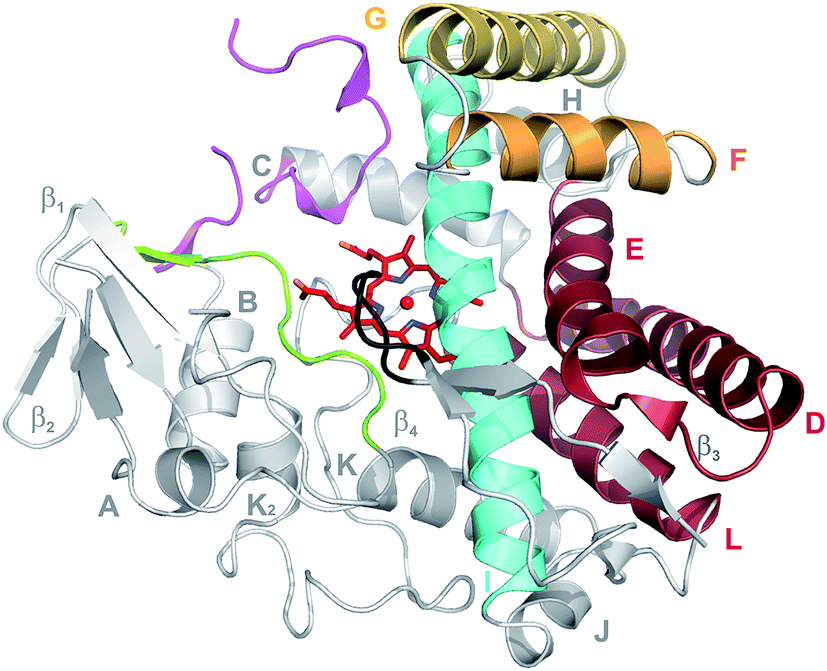 | ||
| Fig. 1 Typical structure of a cytochrome P450, exemplified by OxyBtei (CYP165B7) from teicoplanin biosynthesis showing the preponderance of α-helices in the structure (labelled A–L) together with one β-sheet (β-1/2). The core 4 helix bundle of the P450 is shown in deep red, with the exception of the I-helix that is shown in turquoise. The regions comprising the active site are shown in colours (BC loop region in pink, F-helix shown in orange, the G-helix shown in gold, the I-helix shown in turquoise, the β-strand following the K-helix shown in green and the C-terminal loop shown in black). The central heme moiety is shown as sticks. | ||
2. Chemistry of P450 enzymes
Whilst xenobiotic metabolism in higher eukaryotes highlights the remarkable ability that P450s can exhibit for substrate plasticity, it is in the realm of bacterial P450s – particularly biosynthetic ones – where the truly remarkable range of chemical transformations that P450s are able to mediate is revealed. Furthermore, this is coupled with a remarkable range of different, complex substrates that highlights the amazing potential of these potent biocatalysts. Whilst the archetypal P450-catalysed transformation, hydroxylation of unactivated C–H bonds remains the trademark P450-catalysed reaction, the reported range of chemical reactions able to be performed by P450s is ever increasing, and many examples of these are found in bacterial biosynthetic pathways: such reactions range from aromatic hydroxylation, alkene epoxidation and heteroatom oxidation to processes such as aromatic coupling and multi-step transformations.12,13 One of the most impressive properties of P450s is that these reactions are all performed within a conserved active cycle (Fig. 2).7,14 In this process, a carefully choreographed delivery of two electrons and two protons to molecular oxygen is performed by the P450, whilst the oxidative power of a P450 stems from a highly reactive intermediate generated, known as compound I (Cpd I).15 This highly electrophilic oxidant, which has been identified as an iron-(IV) porphyrin cation radical, is responsible for the vast majority of the oxidation chemistry displayed by P450s. The oxidative power of the compound I intermediate relies on a thiolate ligand for the heme iron in the P450 active site, which in turn requires P450s to be able to maintain this deprotonated thiolate group in spite of the elevated pKa of this moiety.7 In order to generate compound I, the P450 catalytic cycle begins with the displacement of the resting water (or hydroxide ion) from the heme iron upon substrate binding in the active site. The first electron is then transferred from a redox partner protein – typically a ferredoxin – that reduces the iron to Fe2+. If this species is exposed to carbon monoxide, the resultant complex that forms provides a spectrum with a characteristic Soret maximum at 450 nm, for which P450s are named. In the typical active cycle, dioxygen binds to the reduced P450, which then triggers the delivery of a second electron to generate a species equivalent to ferric peroxy anion – a species that has been invoked as the active oxidant in some specific P450-mediated transformations.16 The requirement of the P450-catalytic cycle for electrons is unusual for an oxidative enzyme, with these electrons typically being derived from NAD(P)H via redox partner proteins. Next, the P450 controls the sequential delivery of two protons to the ferric peroxo species, specifically ensuring the protonation of the distal oxygen, through a hydrogen bonded water network established in part through the important acid/alcohol pair of residues typically found in the I-helix above the heme iron.7 This initially leads to the formation of the ferric hydroperoxy intermediate known as compound 0, with the correct delivery of the second proton triggering scission of the O–O bond with concomitant the loss of water and generation of the highly reactive intermediate compound I.14 Instances where protonation of the proximal oxygen occurs during the P450 active cycle lead to the formation of hydrogen peroxide in a process known as uncoupling. This process, whilst largely seen as deleterious, does present the opportunity for some P450s to reverse this process and to utilise hydrogen peroxide directly to bypass the early stages of the P450 active cycle and the necessity for exogenous electron donors. Known as the shunt pathway, this process has been identified as proceeding in several bacterial P450s, although these systems are in the minority compared to those enzymes that utilise a “traditional” P450 active cycle. In the archetypal P450 reaction – C–H hydroxylation – the reaction then proceeds with the abstraction of a hydrogen atom from the substrate, with production of a transient iron-(IV) hydroxyl intermediate known as compound II (Cpd II).17 Rebound of this intermediate onto the substrate radical then affords the hydroxylated product and returns the enzyme to the resting state Fe3+ state. Density function theory calculations suggest that the process of rebound can either proceed with or without a discrete intermediate depending upon the spin state of the intermediates, based upon what is known as two-state reactivity.10,11 Substrate dissociation from the active site and water binding to the heme iron then complete the active cycle – which is one of the more impressive enzyme mechanisms given its complexity and ability to perform such unique oxidative chemistry.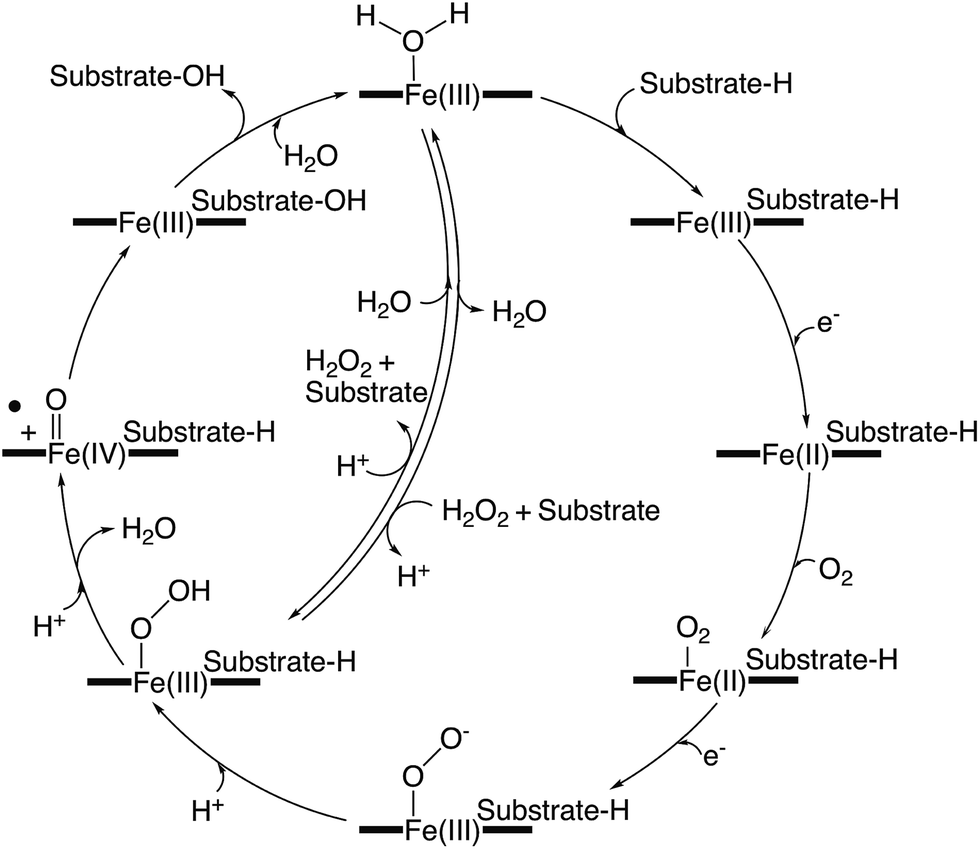 | ||
| Fig. 2 Simplified representation of a generic P450 active cycle resulting in substrate hydroxylation. | ||
3. Bacterial biosynthesis pathways involving P450s
The occurrence of P450s in bacteria is highly variable. Whilst the genome of the model organism E. coli has no identified P450 gene, many secondary metabolite producers like actinomycetes contain multiple P450 homologues. For example, Streptomyces avermitilis putatively contains 33 P450 encoding genes in its genome.18,19 Recently, a comprehensive overview of the sequences, structures and functions of cytochrome P450 enzymes in Streptomyces strains has been published,6 in which the number of homologues stands in contrast to the much lower number of P450s seen, for example, in Bacillus subtilis.20 Bacteria can produce a great range of different secondary metabolites, which provide their bacterial producer strains a selective advantage under specific circumstances. Many of these metabolites have been exploited for use in human therapy for example as antibiotics, immunosuppressants or anticancer agents, which makes understanding their biosynthesis of great importance. In bacteria, cytochrome P450 enzymes catalyse a broad range of physiologically important oxidative reactions with a high degree of chemo-, regio- and stereoselectivity, which are essential for the activity of the final pharmacologically active molecule. P450s are often associated with the major secondary metabolite pathways found in bacteria: polyketide synthesis (performed by polyketide synthases, PKSs); non-ribosomal peptide synthesis (performed by non-ribosomal peptide synthetases, NRPSs); ribosomally synthesised and post-translationally modified peptides (RiPPs); and traditional pathways that produce alkaloids, terpenes, steroids and fatty acids. The genes for the biosynthesis of such bacterial secondary metabolites are often located next to each other on the chromosome, and thus sequences encoding P450 enzymes are typically located within the respective secondary metabolite gene cluster. In some cases, P450s are co-transcribed with their immediate redox partner, typically an Fe-containing ferredoxin, although this is not always the case and many instances have been reported where (multiple) bacterial P450s can accept electrons from a range of endogenous redox partners within the parent bacterial strain.21–24 Even though many P450s identified in bacteria can be assigned to a specific biosynthetic cluster, this does not always make the identification of their biosynthetic role a facile process. One representative example of such a scenario is seen in clavulanic acid biosynthesis, where P450cla (Orf10) has been identified as a part of the gene cluster for more than 15 years.25 In spite of the identification of this P450 within the clavulanic acid biosynthetic cluster, the presence of a presumed ferredoxin redox partner and knockout studies showing the importance of this enzyme in clavulanic acid biosynthesis, no definitive study demonstrating the substrate or catalytic mechanism of this P450 has yet been published. Whilst the likely role of P450cla is the oxidative deamination of clavaminic acid, the exact mechanism of this reaction which affords clavulanate-9-aldehyde via “oxidative enantiomerisation” remains unknown (Fig. 3).25 This serves as a salient reminder that assigning the presumed activity of a P450 enzyme within a biosynthetic pathway can remain challenging.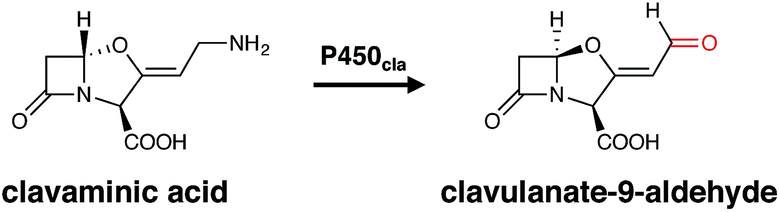 | ||
| Fig. 3 Postulated function of P450cla within clavulanic acid biosynthesis, with the oxidative deamination reaction also proceeding with inversion of stereochemistry of the hemiaminal ether centre of clavaminic acid. The oxygen incorporated as a result of this process is indicated in red. | ||
3.1 Terpene metabolism
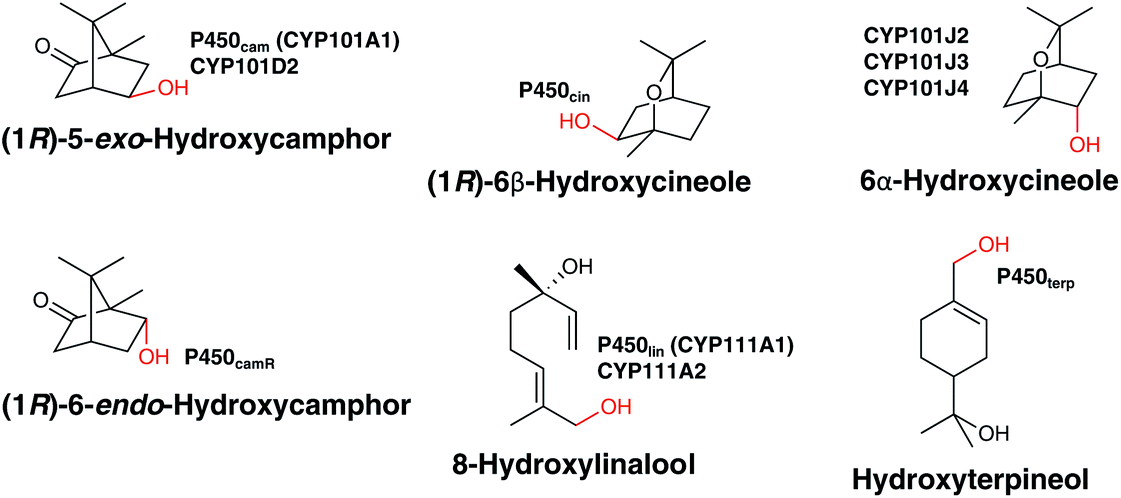
Terpene oxidation mediated by P450s provides the perfect platform from which to appreciate the power and complexity of the chemistry mediated by these enzymes. It ranges from the simple in concept but energetically impressive insertion of oxygen into an unactivated C–H bond through to multistep, intermediate laden transformations that dramatically alter molecular structure and function. P450s have been reported that catalyse the oxidation of terpenes ranging from the relatively simple C10 monoterpenes to the much larger C40 carotenoids in mesophilic and thermophilic bacteria. These oxidation reactions are sometimes associated with the formation of a stable secondary metabolite or the degradation of one to alter its function or provide an energy source for the bacterium. This section will cover the modification of terpenes where this is believed to be the primary, natural function of the P450 in either biosynthesis or biodegradation as the chemistry of these pathways is the same even if the biological context differs.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of cineole alone per year26 – the ability to utilise them as a carbon source is similarly common. The archetypal and best studied P450, P450cam (CYP101A1) from Pseudomonas putida was isolated utilising this strategy and catalyses the hydroxylation of (1R)-camphor to (1R)-5-exo-hydroxycamphor as the first step in its biodegradation.27 P450lin (CYP111A1),28,29 P450terp (CYP108A1)29 and P450cin (CYP176A1)30 were similarly isolated and characterised in organisms that lived on: linalool (Pseudomonas incognita now Acinetobacter baylyi strain ATCC 33305/BD413/ADP1); terpineol (Pseudomonas sp.)); or cineole (reported to be Citrobacter braakii but now believed to be a Rhodococcus sp.). P450cam and P450cin catalyse the enantiospecific hydroxylation of a methylene whilst P450lin and P450terp31 mediated the regiospecific hydroxylation of an allylic methyl group (Fig. 4). In all cases, this oxidation is the first step in an oxidative cascade of reactions that permit conversion of the terpene into fatty acid like molecules that can be taken up into primary cell metabolism with concomitant production of NAD(P)H. Together, these enzymes have had a profound impact on the way P450 chemistry is understood, providing accessible model systems for mechanistic studies and a multitude of X-ray crystal structures32–34 that provided insight into substrate binding and mechanism of oxidation. Interestingly, they were all found to be closely associated with their required redox partners in the bacterial genome, a situation that has not occurred for most subsequently identified bacterial P450s. The redox partners for P450cam (the ferredoxin putidaredoxin and the NADH dependent putidaredoxin reductase) defined class I electron transport in P450s whilst the flavodoxin dependent P450cin defined class III enzymes.35 A number of other monoterpene degrading P450s have subsequently been identified by genome mining and characterised. These include: CYP101D2, a camphor metabolising P450 that also produces (1R)-5-exo-hydroxycamphor;36 CYP111A2 which oxidises linalool in the same way as P450lin;36 CYP101J2, J3 and J4 which oxidise cineole to a stereoisomer of the one produced by P450cin.37,38 Interestingly, none of these enzymes was found closely associated with native redox partners. In the case of CYP102D2 and CYP111A2, a ferredoxin/ferredoxin reductase pair that supported P450 activity was found encoded elsewhere in the genome of the organism (Novosphingobium aromaticivorans) that produces both of these P450s.36 This shared use of redox partners is the strategy commonly found for biosynthetic P450s (vide infra). P450CamR from Rhodococcus sp. NCIMB 9784, for which no DNA sequence information appears available, hydroxylates (1R)-camphor to 6-endo-hydroxycamphor.39 This in turn implies that, as P450camR and P450cam produce structurally isomeric products, biodegradation must proceed by different pathways, unlike the different linalool and cineole oxidising P450s which produce the same or stereoisomeric products. Finally, it is worth noting that not all monoterpene biodegradation relies upon P450s. p-Cymene biodegradation begins with monohydroxylation of the benzylic methyl group but to date in the pathways discovered this is mediated by a non-heme iron oxygenase.40
000 tons of cineole alone per year26 – the ability to utilise them as a carbon source is similarly common. The archetypal and best studied P450, P450cam (CYP101A1) from Pseudomonas putida was isolated utilising this strategy and catalyses the hydroxylation of (1R)-camphor to (1R)-5-exo-hydroxycamphor as the first step in its biodegradation.27 P450lin (CYP111A1),28,29 P450terp (CYP108A1)29 and P450cin (CYP176A1)30 were similarly isolated and characterised in organisms that lived on: linalool (Pseudomonas incognita now Acinetobacter baylyi strain ATCC 33305/BD413/ADP1); terpineol (Pseudomonas sp.)); or cineole (reported to be Citrobacter braakii but now believed to be a Rhodococcus sp.). P450cam and P450cin catalyse the enantiospecific hydroxylation of a methylene whilst P450lin and P450terp31 mediated the regiospecific hydroxylation of an allylic methyl group (Fig. 4). In all cases, this oxidation is the first step in an oxidative cascade of reactions that permit conversion of the terpene into fatty acid like molecules that can be taken up into primary cell metabolism with concomitant production of NAD(P)H. Together, these enzymes have had a profound impact on the way P450 chemistry is understood, providing accessible model systems for mechanistic studies and a multitude of X-ray crystal structures32–34 that provided insight into substrate binding and mechanism of oxidation. Interestingly, they were all found to be closely associated with their required redox partners in the bacterial genome, a situation that has not occurred for most subsequently identified bacterial P450s. The redox partners for P450cam (the ferredoxin putidaredoxin and the NADH dependent putidaredoxin reductase) defined class I electron transport in P450s whilst the flavodoxin dependent P450cin defined class III enzymes.35 A number of other monoterpene degrading P450s have subsequently been identified by genome mining and characterised. These include: CYP101D2, a camphor metabolising P450 that also produces (1R)-5-exo-hydroxycamphor;36 CYP111A2 which oxidises linalool in the same way as P450lin;36 CYP101J2, J3 and J4 which oxidise cineole to a stereoisomer of the one produced by P450cin.37,38 Interestingly, none of these enzymes was found closely associated with native redox partners. In the case of CYP102D2 and CYP111A2, a ferredoxin/ferredoxin reductase pair that supported P450 activity was found encoded elsewhere in the genome of the organism (Novosphingobium aromaticivorans) that produces both of these P450s.36 This shared use of redox partners is the strategy commonly found for biosynthetic P450s (vide infra). P450CamR from Rhodococcus sp. NCIMB 9784, for which no DNA sequence information appears available, hydroxylates (1R)-camphor to 6-endo-hydroxycamphor.39 This in turn implies that, as P450camR and P450cam produce structurally isomeric products, biodegradation must proceed by different pathways, unlike the different linalool and cineole oxidising P450s which produce the same or stereoisomeric products. Finally, it is worth noting that not all monoterpene biodegradation relies upon P450s. p-Cymene biodegradation begins with monohydroxylation of the benzylic methyl group but to date in the pathways discovered this is mediated by a non-heme iron oxygenase.40
 | ||
| Fig. 4 Important examples of bacterial P450-mediated hydroxylation of monoterpenes, leading to the eventual biodegradation of these compounds. P450 generated hydroxyl groups are indicated in red. | ||
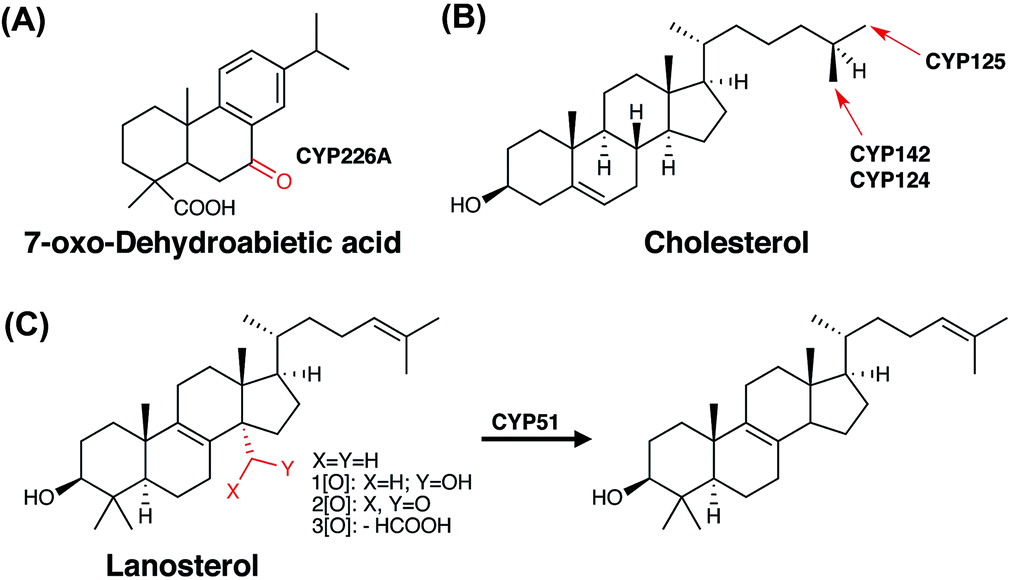 | ||
| Fig. 5 Diterpene and steroid oxidation in bacterial P450 catalysed biodegradation. Examples shown for a diterpene is abietic acid (A) and for steroids cholesterol (B) and lanosterol (C). | ||
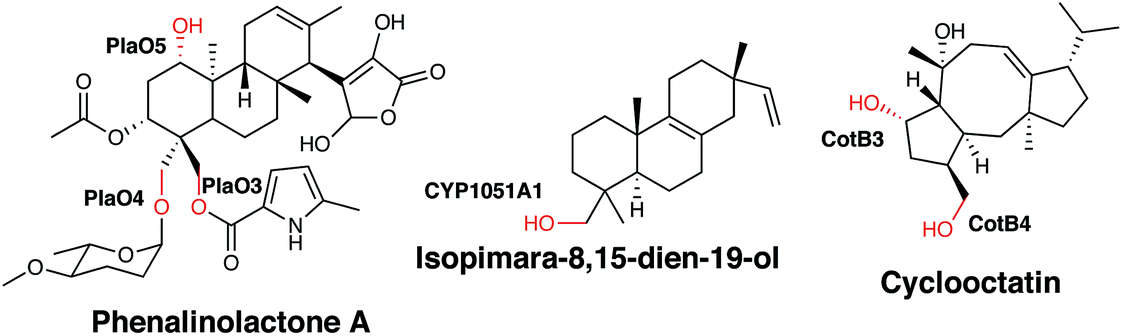 | ||
| Fig. 6 Biosynthetic roles of bacterial P450s in diterpene biosynthesis. Hydroxyl groups generated by P450 action are indicated in red. | ||
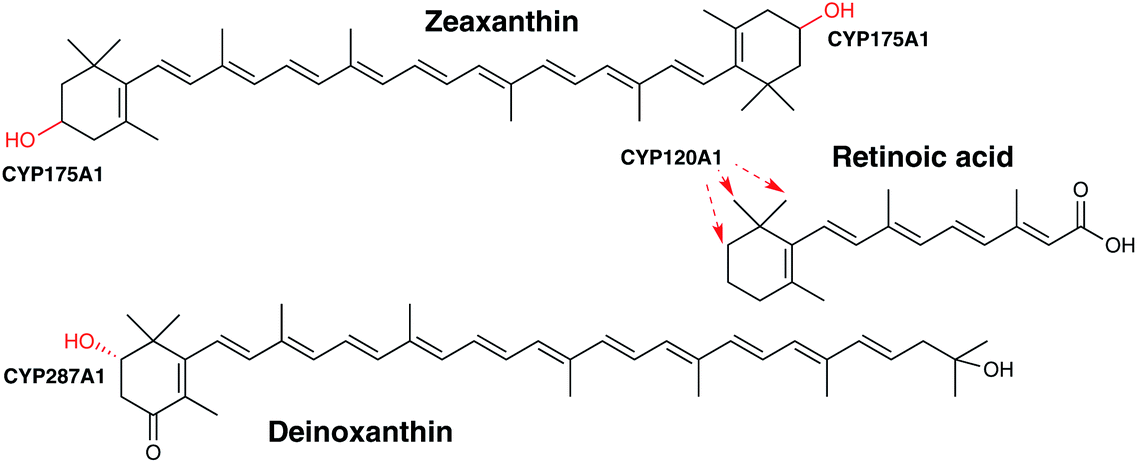 | ||
| Fig. 7 Biosynthetic roles of bacterial P450s in carotenoid biosynthesis. | ||
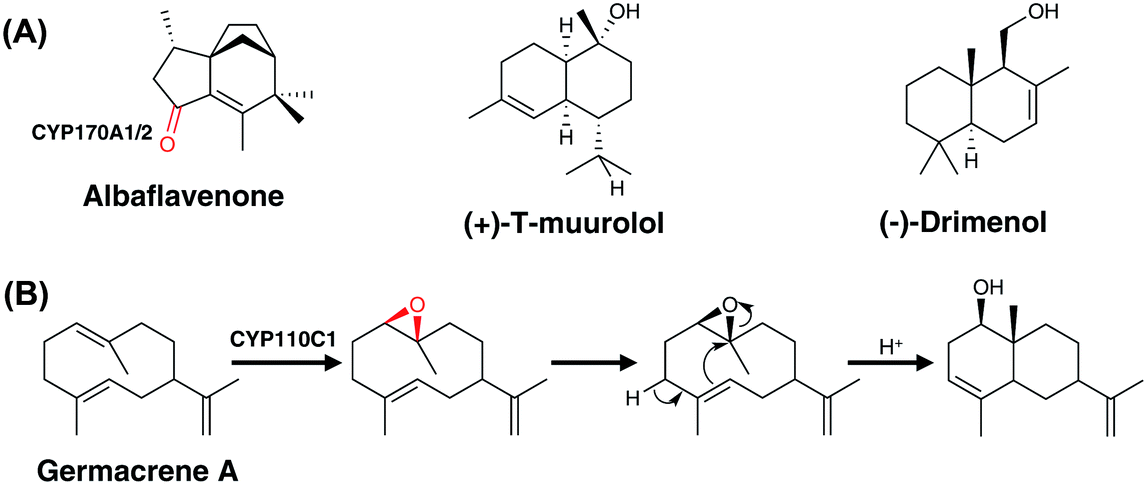 | ||
| Fig. 8 Biosynthetic roles of bacterial P450s in sesquiterpene biosynthesis. Examples of P450 modified sesquiterpene (A) and proposed oxidative cyclisation of germacrene A by CYP110C1 (B). Oxygens incorporated through the actions of P450s are indicated in red. | ||
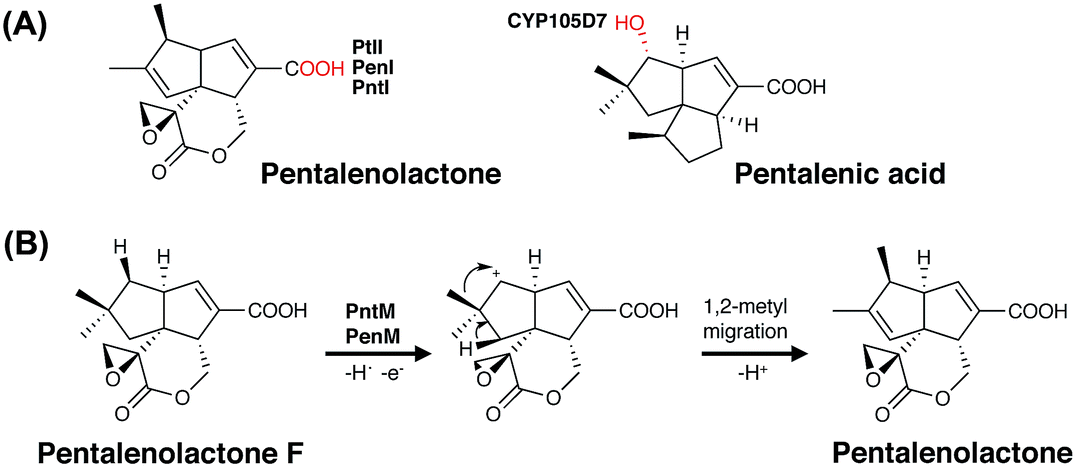 | ||
| Fig. 9 Examples of P450-mediated transformations in the biosynthesis of sesquiterpenes (A) and the proposed mechanism of the P450-mediated oxidative rearrangement in the biosynthesis of the sesquiterpene antibiotic pentalenolactone (B). P450 incorporated oxygens are indicated in red. | ||
Perhaps no biosynthetic pathway better displays the catalytic potential of P450 mediated oxidations than that of the gibberellins.72 The pathway to these important signalling molecules has independently evolved in plants, fungi and in some bacteria, the latter of which are plant symbionts or pathogens.72 These diterpene derived plant phytohormones undergo a number of remarkable P450 mediated transformations (Fig. 10). CYP117 mediates the early regio- and chemospecific transformation of the C19 methyl group into a carboxylic acid moiety by three sequential oxidations proceeding via alcohol and aldehyde intermediates.72 Next CYP114, utilising a co-encoded ferredoxin redox partner, converts a cyclohexane into a carboxy cyclopentane.72 This unusual, three-step reaction is thought to proceed via an initial hydroxylation followed by a semi-pinacol rearrangement of an α-hydroxy cation. Such a species might arise from the usual H atom abstraction by compound I, with steric inhibition of the rebound step as seen in pentalenolactone (vide supra) formation leading to electron transfer and cation generation. Conversion of the immediate formyl product to the acid then follows a standard pathway. It is interesting to note that the C19 carboxylate formed previously is thought to be important in this transformation despite its distal nature. CYP112 then catalyses another two-step regiospecific conversion of a methyl group into an aldehyde intermediate.72 This compound then presumably undergoes oxidative loss of the carboxylic acid moiety catalysed by CYP112, perhaps mediated by a ferric peroxo P450, to yield a carbocationic intermediate which then forms a lactone with the C19 carboxylate installed by CYP117. Such a loss of the carboxy moiety may initially generate a radical intermediate but again steric inhibition of rebound would be required to could promote cation formation. The final P450 involved in the gibberellin pathway is CYP115 that is required to form gibberellin 4, the bioactive hormone, and it catalyses the last step in the pathway. Almost anticlimactically, this is a simple regio- and stereospecific hydroxylation of a methylene group.73
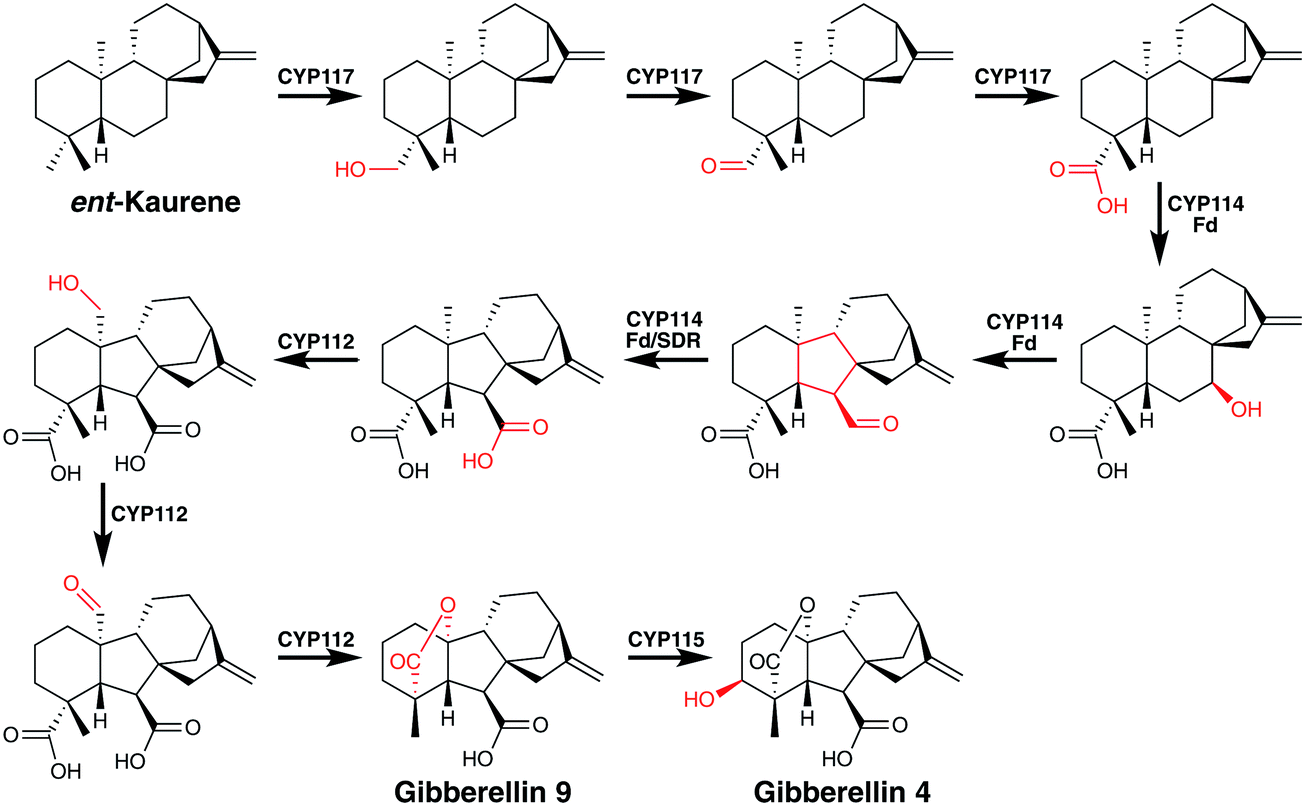 | ||
| Fig. 10 Remarkable P450-mediated transformations found in gibberellin biosynthesis. Fd = gibberellin ferredoxin, SDR = gibberellin short-chain dehydrogenase/reductase. P450-catalysed reactions are indicated in red. | ||
3.2 Fatty acid metabolism
Fatty acid metabolism by cytochrome P450s is one of the most commonly reported reactions for this enzyme class. The relevance of such transformations to biosynthetic pathways and even the actual role of the P450 enzyme in question is not always clear, however, as fatty acids make excellent, flexible and hydrophobic substrates that can interact with many P450 active sites. None the less, there are several well characterised examples of bacterial P450 enzymes that specifically metabolise fatty acids, and even in cases where the relevance of the reaction to the organism is unclear, significant mechanistic insights into P450 function have been gained.The hydroxylation of fatty acids remains the major reaction that is seen in bacterial P450s that act against such substrates (Fig. 11).74,75 Due to the relative scarcity of structural motifs within fatty acids, the selectivity of hydroxylation in such cases is often lower than is seen for most biosynthetic transformations, with the exceptions to this rule being if the site to be hydroxylated is close to either the carboxylic acid moiety (α/β-positions) or the terminal methyl group (ω-position) of the fatty acid. In P450s with the requirement to specifically perform the oxidation of the energetically more challenging methyl group over neighbouring methylene groups, a highly specific active site architecture is required to restrict access of the fatty acid chain to the active site oxidant. Such an active site architecture has been resolved for CYP124 from M. tuberculosis, where selective hydroxylation of a fatty acid methyl group in iso-branched chain fatty acids is enabled by a highly specific fatty acid binding channel that orients the fatty acid perpendicular to the heme plane combined with significant conformational restriction in the vicinity of the heme itself.76 CYP124 does not display as high a selectivity for ω-oxidation of straight chain fatty acids, although the activity for these substrates is several orders of magnitude below that of branched chain substrates and implicates a role of CYP124 in branched chain fatty acid metabolism.76
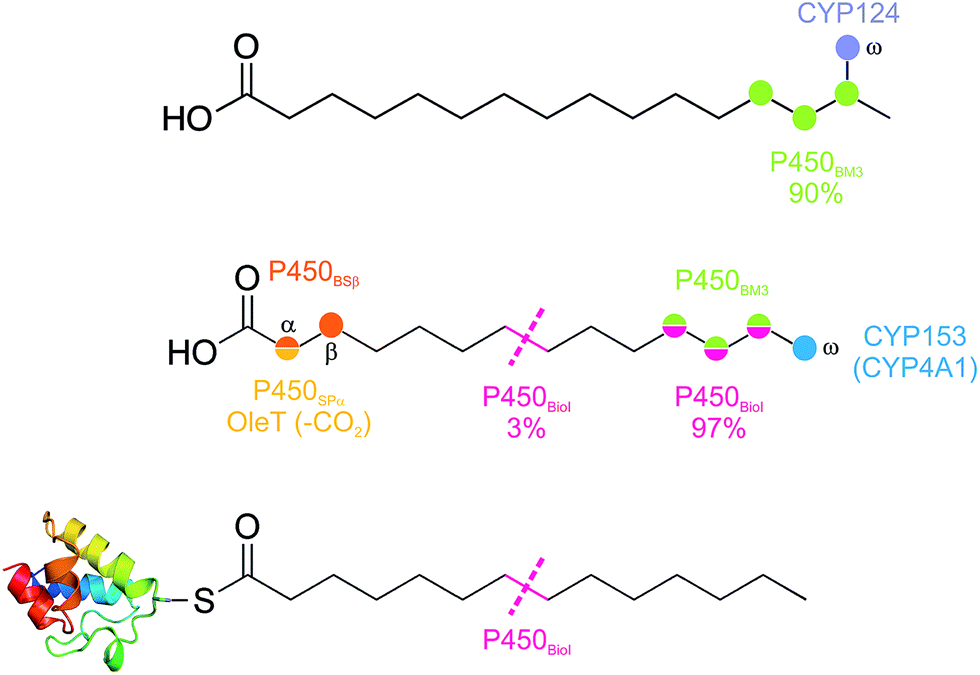 | ||
| Fig. 11 Examples of P450 enzymes involved in bacterial fatty acid metabolism, indicating the regiospecificity of oxidation displayed by these P450 in the metabolism of fatty acid substrates. Colours indicate the P450 enzyme responsible to the oxidation of the fatty acid chain at these specific positions. | ||
Possibly the most well studied examples of ω-hydroxylating P450s are the mammalian CYP4A class, and studies on these enzymes have shown that steric restriction around the active site account for the selectivity of this subfamily.77 One bacterial P450 subfamily that displays a similar level of selectivity for the ω-oxidation of straight chain fatty acids is the CYP153A subfamily, with subfamily members characterised as having very high selectivity for methyl oxidation over that of neighbouring methylene groups.78 CYP153A enzymes have been investigated by a number of groups due to potential interest from a biotechnological perspective, which is also heightened by observations that these P450s can also accept alkanes as substrates and produce terminal alcohols as well as α,ω-diols.79–82 In the case of diol production, further oxidation has also been detected (i.e. to the aldehyde), which appears to increase with chain length of the substrates and probably stems from higher substrate affinity in these cases.80 Production of the aldehyde has also been reported as a minor side product in experiments with CYP4A enzymes,77 and demonstrates the effects that even minor side reactions can have over long reactions times when all of the initial fatty acid substrate has been converted into an intermediate species.
One of the best known examples of a cytochrome P450 is P450BM3 from Bacillus megaterium.83,84 This P450 has received a great deal of attention due to its substrate promiscuity and rapid turnover rate, making it effectively a bacterial equivalent of a xenobiotic metabolising P450 enzyme – albeit a very fast enzyme that is easily amenable to expression, mutation and scale up. Many of the favourable properties of P450BM3 stem from the fact that it is a naturally existing fusion protein, where the FMN and FAD containing redox partners needed to supply electrons to the P450 domain (known as BMP) are fused into one polypeptide chain.85 This arrangement leads to the rapid rates of catalysis for P450BM3, and has inspired significant work to identify more natural P450 fusion proteins as well as attempts to generate synthetic P450-fusion proteins to obtain enzymes with more favourable catalytic properties. One consequence of this fusion arrangement is that P450BM3 appears to be active as a dimer, with electron transfer occurring in an intermolecular fashion.86–88 P450BM3 has long been reported to oxidise a range of fatty acid substrates, with hydroxylation focussed on the methylene groups at the terminus of the fatty acid chain (ω-1 to ω-3).89–91 Increasing chain length alters this distribution and leads to increased oxidation at sites more remote from the ω-position. The stereochemical purity of these alcohol products can be very high (>99% ee) favouring the R-enantiomer, although the enantiopurity of the product alcohols decreases with increasing distance from the terminal methyl group.92–94 The mechanism of P450BM3-mediated hydroxylation has been probed using so-called radical clocks, in which the ring opening of cyclopropyl probes in response to an intermediate radical species prior to oxygen rebound provided strong evidence for a distinct substrate centred radical as opposed to a concerted process.95–97 Long incubations of fatty acids with P450BM3 can lead to the isolation of products that have undergone several rounds of oxidation, producing ketone, diol and hydroxyketone products,89,90 but unlike systems in which sequential oxidations are required (such as P450scc or P450BioI, vide infra) the affinity of the enzymes for hydroxylated fatty acids is significantly reduced over the parent fatty acids themselves. P450BM3 is also able to epoxidise unsaturated fatty acid substrates, with the relative positioning of the alkene with regards the ω-position of the fatty acid either dictating complete epoxidation, a mixture of epoxidation and hydroxylation or complete hydroxylation.98 Such effects have also been observed when thiafatty acid probes were used to investigate the effect that ease of oxidation has on both product regiospecificity and stereospecificity, which show that centres that are more easily oxidised can significantly influence both these parameters.99 The inclusion of methyl groups within a fatty acid chain also provoke alterations to the regiochemistry of P450BM3 oxidation, which is due to a combination of steric and electronic effects in these cases.100 The oxidation of such substrates has been suggested as a possible natural reaction for this P450, although as it stands it is still not completely clear what role this remarkable P450 plays within B. megaterium.83
Other bacterial P450s that utilise fatty acid substrates display important examples of diversity within such enzymes. CYP105D5 from Streptomyces coelicolor has also been demonstrated to perform the hydroxylation of fatty acids in a similar manner to that seen with P450BM3.21 In the case of this P450, reconstitution of activity could be achieved using native ferredoxin and flavoprotein ferredoxin reductase enzymes from S. coelicolor itself,21 which serves to highlight the ability of P450s to interact and accept electrons from redox partners within the cell that may not always be closely associated with the P450. CYP119 is an example of an orphan P450 like P450BM3 whose physiological role is unclear. What makes this P450 of great interest is that it was isolated from the thermophilic archaea Sulfolobus acidocaldarius and as such displays a much higher tolerance to elevated temperatures compared to mesophilic P450s (this P450 is included here in this review partially due to the widespread misconception that this is a bacteria).101,102 The origins of the high thermal stability of this P450 appear strongly connected with aromatic stacking interactions within the protein,103 whilst the ability to combine CYP119 with a thermostable redox system (comprising a ferredoxin and a 2-oxoacid:ferredoxin oxidoreductase) has been shown to significantly boost activity at 70 °C.104 CYP119 is able to oxidise substrates other than fatty acids and has also demonstrated the ability to directly use hydrogen peroxide to support such oxidations.105 CYP119 has also been highly useful in mechanistic investigations into P450 mechanism and reactivity, with the Green group first demonstrating the conclusive identification and characterisation of the key P450 reactive intermediate – the central iron-oxo/ferryl intermediate known as compound I – using this enzyme.15 Further examples of P450s isolated from bacteria that inhabit extreme environments include CYP261C1/2D1 that were isolated from Photobacterium profundum SS9 (ref. 106) and Moritella sp. PE36 (ref. 107) that live in a deep-sea environment. These P450s display a pressure-dependent equilibrium between two enzymatic states that appear to be related to control of water access to the P450 active site, with the “restricted access state” hypothesised to be transient in P450s not subjected to such extreme pressures in which protein hydration is favoured. These examples underline the importance of examining enzymes from extremophiles, as their properties not only inform on the adaptions required to tolerate such conditions but also provide insights into the mechanism of mesophilic enzymes.
Several bacterial P450s that oxidise fatty acids have been identified that can use hydrogen peroxide as an oxidant. Both P450SPα (CYP152B1) from Sphingomonas paucimobilis, which regio- and enantioselectively catalyses the S-hydroxylation of the α-position of long chain fatty acids,108–110 and P450BSβ (CYP152A1) from Bacillus subtilis, which catalyses the hydroxylation of long chain fatty acids to produce a 3:2 mixture of β-R- and α-S-hydroxylated products, utilise hydrogen peroxide as an oxidant. Specific arginine (R241/R242) residues in P450SPα and P450BSβ are crucial as they bind the carboxylate of the fatty acid to position the substrate above the heme for oxidation in both cases.111,112 Moreover, the carboxylic acid moiety appears to play a role in the generation of compound I from hydrogen peroxide in these P450s, which is an important example of substrate-assistance in the P450 active cycle.111–113 The regioselectivity of P450SPα appears to be maintained through the channel that orients the fatty acid within the active site, as mutations corresponding the P450BSβ active site are unable to switch regioselectivity towards the β-position.111 The opposite is true for mutations to the P450BSβ active site, which are capable of switching the regiochemistry of oxidation by this P450 towards the α-position.111 The use of hydrogen peroxide as an oxidant means that there is not the need to interact with redox partner proteins, and this is reflected in an extended loop prior to the axial cysteine ligand on the proximal face of the heme coupled with a predominant negative charge, which differs to P450s that interact with redox partner proteins.111,112 The use of peroxide by these P450s accounts for their rapid rate of turnover and also serves to explain the high levels of interest in such enzymes as potential biocatalysts, although inactivation by high levels of peroxide can limit the usefulness of this approach.114 An alternate solution has been demonstrated in the case of the related α-hydroxylase CYP152A2 from Clostridium acetobutylicum, in which reconstitution with a functional redox partner chain was able to improve the catalytic properties of this enzyme.114 Thus, the relevance and use of hydrogen peroxide as an oxidant in P450-mediated transformations always needs to be tested in comparison to a redox enzyme pathway in order to deliver the optimal system for study.
A further example of P450 diversity in bacterial fatty acid metabolism is provided by OleT (CYP152L1) from Jeotgalicoccus sp. ATCC 8456, which catalyses the oxidative decarboxylation of fatty acids to generate terminal alkenes.115,116 OleT has a similar structure to P450SPα and P450BSβ, including the extended loop near the conserved cysteine ligand and the presence of an arginine residue in the active site to coordinate to the carboxylate of the fatty acid.116,117 The decarboxylation of fatty acids by OleT is supported both by peroxide and electron transfer partners, and has been extensively investigated due to the unusual nature of the reaction and the potential to use such an enzyme for biofuel production.115–121 Mechanistically, it has been demonstrated that this enzyme utilises the highly reactive compound I as an intermediate in the oxidation, with a comparatively long-lived compound II species also being detected during the course of the reaction rather than the typical oxygen rebound pathway.120,121 OleT serves as an important example of how bacterial P450s can perform atypical P450 chemistry despite high levels of structural and sequence similarity to related P450s that perform more standard hydroxylation chemistry.
One example of fatty acid oxidation that relies upon the selective oxidation of the middle of the fatty acid chain is that performed by P450BioI, which is found in the biotin operon of various species of Bacillus.122,123 The role of this P450 is to generate pimelic acid (or a biological equivalent), a C7 diacid precursor of biotin biosynthesis, from long chain fatty acids.74,123,124 Mechanistically, this P450-catalysed multiple step oxidation proceeds via two sequential hydroxylations to generate a vicinal diol, which is then cleaved by the P450 to generate a C7 acid/aldehyde product that then undergoes rapid oxidation to the desired diacid.125 This mechanism resembles that utilised by P450scc, the human enzyme that cleaves the side chain of cholesterol during steroid biosynthesis, which has been investigated in detail due to the importance of steroid biosynthesis for human health.124 The activity of P450BioI against free fatty acids can be reconstituted, but this oxidation proceeds with low selectivity for in chain cleavage: rather, such reactions afford R-hydroxylated fatty acids with the site of oxidation close to (but not including) the ω-terminus of the fatty acid.125,126 The situation dramatically alters, however, when P450BioI catalyses the oxidation of fatty acids bound to acyl carrier protein (ACP).123 In these cases, the production of the desired diacid product proceeds with complete selectivity. A crystal structure of the P450BioI–ACP complex – the first such complex to be isolated and structurally characterised – clearly demonstrated how the selective oxidation of ACP-bound fatty acids by P450BioI occurs.127 In the complex structure, the phosphopantetheine arm of the ACP upon which the fatty acid is bound projects in towards the heme of the active site, and is closely coordinated via electrostatic interactions that lock the position of this arm in place like a ruler above the heme.127 The active site, which is almost exclusively comprised of large, hydrophobic amino acid residues, then ensures that the fatty acid chain adopts a U-shaped conformation over the heme, with the bottom of the “U” comprising the C7 and C8 atoms of the fatty acid that are then closest to the heme for subsequent oxidation.127 The utilisation of a carrier protein in the substrate of P450BioI demonstrates that P450s can act against a wide range of substrates – even protein bound substrates – and that carrier protein bound substrates need to be considered when investigating P450s that are involved in systems that contain carrier proteins (such as polyketide synthases and non-ribosomal peptide synthetases (vide infra)).74,128
3.3 Alkaloid biosynthesis pathways
Alkaloids are secondary metabolites, generally biosynthesised from amino acids, which contain at least one nitrogen atom that is generally part of a heterocycle. Interest in alkaloids is often dependant on their pharmacological applications and these frequently drive investigations into their biosynthesis. Historically, alkaloids were commonly derived from plant sources, but are now known to be generated by a number of different organisms, including bacteria and fungi. Nevertheless, there are still limited examples of alkaloids biosynthesised by bacterial species and consequently, there are very few instances of P450s involved in the alkaloid biosynthesis in these organisms. However, this scarcity has not limited the unusual reactions that can be performed these P450s. Alongside the typical carbon hydroxylation reactions, bacterial P450s can execute N-hydroxylations and form both carbon–carbon and nitrogen–carbon bonds in the generation of alkaloids.NzsA is an example of a P450 that catalyses a classical carbon hydroxylation reaction (Fig. 12). In Streptomyces sp. MA37, neocarazostatin B, a tricyclic carbazole alkaloid derived from L-tryptophan, is converted to neocarazostatin A triol via hydroxylation at C10.129 Two genes (NozD and E) encoding putative P450s have also been identified in the genome of Norcardiopsis sp. CMB-M0232 and are proposed to be involved in the biosynthesis of nocardioazines A and B. Like neocarazostatin, these alkaloids are also derived from L-tryptophan, but in this case two L-tryptophan molecules combine to produce a cyclodipeptide.130 NozD and E are proposed to catalyse carbon hydroxylation and epoxidation in the final stages of nocardioazine biosynthesis,130,131 but further biochemical studies are necessary to determine the exact function of these enzymes (Fig. 12).
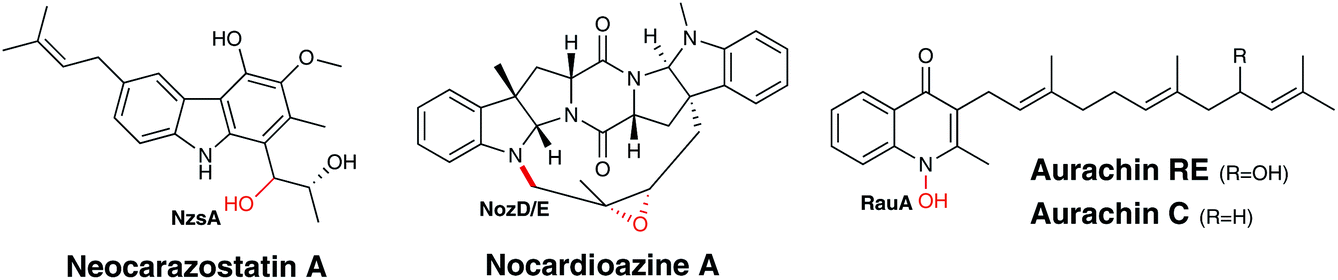 | ||
| Fig. 12 Biosynthetic roles of bacterial P450s in alkaloid biosynthesis. Oxygen atoms incorporated as a result of P450 activity are indicated in red. | ||
RauA (CYP1050) provides a rare example of a P450-catalysed N-hydroxylation in the production of an aurachin alkaloid antibiotic, aurachin RE from Rhodococcus erythropolis JCM 6824 (Fig. 12). The hydroxylation on the nitrogen of the quinolone ring is crucial as it is essential for the antibacterial activity of the molecule and is the final step of its biosynthesis.132,133 Conversely, in other species that produce these alkaloids this oxidation is not performed by a P450. Stigmatella aurantiaca Sg a15 generates aurachin C, which is structurally very similar to aurachin RE, lacking only one hydroxyl moiety in comparison. The absence of a P450 in the genome of S. aurantiaca and genetic knockout studies suggest a Rieske [2Fe–2S] oxygenase is utilised for this oxidation rather than a P450.134,135
The most intriguing alkaloid transformations are those catalysed by the P450s involved in the biosynthesis of the indolocarbazole alkaloids, rebeccamycin and staurosporine. These include the StaP (CYP245A1) and RebP (CYP245A2), enzymes that catalyse an intramolecular carbon–carbon coupling reaction, and StaN (CYP244A1), which is responsible for the formation of a carbon–nitrogen bond in staurosporine (Fig. 13). The biosynthesis of both highly similar indolocarbazoles begins with the dimerisation of two imines derived from L-tryptophan (or the dichlorinated version in the case of rebeccamycin) catalysed by the hemoproteins StaD or RebD.136 The P450s StaP or RebP are then responsible for catalysing the aryl–aryl coupling reaction to create a new central ring.137,138 Crystal studies have suggested, due to the potentially limited movement of the substrate within the active site, that a one-electron oxidation would be favoured over the typical P450 hydroxylation.139 This would facilitate proton abstraction from the indole nitrogen, which is followed by a second one-electron oxidation that triggers radical migration resulting in a formation of a new C–C bond. The required second indole proton abstraction could occur either before or after the formation of the new bond. Further work has also suggested that if the substrate does indeed have reduced access to the heme iron it could employ a histidine (H250) in conjunction with two water molecules to catalyse this C–C bond formation.140 Initial studies originally proposed that the subsequent oxidative decarboxylations were also catalysed by StaP or RebP. However, although it has now been established that these reactions can occur non-enzymatically137 they must be enzymatically-controlled in order to generate the desired product.137,141,142 For rebeccamycin the oxidation is facilitated by RebC, which produces 1,11-dichloroarcyriaflavin A and excludes the generation of all other products that can be formed in the absence of the enzyme. In contrast, StaC mediates a 4-electron oxidation to generate exclusively K252c the aglycone core that forms staurosporine. Finally, the biosynthesis of staurosporine also requires a second P450, StaN to initiate the formation of the second carbon–nitrogen bond between the sugar and the aglycone core.143,144 First, a glycosyltransferase StaG catalyses formation of the N-glycosidic bond that attaches the deoxysugar to the aglycone K252c to produce holyrine A. It is proposed that a StaN-mediated hydroxylation of the sugar moiety of holyrine A, followed by elimination of water creates an oxonium cation primed for attack by the nitrogen and consequently the formation of the second carbon–nitrogen bond. Alternatively, the oxonium ion may be formed directly from the radical intermediate generated by compound I H atom abstraction.
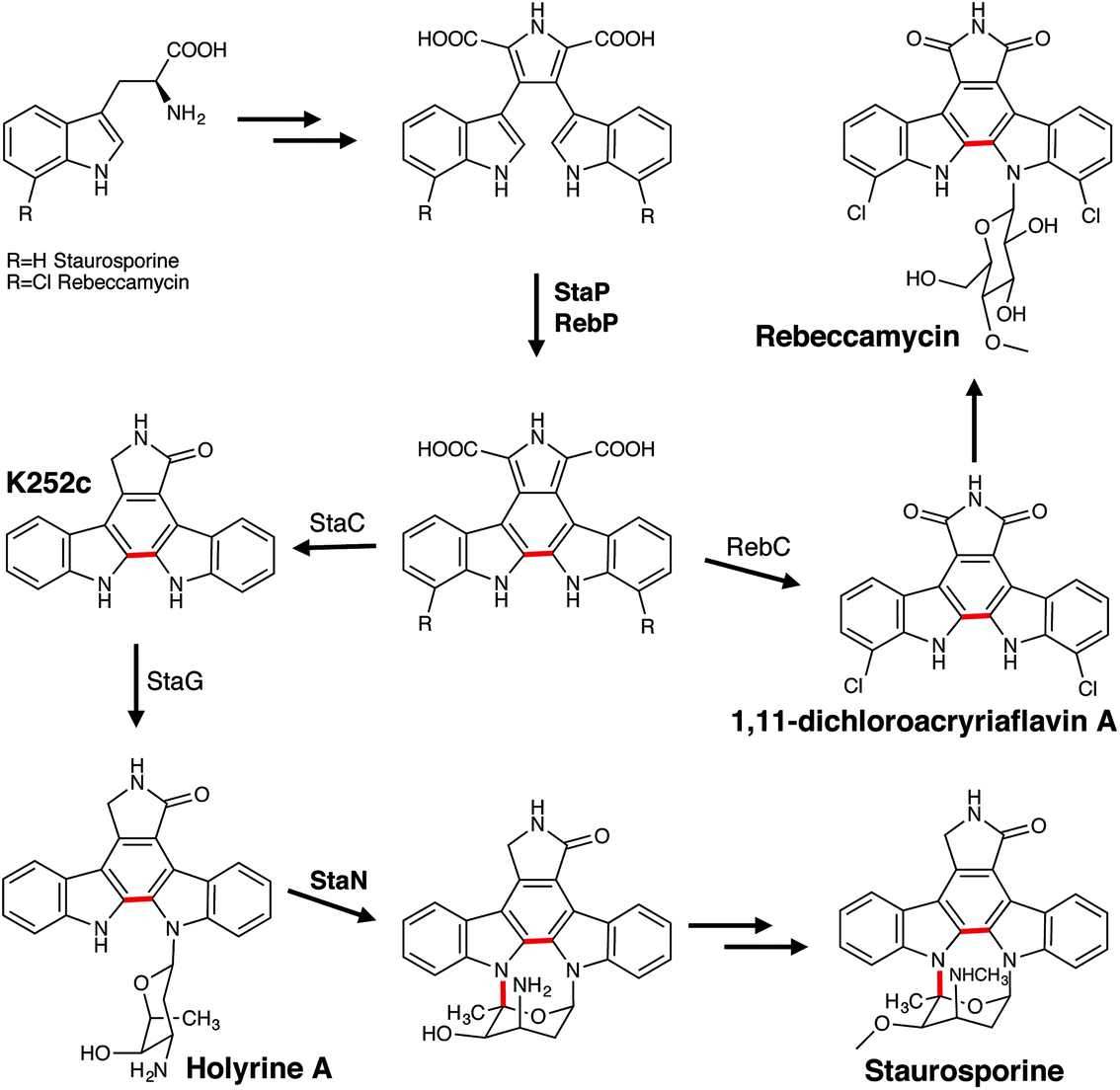 | ||
| Fig. 13 Roles of P450 enzymes in the biosynthesis of the indolocarbazole alkaloids rebeccamycin and staurosporine. P450 enzymes are in bold, the P450-catalysed reactions are indicated in red. | ||
3.4 Shikimate biosynthesis pathways
Shikimate biosynthesis involves the production of many compounds that contain aromatic amino acids and other molecules containing polyphenols. Traditionally, the shikimate pathway is associated with the production of lignins in plants and it has been proposed that bacteria are essential in the breakdown of these molecules in nature. Preliminary genomic analysis of bacteria-mediated lignin degradation has indicated that P450s in Rhizobium sp. strain YS-1r maybe be involved in this process.145 Bacterial P450s involved in biosynthetic pathways that include shikimate metabolites have been found associated with non-ribosomal peptide synthetases (NRPS; vide infra) and in alkaloid biosynthesis (vide supra). These enzymes catalyse the hydroxylation of aromatic amino acids such as tyrosine and tryptophan. However, more complex P450 reactions catalysing oxidative biaryl coupling during the generation of melanin are also known.P450mel (CYP107F1) from Streptomyces griseus, named for its role in the production of melanin, is an example of one of a P450 that catalyses one of these more complex reactions. It catalyses the oxidative coupling of two 1,3,6,8-tetrahydronaphthalene (THN) subunits to create 1,4,6,7,9,12-hexahydroxyperylene-3,10-quinone (HPQ) (Fig. 14).146 These HPQ units then autopolymerise to form HPQ-derived melanin, which is unlike other melanins that are typically formed from dihydroxyphenylalanine (DOPA) subunits. Again, like P450s such as CYP158 (vide infra), the formation of this new aryl–aryl bond is thought to occur via the creation of a phenolate radical and subsequent proton coupled electron transfer.
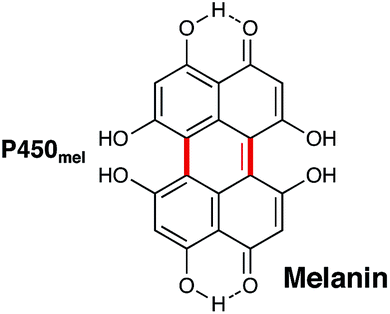 | ||
| Fig. 14 Structure of melanin, the biosynthesis of which involves P450mel in the oxidative dimerization of two 1,3,6,8-tetrahydronaphthalene subunits. The bonds introduced through P450-catalysed dimerisation are indicated in red. | ||
3.5 Polyketide (PKS) biosynthesis
Polyketides form one major class of secondary metabolites, and include many important examples such as the antibiotic erythromycin, the antifungal amphotericin, the immunosuppressant tacrolimus or the cholesterol-lowering agent lovastatin. They are synthesised by the sequential incorporation of malonyl-CoA extender units or derivatives into a growing polyketide chain by polyketide synthases (PKSs) through repetitive Claisen condensation reactions. Based on the architecture of the PKS, these assembly lines are divided into type I–III systems.147 Type I systems are very large, multifunctional proteins, where a set of different enzymatic domains build up one module and this module is responsible for extension by one extender unit. A minimal module consists of an acyltransferase (AT), an acyl carrier protein (ACP) and a ketosynthase (KS) domain. Further reducing domains may be present within a module, which modify the β-keto ester intermediate during assembly. The products are usually long polyketide chains, which remain linear or undergo further (macro)cyclisation after cleavage from the multi-enzymatic complex. In contrast, type II PKS are iteratively working enzymes, consisting of a ketosynthase, a chain length factor and an ACP. The final polyketide is then modified by cyclases/aromatases, leading basically to multicyclic compounds. PKS type III are small, multifunctional enzymes building small aromatic polyketides by the condensation of one starter unit with 2–3 extender units; furthermore, such systems are dependent on coenzyme A rather than the carrier protein domains found in other PKS systems.148Following the biosynthesis of the polyketide backbone of the PKS system, further modifications often take place: these modifications are generally related to glycosylation, the incorporation of other functional groups or further reduction/oxidation reactions. Due to the different length of the polyketide, their various cyclisation patterns, as well as further modifications, polyketides form a very large group of structurally diverse natural products. Within their biosynthesis, P450 enzymes play crucial roles, typically related to late-stage modifications, which can have a dramatic impact on the final bioactivity of the compound. Understanding P450-catalysed reactions within PKS biosynthesis (Fig. 15) is of great interest for efforts to modify existing compounds or develop new chemotherapeutic agents as, given their structural complexity, many of these medically important compounds have to be produced via fermentation of producer strains rather than synthesis.
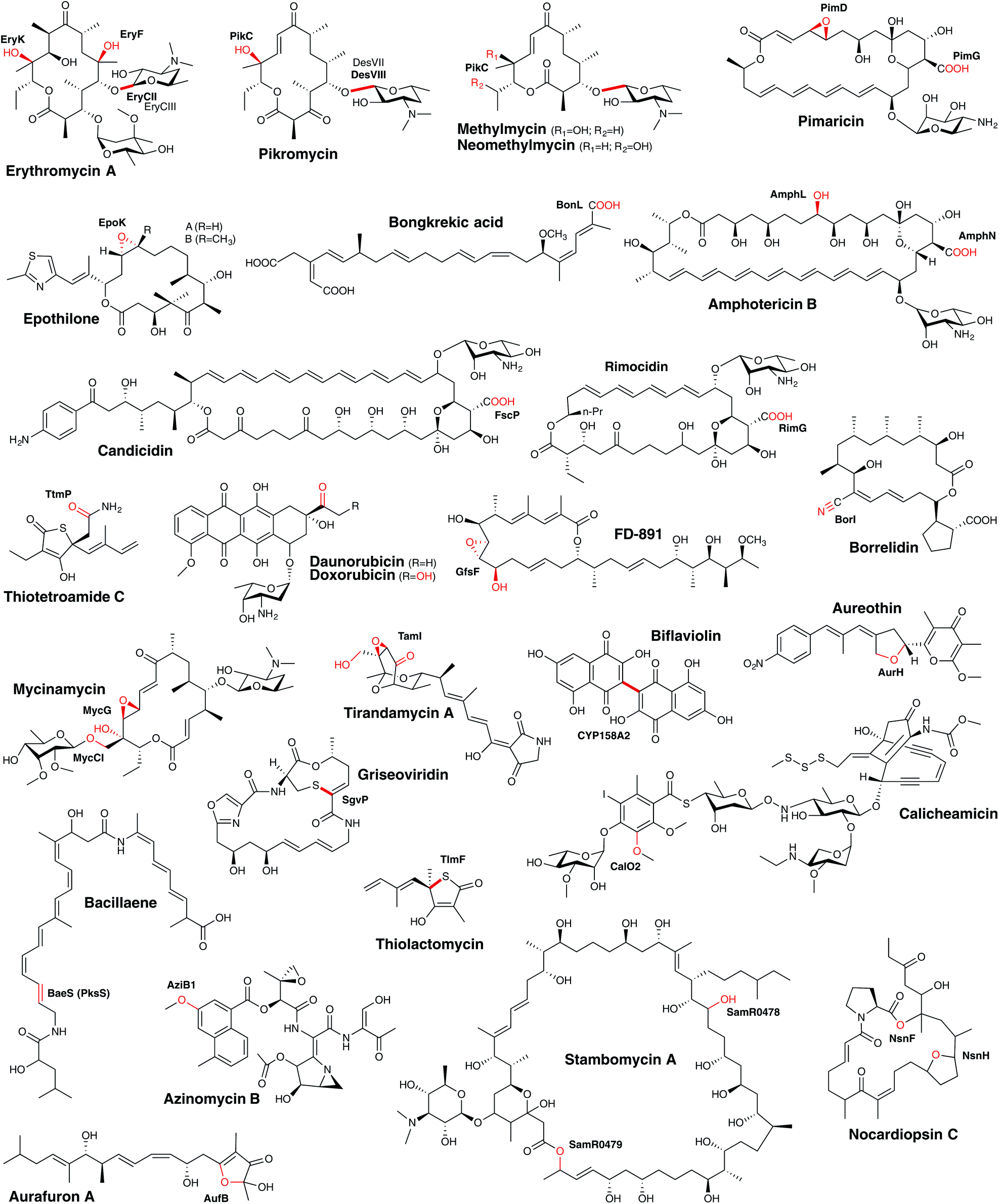 | ||
| Fig. 15 Examples of bacterial cytochrome P450 enzymes involved in PKS metabolism. The products of P450-catalysed reactions are indicated in red. | ||
Single P450 enzymes are also able to act on multiple sites within a substrate during the course of PKS biosynthesis. In the biosynthesis of daunorubicin and doxorubicin, DoxA is capable of installing an α-hydroxyketone moiety during the biosynthesis of these polycyclic aromatic compounds (Fig. 16A). Mechanistically, it has been shown that DoxA prefers substrates bearing 4-methoxy groups over 4-hydroxy groups and that the reaction proceeds initially by hydroxylation at the C13 methylene position. Subsequent oxidation at this position to generate the ketone affords daunorubicin, with further oxidation of the C14 methyl group to the α-hydroxyketone then giving rise to doxorubicin.166–168 Other complex examples of multi-step P450 reactions (combining hydroxylation and epoxidation) have been reported in the biosynthesis of the macrolides FD-891,169 mycinamycin (MycG)170 and tirandamycin (TamI).171 In the case of MycG, the two-step transformation preferentially begins with hydroxylation at C14, with subsequent epoxidation of the C12-C13 double bond (Fig. 16B). Substrates that have undergone epoxidation before hydroxylation are no longer able to be further processed by MycG, which means that in order to efficiently perform both transformations MycG must carefully control the orientation of the substrate such that the hydroxylation step – the chemically more challenging step – takes place before epoxidation of the double bond occurs. An even more complex process is catalysed by the P450 TamI in tirandamycin biosynthesis (Fig. 16C). In this case, TamI performs three oxidation steps that are coordinated with the action of the flavoprotein TamL. TamI performs the first reaction in the cascade, which is the hydroxylation of C10 within the TirC intermediate. This is then further oxidised by TamL at C10 to generate the ketone, which then undergoes two further TamI-mediated reactions: first, epoxidation of the C11–C12 double bond, and finally hydroxylation of the C18 methyl group to generate TirB. This example is surely one of the more impressive examples of the power and control that P450s can bring to biosynthetic transformations and also serves to demonstrate why fully unravelling such pathways can require extensive experimental endeavour.
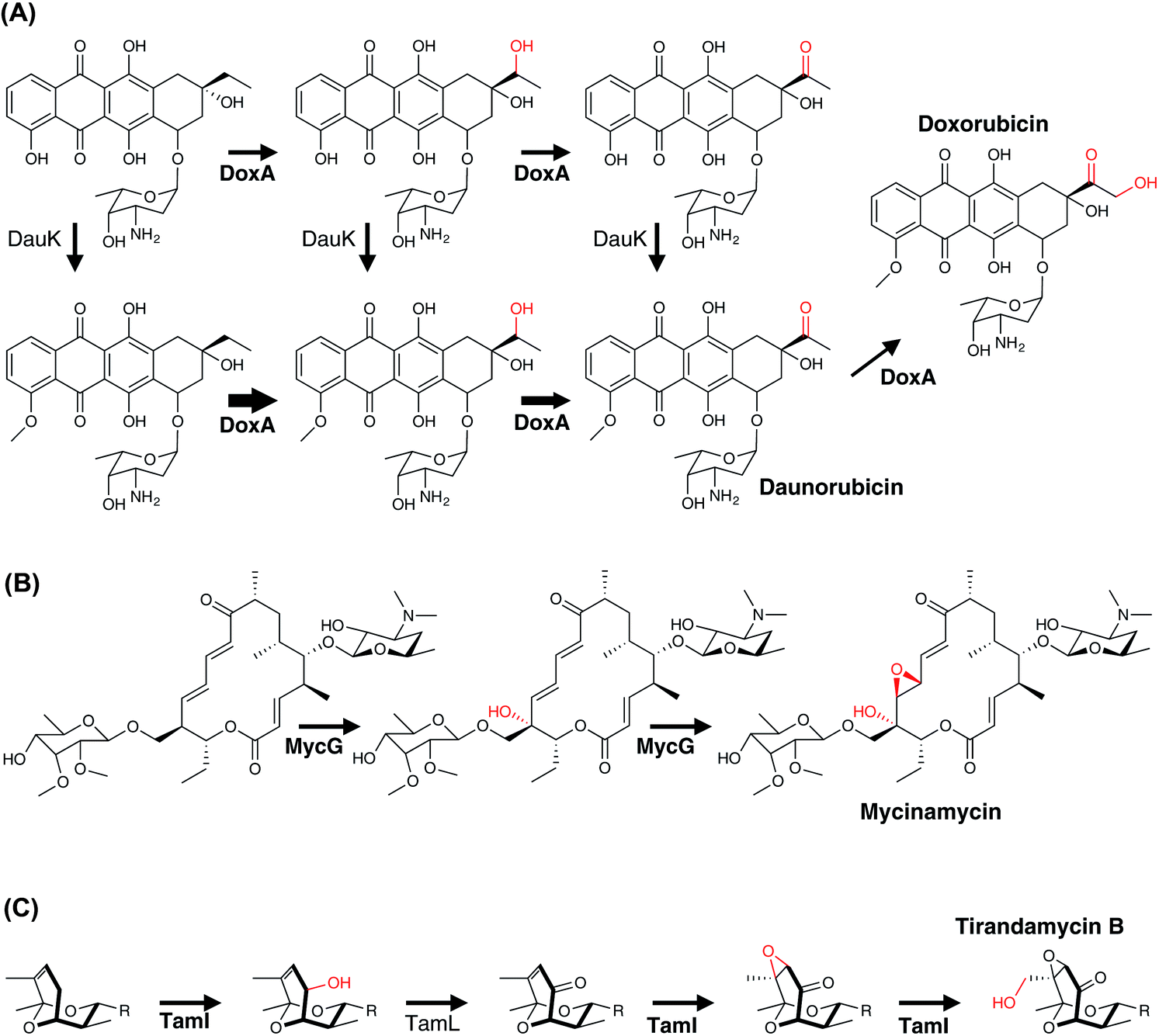 | ||
| Fig. 16 Examples of multi-step P450 reactions found in bacterial polyketide biosynthetic pathways. (A) Installation of an α-hydroxyketone moiety during the biosynthesis of doxorubicin by the P450 DoxA. The thickness of the arrow indicates the velocity of the reaction being catalysed. (B) Hydroxylation and subsequent epoxidation by the P450 MycG involved in mycinamycin biosynthesis. (C) Three-step oxidation by P450 TamI during tirandamycin B biosynthesis. P450 enzymes are indicated in bold and the oxygen atoms introduced by P450 activity are shown in red. | ||
3.5.3.1 Dehydrogenation – bacillaene biosynthesis. Bacillaene is an antibiotic produced by Bacillus subtilis and is produced by a large hybrid PKS/NRPS machinery that includes 16 modules (13/3, respectively) and a single cytochrome P450 (PksS/BaeS).172 Characterisation of both the molecules itself and the biosynthetic pathway responsible for production of this molecule were difficult, in part due to the instability of the compound and also the challenge of assigning the biosynthetic machinery to the structure of bacillaene itself. Whilst bacillaene contains two hydroxyl groups and nine double bonds, no obvious role for the P450 could be identified (either hydroxylation or epoxidation). Instead, reconstitution of the activity of this P450 using a cytosolic extract of B. subtilis and dihydrobacillaene indicated that the role of PksS is to introduce the double bond at C14, C15 into dihydrobacillaene (Fig. 17A), affording bacillaene itself.173 Such a system is naturally highly challenging to investigate due to the difficulty in obtaining modified substrates to probe the activity of this P450, however recent work on other systems has shown that P450s are able to directly catalyse the insertion of double bonds into substrates without requiring an intermediate hydroxylation step.174 Whilst the elimination of an intermediate hydroxylated species could also represent a viable route towards the formation of hydrobacillaene (especially due to the facile nature of dehydration and the stability of the resultant conjugated alkene), the possibility remains that PksS is another example of a P450 that directly generates alkene products: it is also important to recognise that the appearance of double bonds within a final compound can also arise from the activity of P450 enzymes.
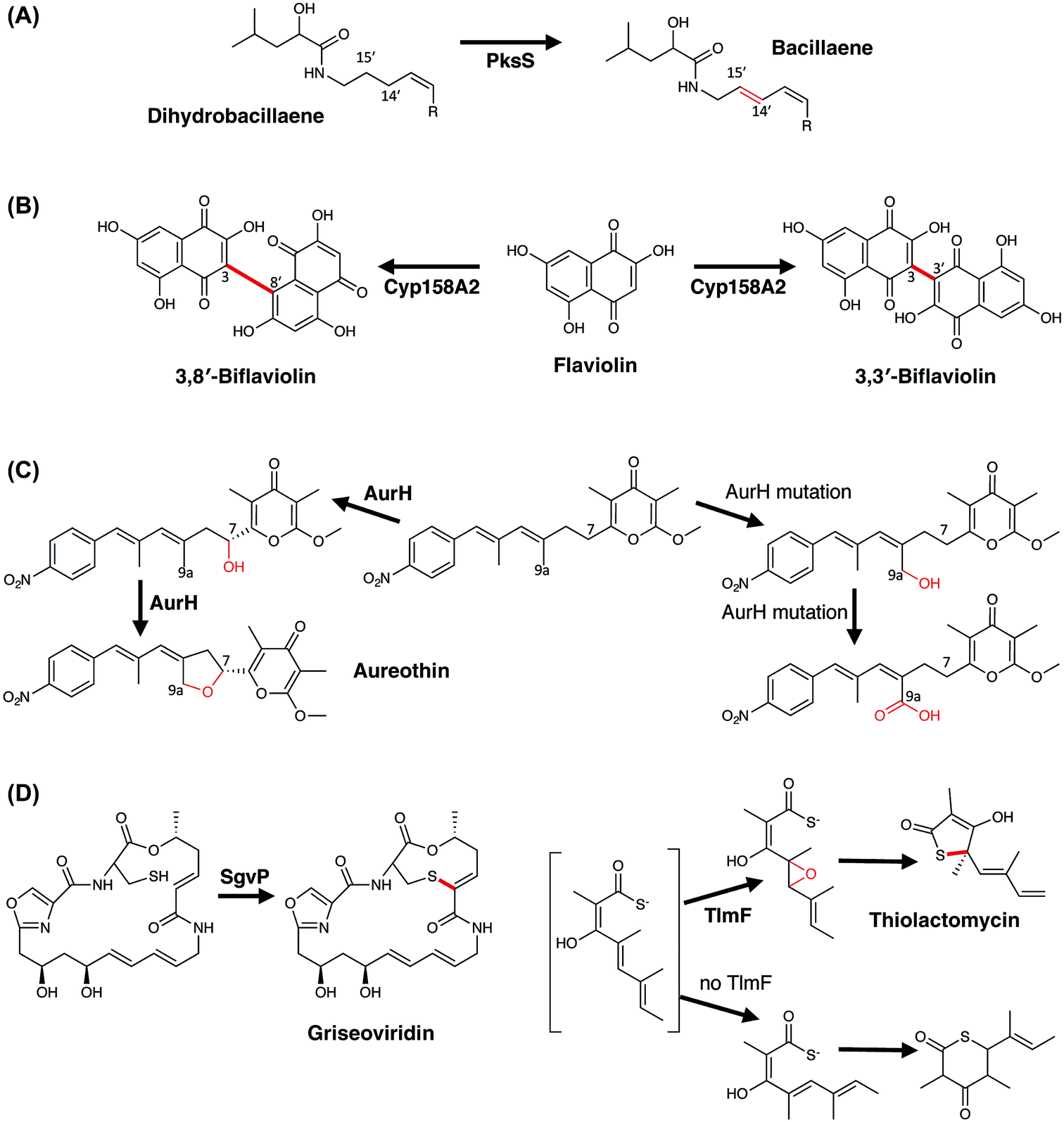 | ||
| Fig. 17 Examples of less common P450-catalysed transformations found in bacterial PKS biosynthesis. (A) Introduction of a double-bond into bacillaene by the P450 PksS. (B) Dimerisation of flaviolin by the P450 CYP158A2 via the 3,3′-positions and 3,8′-positions. (C) tetrahydrofuran ring formation in aureothin biosynthesis by P450 AurH. (D) C–S bond formation in griseoviridin (left) and thiolactomycin (right) catalysed by P450s SgvP and TlmF, respectively. In the absence of TlmF, a six-membered thiolactone shunt product is formed. P450 catalysed reactions are shown in red. | ||
3.5.3.2 Aromatic crosslinking – oxidative dimerisation of flaviolin. Amongst their oxidative chemical repertoire, P450s are also capable of performing the oxidative crosslinking of aromatic rings. In PKS biosynthesis, the most well studied examples are found in the biosynthesis of pigments, which in the case of Streptomyces coelicolor includes the dimerisation of flaviolin performed by CYP158A2 (ref. 175) and likely by CYP158A1.176 The biochemical characterisation of CYP158A2 showed that flaviolin binds tightly to this P450 and causes a type-I (activation) spectral shift in the P450.175 Furthermore, the incubation of CYP158A2 and flaviolin together with a functional electron transfer chain (composed of E. coli flavodoxin and flavodoxin reductase) demonstrated that this P450 could produce species with a mass that corresponded to flaviolin dimers and trimers: two of the dimeric species were further characterised and this revealed that the differences between these molecules were the positions through which the monomers were joined (via the 3,3′-positions and 3,8′-positions, respectively) (Fig. 17B).175 The structural characterisation of this enzyme in complex with flaviolin revealed two molecules of flaviolin in the enzyme active site, with these stacked over the heme within the P450 active site. From a mechanistic perspective, the crosslinking reaction likely occurs by hydrogen abstraction from the ring closest to the heme iron, with subsequent generation of a radical species in the distal flaviolin occurring either via proton coupled electron transfer or electron tunnelling as has been postulated for the crosslinking reaction performed by CYP121 (vide infra).177 A further interesting note in the investigation of CYP158 enzymes has been their use in mechanistic studies, where the Green group has been able to use these enzymes to investigate the highly reactive intermediates that are found within the P450 active cycle.17
3.5.3.3 Tetrahydrofuran ring formation – aureothin biosynthesis. Aureothin is a polyketide metabolite of Streptomyces thioluteus that displays various activities, including antifungal, cytotoxic, and insecticidal activity. Within the biosynthesis of this compound the major mechanistic question concerns the formation of the tetrahydrofuran in the final product,178,179 with comparable processes also implicated in the biosyntheses of avermectin180 and platensimycin.181 Several studies have now shown that this moiety is installed by the P450 enzyme AurH, which effectively is able to install two C–O bonds in order to achieve this transformation.178,179 The reaction mechanism adopted by this P450 has been shown to commence with the specific R-hydroxylation of the C-7 methylene group, with the stereochemistry of this position then maintained in the final tetrahydrofuran ring.178 Subsequently, oxidation of the C-9a methyl group occurs together with concomitant production of the tetrahydrofuran ring (Fig. 17C): the ability of AurH to oxidise C-9a has been demonstrated in vitro, where active site mutants of AurH that prevent oxidation of the more hindered C-7 position then lead to the isolation of C-9a oxidised derivatives not normally identified in the biosynthetic pathway of aureothin, bearing hydroxyl, aldehyde or acid moieties.182 Alteration of the starting material for AurH by changing the methylation of the precursor has shown that this P450 will perform sequential oxidation at C-7, with resultant electrocyclic formation of the pyran ring in aureopyran.183 Such experiments also show the importance of substrate binding and orientation in P450 enzymes, as in the case of AurH multiple oxidations at C-7 and C-9a are both clearly possible and would lead to complex mixtures of various products if the oxidative cascade was not carefully optimized for the natural 2-step reaction.
3.5.3.4 C–S bond formation – griseoviridin and thiolactomycin biosynthesis. C–S bond formation is a relatively rare P450-catalysed reaction; however, a number of examples have recently been identified including two examples from bacterial PKS (mixed NRPS/PKS) biosynthesis. Griseoviridin is a streptogramin-type antibiotic produced by Streptomyces griseoviridis and contains a bicyclic structure that is formed by a thioether bond between the side chain of the terminal cysteine residue and the double bond of the alkene installed by the second PKS module.184 A P450 (SgvP) has been implicated in the synthesis of this thioether bridge,185,186 with gene disruption experiments showing the essential nature of this P450 in thioether formation (Fig. 17D).184 Mechanistically, it remains unclear whether this is a diradical coupling mediated process, proceeds by initial epoxidation of the C2–C3 alkene or perhaps involves a sulfenic acid intermediate, and clarification of this pathway awaits further investigation.185 The mechanism of C–S bond formation in the mixed NRPS/PKS biosynthesis of thiolactomycin – a thiotetronate antibiotic produced by several strains of Streptomyces and Salinispora – presents an even more complex case.187,188 Here, the P450 TlmF has been shown to be required for the formation of the five-membered thiolactone ring present in thiolactomycin.188,189 This ring, found at the C-terminus of the molecule, contains a sulfur atom that originates from the side chain of cysteine that is selected and activated by the final module of the PKS/NRPS. Following this, the exact mechanism by which the sulfur is inserted into the molecule is unclear, although the role of the P450 is believed to centre on the epoxidation of the C4–C5 double bond (Fig. 17D). Lack of P450 activity leads to an alternate product being produced, in which a six-membered thiolactone ring is formed. Two postulates then have been proposed for the fate of the intermediate epoxide: direct attack of a thiaacid to give the thiolactone; or the intermediate generation of a thiirane that then attacks the PKS bound thioester leading to the formation of thiolactomycin. Whilst further studies are clearly warranted in this highly interesting biosynthetic pathway, some evidence has been gained in support of the thiirane intermediate from the use of synthetic probes which have identified an intermediate that could result from thiirane hydrolysis.190
From these examples, it is clear that the role of P450s within PKS biosynthesis in bacteria can significantly diverge from what would be considered standard P450 hydroxylation and epoxidation chemistry. Beyond the important implications that such reactions have for potential biotechnological applications, these reactions also serve as a salient reminder for those wishing to assign the roles of P450 identified in PKS biosynthesis clusters in silico to be aware of the chemical diversity that P450s can display and also highlights the importance of testing such hypotheses using experimental techniques.
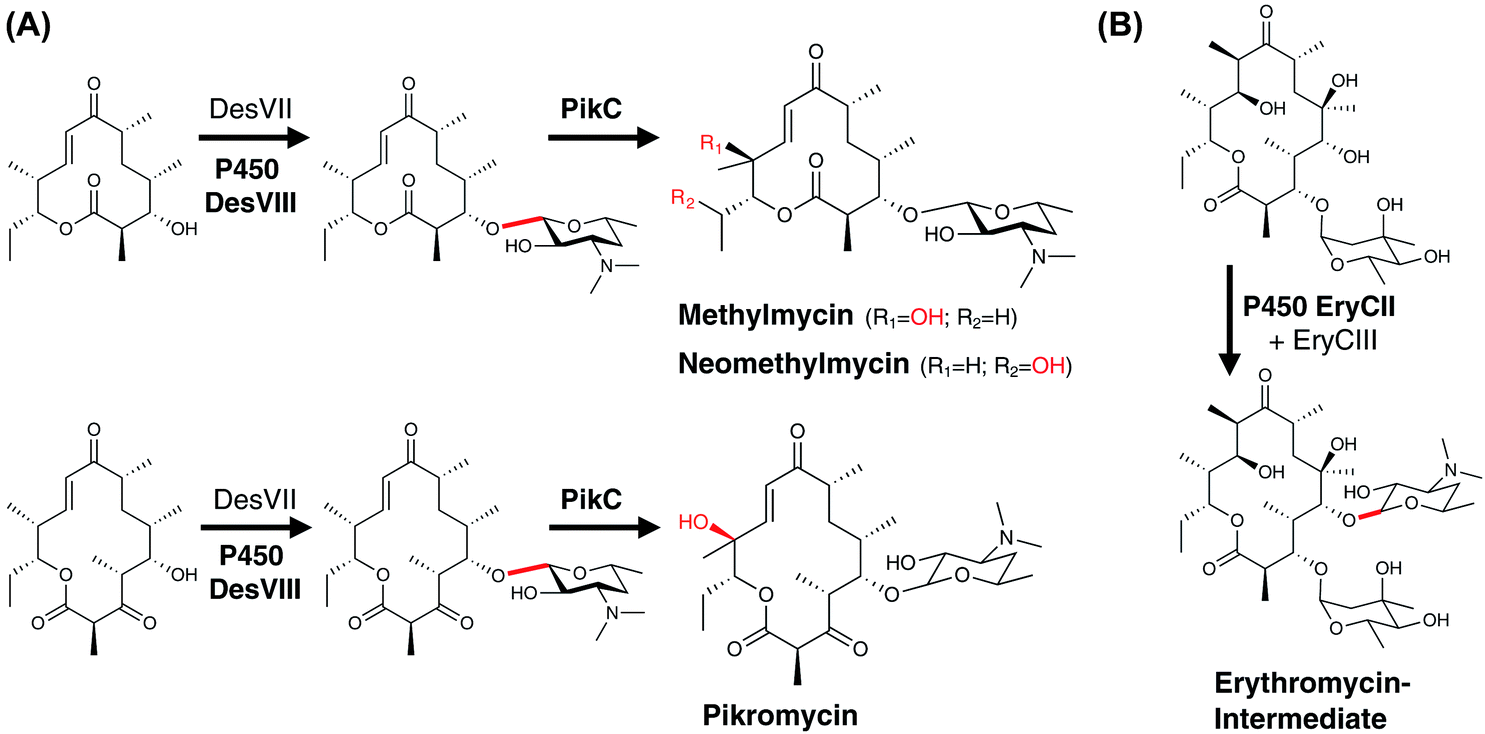 | ||
| Fig. 18 Examples of P450-mediated protein recruitment during PKS modification. (A) The activity of the glycosyltransferase DesVII in methylmycin/pikromycin biosynthesis is dependent upon the presence of the P450-like protein DesVIII. Subsequently, the compounds are then hydroxylated by the P450 PikC. (B) In erythromycin biosynthesis, the glycosyltransferase EryCIII interacts with the P450-like protein EryCII. P450 catalysed reactions are shown in red and the P450 enzymes are indicated in bold. | ||
3.6 Peptide biosynthesis pathways
Polypeptides are another major group of secondary metabolites in which P450s are implicated in their biosynthesis. Such peptides can be biosynthesised in a number of ways: in addition to ribosomally synthesised and post-translationally modified peptides (RiPPs), small peptides can be synthesised in a tRNA-dependent manner without the involvement of the ribosome, whilst another major class of peptide biosynthesis is performed by non-ribosomal peptide synthetases (NRPS) – megaenzymes that have no reliance on tRNA or the ribosome for peptide synthesis. In all cases, P450s in bacterial biosynthetic pathways play important roles in modifying these peptide natural products. | ||
| Fig. 19 Examples of P450 enzymes involved in bacterial RiPP metabolism. P450 enzymes are indicated in bold and the P450-catalysed reactions are shown in red. | ||
3.6.2.1 NRPS-bound substrates.
3.6.2.1.1 Modification of PCP-bound amino acids. In contrast to the ribosomal synthesised peptides, NRPS machineries are not limited to accepting proteinogenic amino acids, but can also activate and incorporate a wide range of unusual, non-proteinogenic amino acids (Fig. 20).215 Furthermore, modified amino acids such as β-hydroxy amino acids have been identified in a number of non-ribosomal peptides, including the vancomycin-type antibiotics (Tyr),216–219 the aminocoumarin-containing angucycline antibiotics (Tyr; novobiocin, simocyclinone, coumermycin, clorobiocin),220 echinomycin (plus other compounds that contain the intercalative chromophores quinoxaline-2-carboxylic acid (QXC) or 3-hydroxyquinaldic acid (HQA);221 Trp),222,223 zorbamycin (Val)224 and nikkomycin (His).225,226 Common to all these pathways is an NRPS/P450 dependant process for generating these β-hydroxy amino acids for incorporation into the final natural product. This system comprises a di-domain NRPS module of an adenylation domain and subsequent PCP domain, which is responsible for selecting and activating the requisite amino acid and loading it onto the PCP-domain. This PCP-domain is then recognised by the downstream P450 enzyme, which performs the β-hydroxylation of the tethered amino acid. Subsequently, the modified amino acid can either be hydrolysed by a separate thioesterase domain for selection by separate NRPS machinery (as occurs for example in echinomycin or glycopeptide antibiotic (GPA) biosynthesis (Fig. 21A)227 or utilised directly for further modification (as occurs in aminocoumarin biosynthetic pathways).228 The P450 enzymes in these systems recognise the PCP-domain of the di-domain NRPS module rather than the loaded amino acid, with such P450s also maintaining a rigid PCP-binding site that consists of a small number of highly conserved residues, allowing the function of such P450s to be predicted largely on the basis of sequence comparison alone. A similar process is believed to be at play during the biosynthesis of hectochlorin (mixed NRPS/PKS) where the PCP-bound substrates are believed to be 2-oxo-isovaleric acid.229 An example of a two-step transformation of a PCP-bound amino acid has been suggested to occur in the biosynthesis of the cyclic peptides JBIR-34 and JBIR-35.230 In this pathway, the di-domain NRPS module FmoA1 presents a tryptophan residue (or chlorinated derivative) to the P450 FmoC, which performs two oxidation reactions; hydroxylation of the indole ring as well as β-hydroxylation.230 Cleavage of the dihydroxylated product would then be performed by the thioesterase FmoB (Fig. 25A). This di-hydroxylation shares similarity to that catalysed by TxtE in thaxtomin A biosynthesis (vide infra), although in the case of FmoC the substrate amino acid is PCP-bound.231
P450-mediated hydroxylation of NRPS-produced peptides can also occur via direct hydroxylation of PCP-bound amino acids during their incorporation into the parent NRP: in the first identified case – that of skyllamycin biosynthesis232,233 – P450sky (CYP163B3) was found to be responsible for the β-hydroxylation of L-Phe, OMe–L-Tyr and L-Leu residues attached to NRPS modules 5, 7 and 11, respectively.234 P450 selectivity in this system is again based on the PCP domains involved, with tolerance for different amino acids as substrates when these were loaded onto PCP domains that are recognised by this P450.234–236 A further example of this amino acid hydroxylation strategy has been identified in telomycin biosynthesis, in which the P450 enzyme Tem23 hydroxylates the Leu-10 residue during NRPS-mediated biosynthesis of the peptide.237 A related strategy to this has also been invoked in hydroxylation of a PCP-bound threonine residue during actinomycin G biosynthesis, although in this case it appears as though there is a relationship between the P450 (AcmG8) and a halogenase that modifies the same position (AcmG9) that needs further clarification.238 A potential modification of this strategy of targeting PCP-bound amino acids has been identified in the biosynthesis of the mixed NRPS/PKS α,β-epoxyketone proteasome inhibitor TMC-86A.239 In this case, evidence suggests that P450 TmcK introduces a double bond into the side chain of Leu-2 after the amino acid is PCP-bound, with further experiments no doubt required to probe this unusual PCP-bound transformation.239
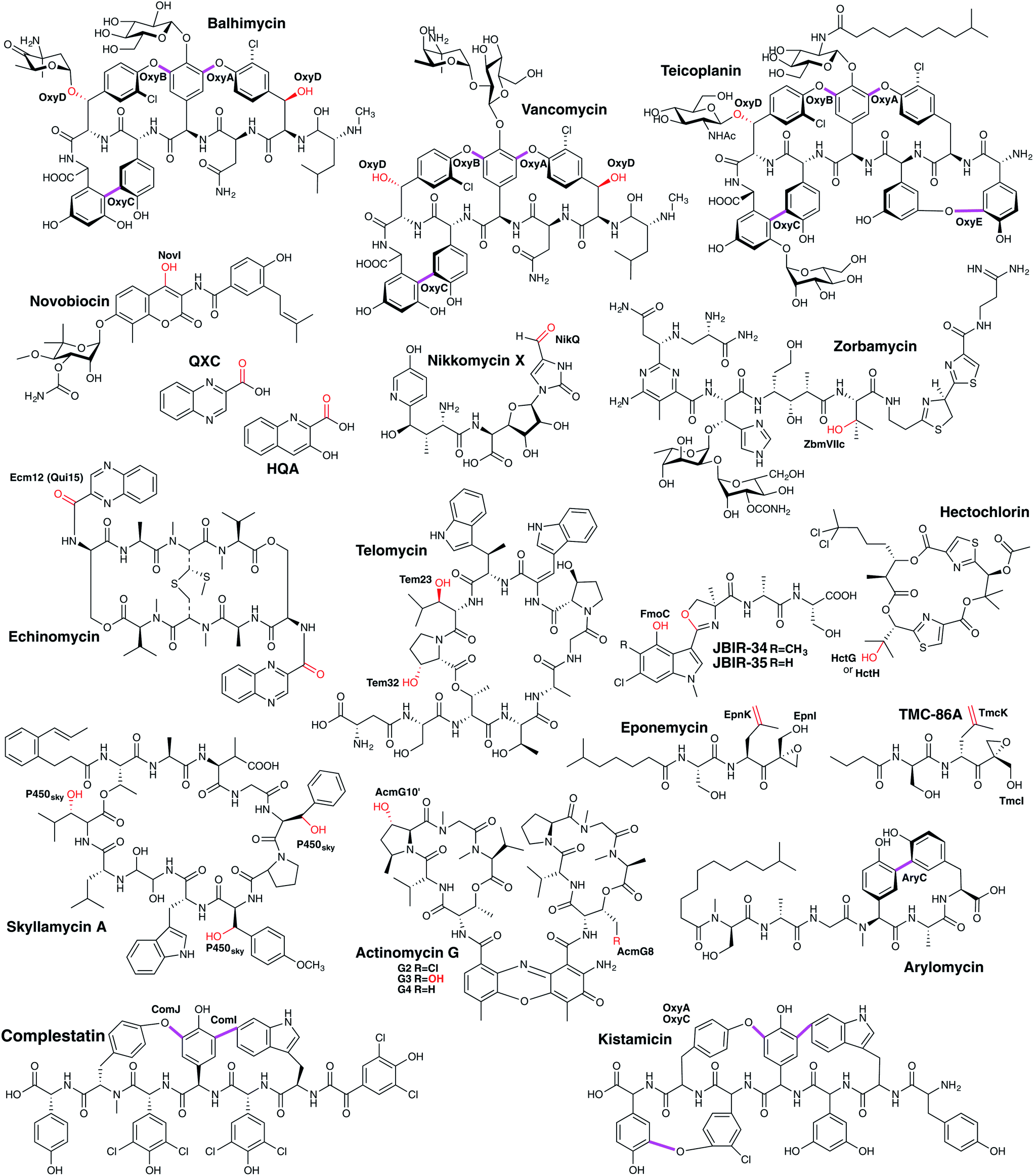 | ||
| Fig. 20 Examples of P450 enzymes involved in the modification of PCP-bound amino acids and peptides in NRPS metabolism. P450 enzymes are indicated in bold, the P450-catalysed reactions are shown in red and the phenolic crosslinking of PCP-bound peptides are shown in magenta. | ||
3.6.2.1.2 Phenolic coupling of PCP-bound peptides. In addition to the relatively common P450-catalysed β-hydroxylation of PCP-bound amino acids, one further major class of P450-mediated transformations against PCP-bound substrates is the oxidative crosslinking of aromatic side chains within PCP-bound NRPS peptides. The most well-known compound class that exhibits multiple aromatic crosslinks that are installed by P450 enzymes are the glycopeptide antibiotics (GPAs), which include the representative examples vancomycin and teicoplanin.240–243 The intramolecular cyclisation reactions catalysed by cytochrome P450 enzymes lead to a specific tertiary structure able to bind to the terminal D-Ala–D-Ala motif of the bacterial cell wall precursor lipid II and thereby blocking cell growth. As last-line antibiotics, they are the major treatment against multiresistant gram-positive pathogens like methicillin-resistant Staphylococcus aureus (MRSA). In these NRPS-biosynthesised heptapeptides, the three (vancomycin-type) or four (teicoplanin-type) crosslinks have been demonstrated to be inserted in vivo by cytochrome P450 enzymes, with each ring installed by a specific P450.244–248 Furthermore, gene disruption experiments determined that a specific order of ring installation occurs during GPA biosynthesis (Fig. 21B). Ring insertion commences with generation of the C–O–D ring by OxyB, with subsequent activity of the optional OxyE enzyme (installing the teicoplanin-type F–O–G ring) and the essential OxyA enzyme (installing the D–O–E ring), before terminating with insertion of the AB crosslink that is catalysed by OxyC.244–249 These experiments also led to the hypothesis that the crosslinking performed by these P450 enzymes was likely to occur against NRPS bound (i.e. PCP-bound) peptides, which was confirmed for the OxyB enzyme from vancomycin biosynthesis by the work of Robinson and co-workers.250–252 More recent work has shown that the process of P450 recruitment to the NRPS-bound peptide is more complicated than is the case for PCP-bound amino acids, with GPA biosynthesis relying on the X-domain, a conserved (albeit modified) condensation domain found between the PCP253 and TE-domains in the final NRPS module.243,254–257 The X-domain has an interaction interface that specifically binds to the Oxy enzymes, with these continually binding to and releasing from the X-domain to ensure that complete crosslinking of the peptide occurs.254 The X-domain is also required in the majority of cases to support efficient P450-catalysed crosslinking in vitro, with this requirement strictly enforced for OxyA and OxyE that act after OxyB.243,255,256,258–263 Complestatin264 and kistamicin265 are structurally similar to GPAs, and also display several oxidative crosslinks whose generation has been ascribed to P450 enzymes, i.e. NRPS-bound modification with X-domain mediated P450 recruitment. The initial ring inserted in complestatin is the equivalent of the GPA C–O–D ring and is performed by the P450 enzyme ComJ, with the C–C bond formed between and Trp and Hpg residues in complestatin installed by P450 enzyme ComI.266,267 Kistamicin is similar in structure to complestatin although it contains an additional A–O–B ring: curiously, only two P450 enzymes were identified in the kistamicin gene cluster, implying a dual role for one of these P450 enzymes.265 Another example of a P450-mediated crosslinking of aromatic side chains within a peptide is found in arylomycin biosynthesis, in which a C–C bond is formed between the Hpg-4 and Tyr-6 residues whilst the peptide remains bound to the NRPS.268,269 Here, there is no X-domain identified in the NRPS machinery, presumably due to the single P450-catalysed modification occurring in this case.268,269
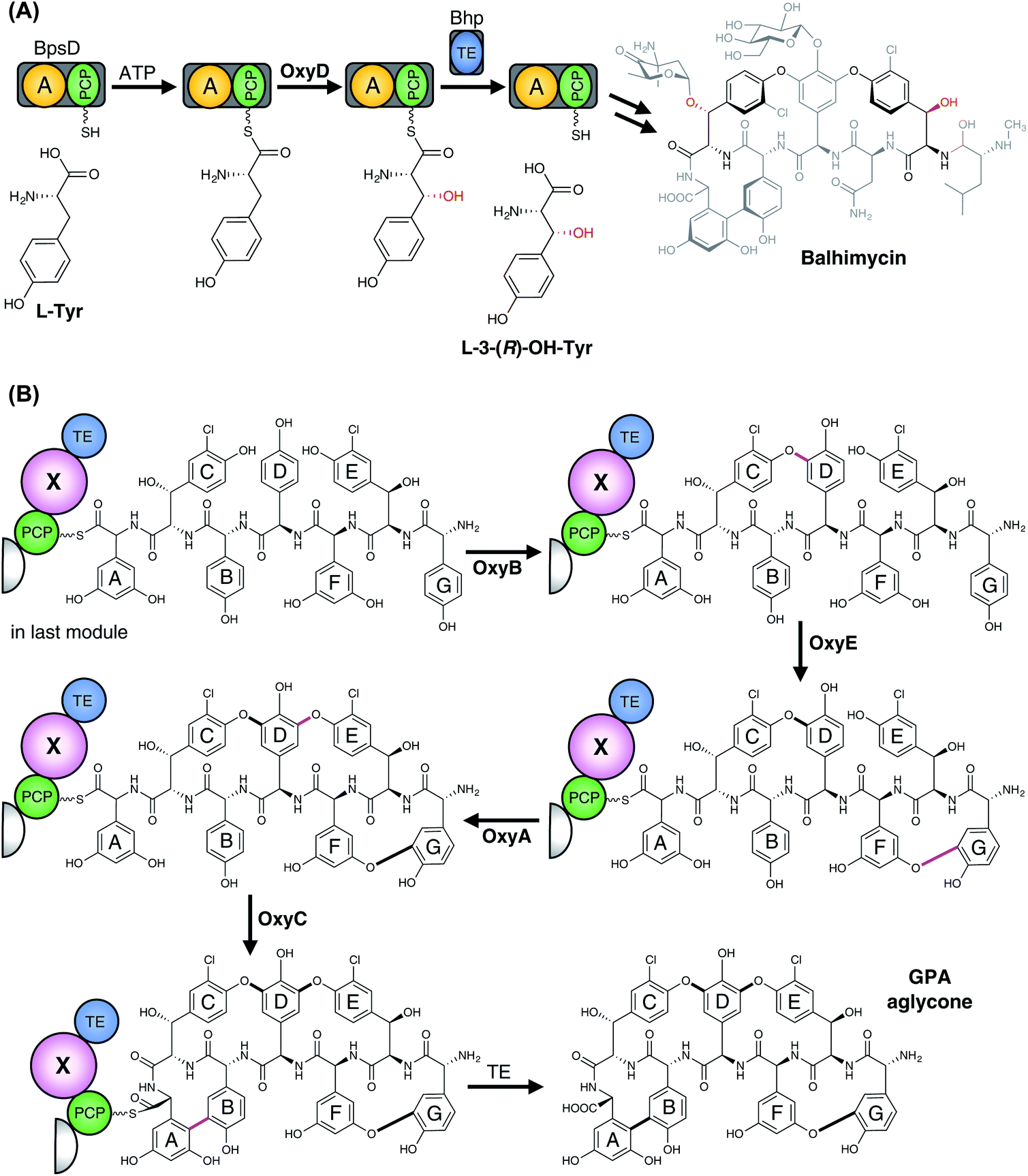 | ||
| Fig. 21 P450 enzymes involved in glycopeptide antibiotics biosynthesis. (A) In balhimycin biosynthesis, two β-hydroxytyrosine residues are incorporated into the heptapeptide compound through the actions of an NRPS. The amino acid (L-Tyr) is initially selected, activated and loaded onto the di-domain BpsD, which consists of an A- and a PCP-domain. After β-hydroxylation of the tethered amino acid, this is then hydrolysed from the PCP-domain by the separate thioesterase Bhp and subsequently used by the NRPS system for assembly of the heptapeptide. Incorporated OH–Tyr are indicated in the final balhimycin compound (grey). (B) Following assembly of the heptapeptide chain in GPA biosynthesis, the three (vancomycin-type GPA) or four (teicoplanin-type GPA) crosslinks of the aglycone are installed by different P450 enzymes, which occurs in a specific order when the peptide is still bound to the PCP of the last module of the NRPS assembly line. The X-domain present within the final module of GPA NRPS assembly lines is a unique interaction platform for these P450 enzymes. The P450 cyclisation cascade first proceeds with installation of the C–O–D ring by OxyB, then F–O–G ring (teicoplanin-type GPAs only) insertion catalysed by OxyE, the D–O–E catalysed by OxyA and finally the A–B ring formation catalysed by OxyC. The P450-catalysed crosslinkings are indicated in purple. A, adenylation domain; PCP, peptidyl carrier protein; TE, thioesterase domain; X, P450 recruitment domain. | ||
3.6.2.2 Peptide modification. P450-catalysed modification of peptides within NRPS pathways have also been identified in numerous systems (Fig. 22), some of which share similarities with those identified in RiPP pathways.
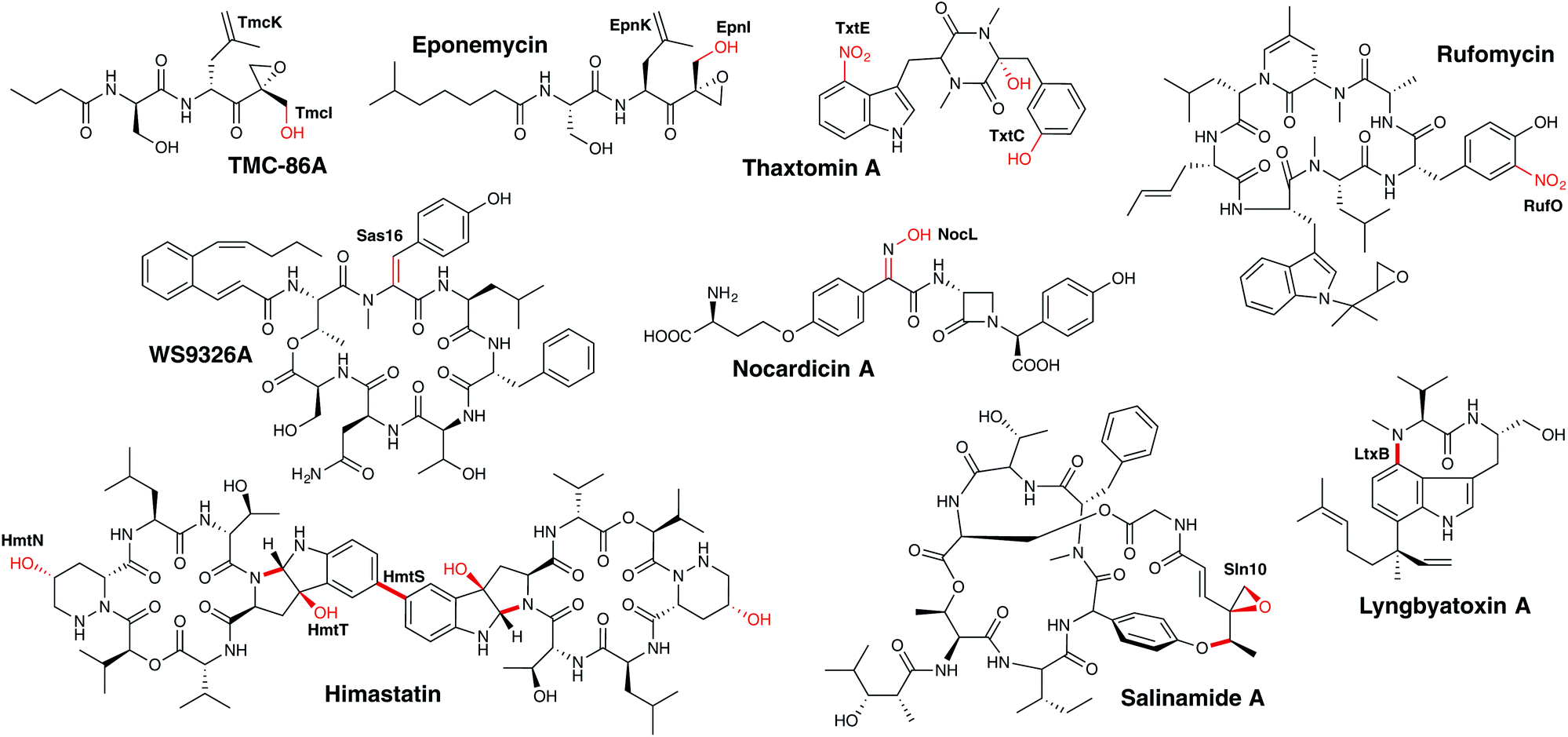 | ||
| Fig. 22 Examples of P450 enzymes involved in the modification of peptides (PCP-independent) in NRPS metabolism. P450 enzymes are indicated in bold, the P450-catalysed reactions are shown in red. | ||
3.6.2.2.1 Peptide hydroxylation – TMC-86A biosynthesis. The archetypal P450-catalysed reaction – the selective hydroxylation of unactivated C–H bonds – has also been identified in the modification of NRPS-produced peptides that occurs after cleavage of the peptide chain from the NRPS machinery (see also himastatin, vide infra).270,271 An example of this chemistry is found in the biosynthesis of the α,β-epoxyketone proteasome inhibitor TMC-86A (a mixed NRPS/PKS pathway), in which the P450 enzyme TmcI performs the hydroxylation of the methyl group adjacent to the epoxy moiety in this compound.239 The biosynthesis of the related compound eponemycin also contains a homologous P450 that is believed to perform the analogous reaction; such a P450 is absent in the epoxomicin gene cluster that lacks this hydroxyl group.272
3.6.2.2.2 Introduction of double bonds – WS9326A biosynthesis. The P450 Sas16 is proposed to be involved in the biosynthesis of the (E)-2,3-dehydrotyrosine residue found in WS9326A and its derivatives (Fig. 22).273 The deletion of the gene led to mutants producing a derivative containing a standard tyrosine residue without the double bond. The exact nature of the substrate of Sas16, for example whether it accepts on free tyrosine or PCP-bound intermediates thereof, requires further investigation.273
3.6.2.2.3 Aromatic nitration – rufomycin biosynthesis. One of the more unusual examples of a P450-catalysed transformation is the nitration of aromatic rings, which was initially identified in thaxtomin A biosynthesis in the nitration of a tryptophan residue (vide infra)274 and has now also been reported in the nitration of a tyrosine residue in rufomycin biosynthesis.275 A cyclic heptapeptide, rufomycin contains a number of unusual amino acid building blocks, with two – 3-nitrotyrosine and trans 2-crotylglycine – utilising P450s in their formation. 3-Nitrotyrosine is formed by the P450 RufO using nitric oxide that is formed by the neighbouring RufN nitric oxide synthase and is believed to occur prior to the amino acid being selected and activated by the NRPS machinery (Fig. 23A).275 Incidentally, two further P450s have also been implicated in oxidative transformations of the cyclic rufomycin peptide, which include the epoxidation of the N-dimethylallyltryptophan residue and the multistep oxidation of a Leu-5 methyl group into a carboxylic acid moiety.275
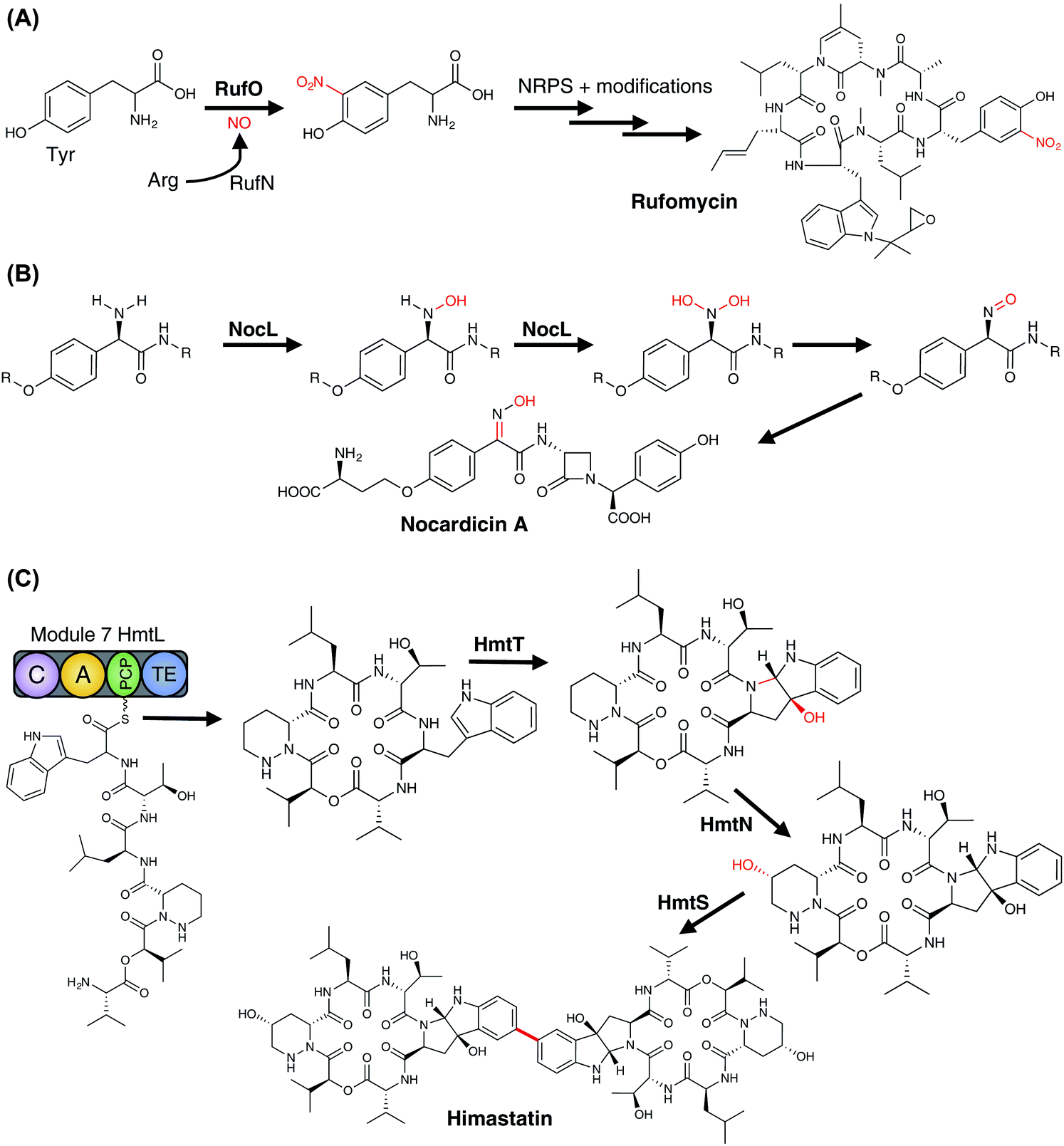 | ||
| Fig. 23 P450 involved in the modification of non-ribosomal peptides. (A) In rufomycin biosynthesis, the P450 RufO catalyses the nitration of a tyrosine residue. This P450 enzyme uses nitric oxide that is formed from arginine by the nitric oxide synthase RufN. (B) In norcardicin biosynthesis, the P450 enzyme NocL performs two N-hydroxylation reactions, followed by spontaneous dehydration to afford the oxime. (C) In himastatin biosynthesis, there are multiple transformations catalysed by P450 enzymes. The P450 HmtT generates hexahydropyrroloindole from the 6-Trp residue in the cyclic hexadepsipeptide precursor. Subsequently, the P450 HmtN catalyses hydroxylation and finally the P450 HmtS performs the symmetrical biaryl coupling of two cyclic hexadepsipeptide precursors to generate himastatin. P450 enzymes are indicated in bold, the P450-catalysed reactions are shown in red. A, adenylation domain; C, condensation domain; PCP, peptidyl carrier protein; TE, thioesterase domain. | ||
3.6.2.2.4 Heteroatom oxidation – nocardicin biosynthesis. Nocardicin biosynthesis contains many examples of novel biosynthetic chemistry,276 including several examples of unusual NRPS-catalysed reactions,277,278 an atypical SAM transferase279 and a cytochrome P450, which has been shown to perform the oxidative transformation of the amino group of the N-terminal Hpg moiety into an oxime.280 The P450 in question, NocL, has been shown to perform this transformation in vitro, with the reaction presumably following a two-step mechanism that begins with the hydroxylation of nocardicin C to 2′-N-hydroxy-nocardicin C. A second hydroxylation to afford 2′-N,N-dihydroxy-nocardicin C is hypothesised to then be followed by dehydration to afford a nitroso species that then tautomerises to provide the oxime of nocardicin A (Fig. 23B).280 This transformation is analogous to the CYP79 catalysed formation of oximes from amino acids in plants, except that in this case it is coupled decarboxylation-dehydration that leads to the nitroso intermediate.281 Heteroatom oxidation by P450 enzymes has been widely reported,12 and although such reactions are often found in xenobiotic metabolism, NocL serves as an important reminder that the role of P450s in heteroatom oxidation during secondary metabolism cannot be discounted.
3.6.2.2.5 Heterocyclic ring formation. P450 activity that generates bonds between heteroatoms and aryl groups within peptides is well known from P450s that interact directly with NRPS-bound peptides, such as occurs in GPA biosynthesis. This type of chemistry has also been identified in the biosynthesis of some examples of peptides produced NRPS machineries. In salinamide A biosynthesis (strictly a mixed NRPS/PKS), the P450 Sln10 is hypothesised to generate the linkage between the phenol of the 4-hydroxyphenylglycine residue and the (4-methylhexa-2,4,-dienoyl)glycine moiety.282 The crosslinking reaction also leads to the generation of an epoxide group that includes the 4-methyl group of the (4-methylhexa-2,4,-dienoyl)glycine moiety, which indicates that this may be the initial site of oxidation by this P450.282 C–N bond formation to generate a new heterocycle has also been identified in the biosynthesis of lyngbyatoxin.283,284 In this case, P450 LtxB has been shown to perform the N-alkylation of the side chain of the Trp-2 residue using the methylated amine group of the Val-1 residue. This reaction has been reconstituted in vitro, and this has shown that there is significant flexibility in the substrate selectivity of this P450, meaning that the valine residue can be exchanged for related amino acid residues with little effect on activity.284 Mechanistically, this reaction has been postulated to include an intermediate epoxide species that – following attack of the valine amine group – undergoes dehydration to re-aromatise the indole ring.283
3.6.2.2.6 Multiple transformations – himastatin biosynthesis. One of the most interesting collections of P450s in NRPS-mediated peptide biosynthesis are those involved in himastatin formation; these also display the biosynthetic potential of these enzymes. In this pathway (Fig. 23C), three P450s (HmtN, HmtS and HmtT) play vital and yet highly different roles in the biogenesis of himastatin, a symmetrical dimeric cyclohexadepsipeptide antibiotic.270,271 The first P450 to act in this pathway is HmtT, which generates hexahydropyrroloindole from the Trp-6 residue in the cyclic hexadepsipeptide precursor, a reaction that has similarities to that of LtxB (vide supra) in lyngbyatoxin biosynthesis283,284 and has been suggested to occur via epoxidation of the indole ring of the tryptophan moiety.270 The second P450, HmtN, subsequently performs a more standard – albeit still impressive – selective hydroxylation of the cyclic hexadepsipeptide precursor to produce the γ-hydroxy moiety of the 3-piperazine residue.270,271 Finally, HmtS then performs the symmetrical biaryl coupling of two cyclic hexadepsipeptide precursors via their hexahydropyrroloindole moieties,270 a reaction with parallels to these found in flaviolin and GPA biosynthesis.175,243 The examples of the diversity of reactions displayed not only within himastatin biosynthesis but within all NRPS-biosynthetic pathways discussed here shown how important P450s are in the production of diverse secondary metabolites, and also gives an indication of the diversity of roles that should be considered when assigning potential roles to P450 enzymes in newly identified examples of such biosynthetic gene clusters.
3.6.3.1 tRNA-dependent DKP biosynthesis pathways. The tRNA-dependent cyclodipeptide synthases (CDPS) use aminoacyl-tRNAs instead of the AMP-activated amino acids that are generated during in NRPS-mediated biosynthesis.285 Thus, the CDPS pathway connects primary and secondary metabolism by “hijacking” aminoacyl-tRNAs, with the result that their substrate selection is restricted to natural proteinogenic amino acids. Following condensation and cyclisation of two amino acids, the cyclic dipeptide can also undergo further modifications, several of which have been identified to be performed by cytochrome P450s. One example from Bacillus subtilis is the biosynthesis of pulcherrimin, which is involved in iron chelation. Commencing from cyclo(L-Leu–L-Leu), the P450 CypX performs a series of sequential oxidation reactions on the DKP ring itself, resulting in the formation of pulcherriminic acid in which both nitrogen atoms have been oxidised to N-oxides and the DKP ring itself has been aromatised.286 Chelation with iron then affords the final red pigment pulcherrimin (Fig. 24A). Mycocyclosin is another example of a P450-modified bacterial DKP that is produced by M. tuberculosis: following the synthesis of cyclo(L-Tyr–L-Tyr), Cyp121 performs an oxidative C–C coupling of the two aromatic side chains to generate mycocyclosin (Fig. 24B).287 Given the essential nature of CYP121 to the survival of M. tuberculosis,288 this P450 has been extensively characterised by a number of groups, which have afforded structural, biochemical and mechanistic insights into the function of this P450.289–291 Mechanistically, the formation of the C–C bond (similar to flaviolin) has been postulated to occur either by via proton coupled electron transfer or electron tunnelling between the two aromatic rings,177,292 which is a needed to avoid having to completely re-orient the substrate within the active site during catalysis, which was an early mechanistic postulate in this case.287 One of the more impressive series of transformations reported to occur during bacterial DKP synthesis is found in the biosynthesis of bicyclomycin.293,294 In this pathway, the initial cyclo(L-Leu–L-Ile) DKP undergoes six oxidation events (catalysed by five α-ketoglutarate/Fe2+-dependant dioxygenases (BcmB/C/E/F/G) and one cytochrome P450 (BcmD)) to afford the final, bicyclic structure of bicyclomycin. Within this pathway, the BcmD has been identified as carrying out the penultimate oxidation reaction, which affords hydroxylation of the C6 position of the DKP ring (Fig. 24C).294 Thus, DKP-modification by cytochrome P450s – whilst relatively rare in bacteria – provides several examples of novel P450-catalysed transformations and such transformations should always be considered when P450s are identified in close proximity to a cyclodipeptide synthase.
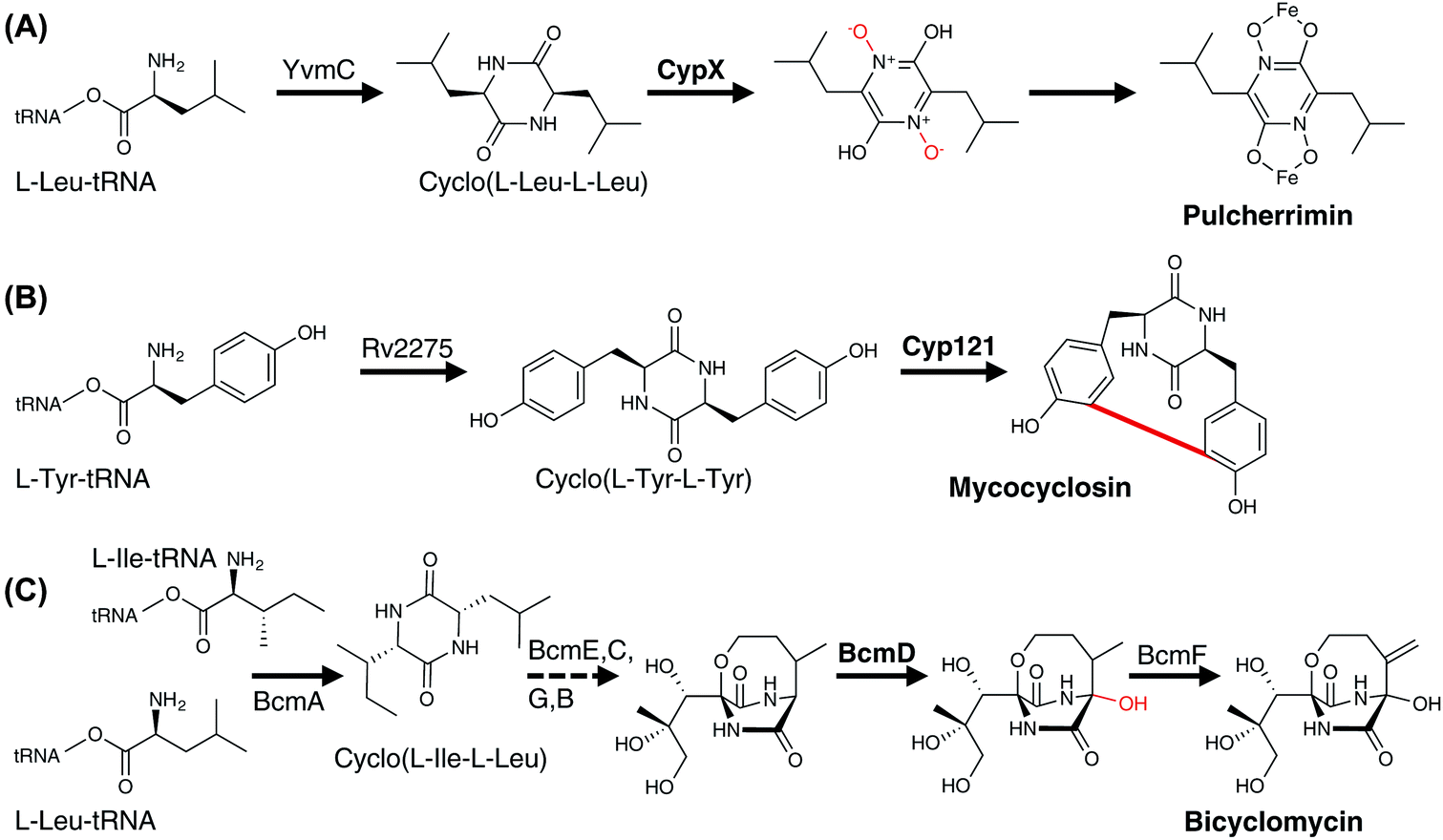 | ||
| Fig. 24 Examples of bacterial P450s involved in tRNA-dependant DKP biosynthesis. (A) In pulcherrimin biosynthesis, following the cyclisation of two leucine residues to form the DKP P450 CypX catalyses the oxidation of both nitrogen atoms into N-oxides as well as aromatisation of the DKP ring, which leads to a compound that is able to chelate Fe ions. (B) In M. tuberculosis, P450 Cyp121 performs an oxidative C–C coupling of the two aromatic side chains of cyclo(Tyr–Tyr). (C) In the biosynthesis of bicyclomycin, following the cyclisation of Ile and Leu and several modifications by α-ketoglutarate/Fe2+-dependant dioxygenases, the oxidation of C6 is catalysed by the P450 enzyme BcnD. P450-catalysed reactions are shown in red, P450 enzymes are indicated in bold. | ||
3.6.3.2 NRPS-dependent DKP biosynthesis pathways. NRPS systems are also able to synthesise cyclic dipeptides, either as the primary function of the NRPS (such as in thaxtomin A biosynthesis) or as a shunt product produced by the spontaneous generation of a DKP from a PCP-bound dipeptide. DKP biosynthesis by NRPSs is relatively rare, with the NRPS machineries responsible for producing such DKP producing peptides containing a specific condensation domain to catalyse the formation of the DKP ring.231,295,296 Whilst not a biosynthetic reaction as such, the production of DKPs via spontaneous cyclisation has proved to be most useful in the in vitro characterisation of complex NRPS machineries, as it allows the initial two modules of an NRPS to be used as a standalone catalytic system without the requirement to reconstitute the entire NRPS.297–300 Thaxtomin A, a DKP natural product produced by a two module NRPS machinery from several species of Streptomyces, contains two P450 enzymes within the biosynthetic cluster (TxtC and TxtE) that both represent intriguing examples of P450-mediated transformations (Fig. 25B). TxtC has been implicated in the installation of two hydroxyl groups on the thaxtomin D precursor, with the first reaction occurring at the α-carbon of the phenylalanine residue and a subsequent aromatic hydroxylation of the aromatic ring generating thaxtomin A.231 Whilst multistep transformations are somewhat common in P450s, this example is intriguing due to the differing reactivities and positions of the two hydroxylation sites, which surely warrants further characterisation of this P450. The second P450, TxtE,274,301 performs an even more intriguing transformation: the regiospecific 4-nitration of the indolyl moiety of L-tryptophan.274 Subsequent studies have also shown that TxtE has tolerance for certain alternate substrates.302 This reaction has been shown to require NO (generated by TxtD from arginine) and O2 in addition to L-Trp, with electrons provided by a competent redox partner system from NAD(P)H (e.g. spinach Fdx/FdR) in order to generate a ferric peroxynitrite species as the reactive intermediate.274 This reaction has similarities to that performed by RufO in the nitration of tyrosine during rufomycin biosynthesis (Fig. 23A),275 and demonstrates yet again the breadth of the catalytic repertoire of cytochrome P450s within secondary metabolism pathways.
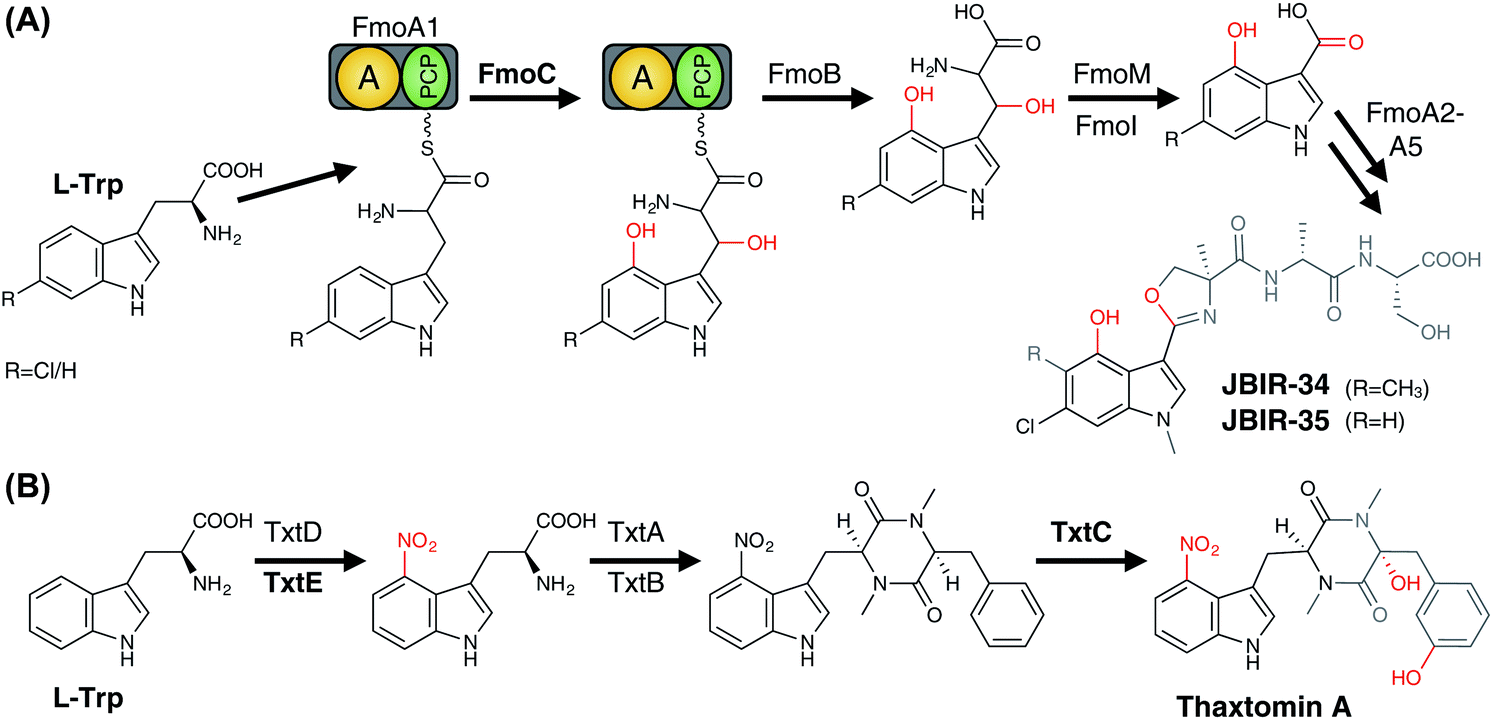 | ||
| Fig. 25 Examples of P450-catalysed reactions using either free tryptophan or PCP-bound tryptophan as a substrate. In JBIR-34/-35 and thaxtomin biosynthesis, modified tryptophan residues are incorporated. (A) In JBIR-34/-35 biosynthesis, PCP-bound tryptophan is twice hydroxylated by the P450 FmoC. (B) In thaxtomin biosynthesis, the P450 enzyme TxtE catalyses the regiospecific 4-nitration of the indolyl moiety of free tryptophan. Subsequently, the P450 TxtC installs two hydroxyl groups to finally produced thaxtomin A. P450 enzymes are indicated in bold, the P450-catalysed reactions are shown in red. The modified Trp residues incorporated are indicated in the final compound (grey). A, adenylation domain; PCP, peptidyl carrier protein. | ||
4. Conclusions
P450 enzymes clearly play a major role in biosynthetic pathways in many bacteria. Although these P450 enzymes all belong to the same superfamily of heme-containing monooxygenases, they are able to catalyse a huge range of different reactions and in doing so provide bacteria with the access to a great variety of different secondary metabolite with altered bioactivities. The diversity of substrates of the P450s found in bacteria also serves as a testament to the impressive ability of P450s to perform diverse, challenging chemistry against a wide range of substrates and yet to do so with selectivity and specificity that is the envy of an organic chemist. The future application of such P450s as specific biocatalysts remains bright, whilst the examples discussed in this review have also demonstrated that there are undoubtedly new examples of bacterial P450 diversity still awaiting discovery within bacterial biosynthesis pathways.5. Conflicts of interest
The authors declare no competing financial interest.6. Acknowledgements
The authors would like to acknowledge the support of: The University of Queensland (J. J. D. V.); Monash University & EMBL Australia (M. J. C); the Australian Research Council (DP170102220 M. J. C. and J. J. D. V); and the National Health and Medical Research Council (APP1140619 M. J. C.). Open Access funding provided by the Max Planck Society.7. References
- D. R. Nelson, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 141–154 CrossRef CAS PubMed.
- D. Sirim, M. Widmann, F. Wagner and J. Pleiss, BMC Struct. Biol., 2010, 10, 34 CrossRef PubMed.
- D. Sirim, F. Wagner, A. Lisitsa and J. Pleiss, BMC Biochem., 2009, 10, 27 CrossRef PubMed.
- D. R. Nelson, Hum. Genomics, 2009, 4, 59 CAS.
- S. C. Moody and E. J. Loveridge, J. Appl. Microbiol., 2014, 117, 1549–1563 CrossRef CAS PubMed.
- J. D. Rudolf, C.-Y. Chang, M. Ma and B. Shen, Nat. Prod. Rep., 2017, 34, 1141–1172 RSC.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- Ł. Gricman, M. J. Weissenborn, S. M. Hoffmann, N. Borlinghaus, B. Hauer and J. Pleiss, ChemistrySelect, 2016, 1, 1243–1251 CrossRef.
- O. Gotoh, J. Biol. Chem., 1992, 267, 83–90 CAS.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS PubMed.
- F. P. Guengerich and A. W. Munro, J. Biol. Chem., 2013, 288, 17065–17073 CrossRef CAS PubMed.
- X. Zhang and S. Li, Nat. Prod. Rep., 2017, 34, 1061–1089 RSC.
- P. R. Ortiz de Montellano and J. J. De Voss, Nat. Prod. Rep., 2002, 19, 477–493 RSC.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- M. J. Coon, A. D. N. Vaz, D. F. McGinnity and H.-M. Peng, Contributions to Specificity in Drug Metabolism, 1998, 26, 1190–1193 CAS.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- D. C. Lamb, H. Ikeda, D. R. Nelson, J. Ishikawa, T. Skaug, C. Jackson, S. Omura, M. R. Waterman and S. L. Kelly, Biochem. Biophys. Res. Commun., 2003, 307, 610–619 CrossRef CAS PubMed.
- H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori and S. Ōmura, Nat. Biotechnol., 2003, 21, 526 CrossRef PubMed.
- F. Kunst, N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessières, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S. K. Choi, J. J. Codani, I. F. Connerton, N. J. Cummings, R. A. Daniel, F. Denizot, K. M. Devine, A. Düsterhöft, S. D. Ehrlich, P. T. Emmerson, K. D. Entian, J. Errington, C. Fabret, E. Ferrari, D. Foulger, C. Fritz, M. Fujita, Y. Fujita, S. Fuma, A. Galizzi, N. Galleron, S. Y. Ghim, P. Glaser, A. Goffeau, E. J. Golightly, G. Grandi, G. Guiseppi, B. J. Guy, K. Haga, J. Haiech, C. R. Harwood, A. Hénaut, H. Hilbert, S. Holsappel, S. Hosono, M. F. Hullo, M. Itaya, L. Jones, B. Joris, D. Karamata, Y. Kasahara, M. Klaerr-Blanchard, C. Klein, Y. Kobayashi, P. Koetter, G. Koningstein, S. Krogh, M. Kumano, K. Kurita, A. Lapidus, S. Lardinois, J. Lauber, V. Lazarevic, S. M. Lee, A. Levine, H. Liu, S. Masuda, C. Mauël, C. Médigue, N. Medina, R. P. Mellado, M. Mizuno, D. Moestl, S. Nakai, M. Noback, D. Noone, M. O'Reilly, K. Ogawa, A. Ogiwara, B. Oudega, S. H. Park, V. Parro, T. M. Pohl, D. Portetelle, S. Porwollik, A. M. Prescott, E. Presecan, P. Pujic, B. Purnelle, G. Rapoport, M. Rey, S. Reynolds, M. Rieger, C. Rivolta, E. Rocha, B. Roche, M. Rose, Y. Sadaie, T. Sato, E. Scanlan, S. Schleich, R. Schroeter, F. Scoffone, J. Sekiguchi, A. Sekowska, S. J. Seror, P. Serror, B. S. Shin, B. Soldo, A. Sorokin, E. Tacconi, T. Takagi, H. Takahashi, K. Takemaru, M. Takeuchi, A. Tamakoshi, T. Tanaka, P. Terpstra, A. Tognoni, V. Tosato, S. Uchiyama, M. Vandenbol, F. Vannier, A. Vassarotti, A. Viari, R. Wambutt, E. Wedler, H. Wedler, T. Weitzenegger, P. Winters, A. Wipat, H. Yamamoto, K. Yamane, K. Yasumoto, K. Yata, K. Yoshida, H. F. Yoshikawa, E. Zumstein, H. Yoshikawa and A. Danchin, Nature, 1997, 390, 249 CrossRef CAS PubMed.
- Y.-J. Chun, T. Shimada, R. Sanchez-Ponce, M. V. Martin, L. Lei, B. Zhao, S. L. Kelly, M. R. Waterman, D. C. Lamb and F. P. Guengerich, J. Biol. Chem., 2007, 282, 17486–17500 CrossRef CAS PubMed.
- R. J. Lawson, C. von Wachenfeldt, I. Haq, J. Perkins and A. W. Munro, Biochemistry, 2004, 43, 12390–12409 CrossRef CAS PubMed.
- A. J. Green, A. W. Munro, M. R. Cheesman, G. A. Reid, C. von Wachenfeldt and S. K. Chapman, J. Inorg. Biochem., 2003, 93, 92–99 CrossRef CAS PubMed.
- N. Geib, T. Weber, T. Wörtz, K. Zerbe, W. Wohlleben and J. A. Robinson, FEMS Microbiol. Lett., 2010, 306, 45–53 CrossRef CAS PubMed.
- R. Li, N. Khaleeli and C. A. Townsend, J. Bacteriol., 2000, 182, 4087–4095 CrossRef CAS PubMed.
- R. M. Carman and M. T. Fletcher, Aust. J. Chem., 1983, 36, 1483–1493 CrossRef CAS.
- P. R. O. de Montellano and J. J. De Voss, in Cytochrome P450 Structure Mechanism and Biochemistry, ed. P. R. Ortiz de Montellano, Springer US, Boston, MA, 2005, pp. 183–245. Search PubMed.
- A. J. Ullah, R. I. Murray, P. K. Bhattacharyya, G. C. Wagner and I. C. Gunsalus, J. Biol. Chem., 1990, 265, 1345–1351 CAS.
- J. A. Peterson, J. Y. Lu, J. Geisselsoder, S. Graham-Lorence, C. Carmona, F. Witney and M. C. Lorence, J. Biol. Chem., 1992, 267, 14193–14203 CAS.
- D. B. Hawkes, J. Biol. Chem., 2002, 277, 27725–27732 CrossRef CAS PubMed.
- J. A. Fruetel, R. L. Mackman, J. A. Peterson and P. R. Ortiz de Montellano, J. Biol. Chem., 1994, 269, 28815–28821 CAS.
- C. A. Hasemann, K. G. Ravichandran, J. A. Peterson and J. Deisenhofer, J. Mol. Biol., 1994, 236, 1169–1185 CrossRef CAS PubMed.
- Y. T. Meharenna, H. Li, D. B. Hawkes, A. G. Pearson, J. De Voss and T. L. Poulos, Biochemistry, 2004, 43, 9487–9494 CrossRef PubMed.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, Science, 2000, 287, 1615–1622 CrossRef CAS PubMed.
- F. Hannemann, A. Bichet, K. M. Ewen and R. Bernhardt, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 330–344 CrossRef CAS PubMed.
- S. G. Bell, A. Dale, N. H. Rees and L.-L. Wong, Appl. Microbiol. Biotechnol., 2010, 86, 163–175 CrossRef CAS PubMed.
- B. Unterweger, D. M. Bulach, J. Scoble, D. J. Midgley, P. Greenfield, D. Lyras, P. Johanesen and G. J. Dumsday, Appl. Environ. Microbiol., 2016, 82, 6507–6517 CrossRef CAS PubMed.
- G. E. Collis, B. Unterweger, G. J. Dumsday and C. M. Forsyth, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2017, 73, 1242–1245 CAS.
- G. Grogan, G. A. Roberts, S. Parsons, N. J. Turner and S. L. Flitsch, Appl. Microbiol. Biotechnol., 2002, 59, 449–454 CrossRef CAS PubMed.
- R. W. Eaton, J. Bacteriol., 1997, 179, 3171–3180 CrossRef CAS PubMed.
- D. J. Smith, V. J. J. Martin and W. W. Mohn, J. Bacteriol., 2004, 186, 3631–3639 CrossRef CAS PubMed.
- D. J. Smith, M. A. Patrauchan, C. Florizone, L. D. Eltis and W. W. Mohn, J. Bacteriol., 2008, 190, 1575–1583 CrossRef CAS PubMed.
- C. A. Morgan and R. C. Wyndham, Can. J. Microbiol., 2002, 48, 49–59 CrossRef CAS PubMed.
- D. J. Frank, Y. Zhao, S. H. Wong, D. Basudhar, J. J. De Voss and P. R. O. de Montellano, J. Biol. Chem., 2016, 291, 7325–7333 CrossRef CAS PubMed.
- J. B. Johnston, H. Ouellet and P. R. Ortiz de Montellano, J. Biol. Chem., 2010, 285, 36352–36360 CrossRef CAS PubMed.
- Y. Aoyama, T. Horiuchi, O. Gotoh, M. Noshiro and Y. Yoshida, J. Biochem., 1998, 124, 694–696 CrossRef CAS PubMed.
- A. Bellamine, A. T. Mangla, W. D. Nes and M. R. Waterman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8937–8942 CrossRef CAS.
- D. C. Lamb, D. E. Kelly, N. J. Manning and S. L. Kelly, FEBS Lett., 1998, 437, 142–144 CrossRef CAS PubMed.
- M. P. Pietila, P. K. Vohra, B. Sanyal, N. L. Wengenack, S. Raghavakaimal and C. F. Thomas Jr, Am. J. Respir. Cell Mol. Biol., 2006, 35, 236–242 CrossRef CAS PubMed.
- X. Ke, G.-J. Ding, B.-X. Ma, Z.-Q. Liu, J.-F. Zhang and Y.-G. Zheng, Process Biochem., 2017, 62, 59–68 CrossRef CAS.
- C. J. Jackson, D. C. Lamb, T. H. Marczylo, A. G. S. Warrilow, N. J. Manning, D. J. Lowe, D. E. Kelly and S. L. Kelly, J. Biol. Chem., 2002, 277, 46959–46965 CrossRef CAS PubMed.
- M. Daum, H.-J. Schnell, S. Herrmann, A. Günther, R. Murillo, R. Müller, P. Bisel, M. Müller and A. Bechthold, ChemBioChem, 2010, 11, 1383–1391 CrossRef CAS PubMed.
- C. Dürr, H.-J. Schnell, A. Luzhetskyy, R. Murillo, M. Weber, K. Welzel, A. Vente and A. Bechthold, Chem. Biol., 2006, 13, 365–377 CrossRef PubMed.
- M. Xu, M. L. Hillwig, A. L. Lane, M. S. Tiernan, B. S. Moore and R. J. Peters, J. Nat. Prod., 2014, 77, 2144–2147 CrossRef CAS PubMed.
- A. Meguro, Y. Motoyoshi, K. Teramoto, S. Ueda, Y. Totsuka, Y. Ando, T. Tomita, S.-Y. Kim, T. Kimura, M. Igarashi, R. Sawa, T. Shinada, M. Nishiyama and T. Kuzuyama, Angew. Chem., 2015, 127, 4427–4430 CrossRef.
- S.-Y. Kim, P. Zhao, M. Igarashi, R. Sawa, T. Tomita, M. Nishiyama and T. Kuzuyama, Chem. Biol., 2009, 16, 736–743 CrossRef CAS PubMed.
- C. Görner, P. Schrepfer, V. Redai, F. Wallrapp, B. Loll, W. Eisenreich, M. Haslbeck and T. Brück, Microb. Cell Fact., 2016, 15, 86 CrossRef PubMed.
- F. Blasco, I. Kauffmann and R. D. Schmid, Appl. Microbiol. Biotechnol., 2004, 64, 671–674 CrossRef CAS PubMed.
- K. Momoi, U. Hofmann, R. D. Schmid and V. B. Urlacher, Biochem. Biophys. Res. Commun., 2006, 339, 331–336 CrossRef CAS PubMed.
- Z. Zhou, W. Zhang, S. Su, M. Chen, W. Lu, M. Lin, I. Molnár and Y. Xu, Appl. Microbiol. Biotechnol., 2015, 99, 10539–10546 CrossRef CAS PubMed.
- B. Zhao, X. Lin, L. Lei, D. C. Lamb, S. L. Kelly, M. R. Waterman and D. E. Cane, J. Biol. Chem., 2008, 283, 8183–8189 CrossRef CAS PubMed.
- B. Zhao, L. Lei, D. G. Vassylyev, X. Lin, D. E. Cane, S. L. Kelly, H. Yuan, D. C. Lamb and M. R. Waterman, J. Biol. Chem., 2009, 284, 36711–36719 CrossRef CAS PubMed.
- S. A. Agger, F. Lopez-Gallego, T. R. Hoye and C. Schmidt-Dannert, J. Bacteriol., 2008, 190, 6084–6096 CrossRef CAS PubMed.
- H. Harada, K. Shindo, K. Iki, A. Teraoka, S. Okamoto, F. Yu, J.-i. Hattan, R. Utsumi and N. Misawa, Appl. Microbiol. Biotechnol., 2011, 90, 467–476 CrossRef CAS PubMed.
- J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87–110 RSC.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-Ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed.
- Y. Hu, W. K. W. Chou, R. Hopson and D. E. Cane, Chem. Biol., 2011, 18, 32–37 CrossRef CAS PubMed.
- R. Quaderer, S. Omura, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 13036–13037 CrossRef CAS PubMed.
- D. Zhu, M.-J. Seo, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2011, 133, 2128–2131 CrossRef CAS PubMed.
- L. Duan, G. Jogl and D. E. Cane, J. Am. Chem. Soc., 2016, 138, 12678–12689 CrossRef CAS PubMed.
- S. Takamatsu, L.-H. Xu, S. Fushinobu, H. Shoun, M. Komatsu, D. E. Cane and H. Ikeda, J. Antibiot., 2011, 64, 65–71 CrossRef CAS PubMed.
- R. S. Nett, M. Montanares, A. Marcassa, X. Lu, R. Nagel, T. C. Charles, P. Hedden, M. C. Rojas and R. J. Peters, Nat. Chem. Biol., 2017, 13, 69–74 CrossRef CAS PubMed.
- R. S. Nett, T. Contreras and R. J. Peters, ACS Chem. Biol., 2017, 12, 912–917 CrossRef CAS PubMed.
- M. J. Cryle, Biochem. Soc. Trans., 2010, 38, 934–939 CrossRef CAS PubMed.
- H. Peter and L. Michael, Curr. Drug Metab., 2010, 11, 85–104 CrossRef.
- J. B. Johnston, P. M. Kells, L. M. Podust and P. R. Ortiz de Montellano, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20687–20692 CrossRef CAS PubMed.
- X. He, M. J. Cryle, J. J. De Voss and P. R. Ortiz de Montellano, J. Biol. Chem., 2005, 280, 22697–22705 CrossRef CAS PubMed.
- S. Honda Malca, D. Scheps, L. Kuhnel, E. Venegas-Venegas, A. Seifert, B. M. Nestl and B. Hauer, Chem. Commun., 2012, 48, 5115–5117 RSC.
- M. Bordeaux, A. Galarneau, F. Fajula and J. Drone, Angew. Chem., 2011, 123, 2123–2127 CrossRef.
- D. Scheps, S. Honda Malca, H. Hoffmann, B. M. Nestl and B. Hauer, Org. Biomol. Chem., 2011, 9, 6727–6733 CAS.
- E. G. Funhoff, U. Bauer, I. García-Rubio, B. Witholt and J. B. van Beilen, J. Bacteriol., 2006, 188, 5220–5227 CrossRef CAS PubMed.
- J. B. van Beilen, E. G. Funhoff, A. van Loon, A. Just, L. Kaysser, M. Bouza, R. Holtackers, M. Röthlisberger, Z. Li and B. Witholt, Appl. Environ. Microbiol., 2006, 72, 59–65 CrossRef CAS PubMed.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- L. P. Wen and A. J. Fulco, J. Biol. Chem., 1987, 262, 6676–6682 CAS.
- K. J. McLean, H. M. Girvan and A. W. Munro, Expert Opin. Drug Metab. Toxicol., 2007, 3, 847–863 CrossRef CAS PubMed.
- R. Neeli, H. M. Girvan, A. Lawrence, M. J. Warren, D. Leys, N. S. Scrutton and A. W. Munro, FEBS Lett., 2005, 579, 5582–5588 CrossRef CAS PubMed.
- T. Kitazume, D. C. Haines, R. W. Estabrook, B. Chen and J. A. Peterson, Biochemistry, 2007, 46, 11892–11901 CrossRef CAS PubMed.
- H. M. Girvan, A. J. Dunford, R. Neeli, I. S. Ekanem, T. N. Waltham, M. G. Joyce, D. Leys, R. A. Curtis, P. Williams, K. Fisher, M. W. Voice and A. W. Munro, Arch. Biochem. Biophys., 2011, 507, 75–85 CrossRef CAS PubMed.
- S. S. Boddupalli, B. C. Pramanik, C. A. Slaughter, R. W. Estabrook and J. A. Peterson, Arch. Biochem. Biophys., 1992, 292, 20–28 CrossRef CAS PubMed.
- S. S. Boddupalli, R. W. Estabrook and J. A. Peterson, J. Biol. Chem., 1990, 265, 4233–4239 CAS.
- M. J. Cryle and J. J. De Voss, ChemBioChem, 2008, 9, 261–266 CrossRef CAS PubMed.
- G. Truan, M. R. Komandla, J. R. Falck and J. A. Peterson, Arch. Biochem. Biophys., 1999, 366, 192–198 CrossRef CAS PubMed.
- M. J. Cryle, N. J. Matovic and J. J. De Voss, Tetrahedron Lett., 2007, 48, 133–136 CrossRef CAS.
- M. J. Cryle and J. J. De Voss, Tetrahedron: Asymmetry, 2007, 18, 547–551 CrossRef CAS.
- M. J. Cryle, P. Y. Hayes and J. J. De Voss, Chem.–Eur. J., 2012, 18, 15994–15999 CrossRef CAS PubMed.
- M. J. Cryle, P. R. Ortiz de Montellano and J. J. De Voss, J. Org. Chem., 2005, 70, 2455–2469 CrossRef CAS PubMed.
- M. J. Cryle, J. M. U. Stuthe, P. R. Ortiz de Montellano and J. J. De Voss, Chem. Commun., 2004, 512–513, 10.1039/b315911f.
- J. H. Capdevila, S. Wei, C. Helvig, J. R. Falck, Y. Belosludtsev, G. Truan, S. E. Graham-Lorence and J. A. Peterson, J. Biol. Chem., 1996, 271, 22663–22671 CrossRef CAS PubMed.
- M. J. Cryle and J. J. De Voss, Angew. Chem., Int. Ed., 2006, 45, 8221–8223 CrossRef CAS PubMed.
- M. J. Cryle, R. D. Espinoza, S. J. Smith, N. J. Matovic and J. J. De Voss, Chem. Commun., 2006, 2353–2355, 10.1039/b601202g.
- R. L. Wright, K. Harris, B. Solow, R. H. White and P. J. Kennelly, FEBS Lett., 1996, 384, 235–239 CrossRef CAS PubMed.
- M. A. McLean, S. A. Maves, K. E. Weiss, S. Krepich and S. G. Sligar, Biochem. Biophys. Res. Commun., 1998, 252, 166–172 CrossRef CAS PubMed.
- A. V. Puchkaev, L. S. Koo and P. R. Ortiz de Montellano, Arch. Biochem. Biophys., 2003, 409, 52–58 CrossRef CAS PubMed.
- A. V. Puchkaev, T. Wakagi and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 2002, 124, 12682–12683 CrossRef CAS PubMed.
- K. S. Rabe, K. Kiko and C. M. Niemeyer, ChemBioChem, 2008, 9, 420–425 CrossRef CAS PubMed.
- E. V. Sineva and D. R. Davydov, Biochemistry, 2010, 49, 10636–10646 CrossRef CAS PubMed.
- D. R. Davydov, E. V. Sineva, N. Y. Davydova, D. H. Bartlett and J. R. Halpert, Biotechnol. Appl. Biochem., 2013, 60, 30–40 CrossRef CAS PubMed.
- I. Matsunaga, E. Kusunose, I. Yano and K. Ichihara, Biochem. Biophys. Res. Commun., 1994, 201, 1554–1560 CrossRef CAS PubMed.
- I. Matsunaga, N. Yokotani, O. Gotoh, E. Kusunose, M. Yamada and K. Ichihara, J. Biol. Chem., 1997, 272, 23592–23596 CrossRef CAS PubMed.
- I. Matsunaga, M. Yamada, E. Kusunose, Y. Nishiuchi, I. Yano and K. Ichihara, FEBS Lett., 1996, 386, 252–254 CrossRef CAS PubMed.
- T. Fujishiro, O. Shoji, S. Nagano, H. Sugimoto, Y. Shiro and Y. Watanabe, J. Biol. Chem., 2011, 286, 29941–29950 CrossRef CAS PubMed.
- D.-S. Lee, A. Yamada, H. Sugimoto, I. Matsunaga, H. Ogura, K. Ichihara, S.-i. Adachi, S.-Y. Park and Y. Shiro, J. Biol. Chem., 2003, 278, 9761–9767 CrossRef CAS PubMed.
- I. Matsunaga, A. Yamada, D.-S. Lee, E. Obayashi, N. Fujiwara, K. Kobayashi, H. Ogura and Y. Shiro, Biochemistry, 2002, 41, 1886–1892 CrossRef CAS PubMed.
- M. Girhard, S. Schuster, M. Dietrich, P. Dürre and V. B. Urlacher, Biochem. Biophys. Res. Commun., 2007, 362, 114–119 CrossRef CAS PubMed.
- Y. Liu, C. Wang, J. Yan, W. Zhang, W. Guan, X. Lu and S. Li, Biotechnol. Biofuels, 2014, 7, 28 CrossRef PubMed.
- J. Belcher, K. J. McLean, S. Matthews, L. S. Woodward, K. Fisher, S. E. J. Rigby, D. R. Nelson, D. Potts, M. T. Baynham, D. A. Parker, D. Leys and A. W. Munro, J. Biol. Chem., 2014, 289, 6535–6550 CrossRef CAS PubMed.
- S. Matthews, J. D. Belcher, K. L. Tee, H. M. Girvan, K. J. McLean, S. E. J. Rigby, C. W. Levy, D. Leys, D. A. Parker, R. T. Blankley and A. W. Munro, J. Biol. Chem., 2017, 292, 5128–5143 CrossRef CAS PubMed.
- S. Matthews, K. L. Tee, N. J. Rattray, K. J. McLean, D. Leys, D. A. Parker, R. T. Blankley and A. W. Munro, FEBS Lett., 2017, 591, 737–750 CrossRef CAS PubMed.
- B. Fang, H. Xu, Y. Liu, F. Qi, W. Zhang, H. Chen, C. Wang, Y. Wang, W. Yang and S. Li, Sci. Rep., 2017, 7, 44258 CrossRef PubMed.
- J. L. Grant, M. E. Mitchell and T. M. Makris, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10049–10054 CrossRef CAS PubMed.
- J. L. Grant, C. H. Hsieh and T. M. Makris, J. Am. Chem. Soc., 2015, 137, 4940–4943 CrossRef CAS PubMed.
- R. J. Lawson, D. Leys, M. J. Sutcliffe, C. A. Kemp, M. R. Cheesman, S. J. Smith, J. Clarkson, W. E. Smith, I. Haq, J. B. Perkins and A. W. Munro, Biochemistry, 2004, 43, 12410–12426 CrossRef CAS PubMed.
- J. E. Stok and J. J. De Voss, Arch. Biochem. Biophys., 2000, 384, 351–360 CrossRef CAS PubMed.
- J. J. De Voss and M. J. Cryle, Met. Ions Life Sci., 2007, 3, 397–435 CAS.
- M. J. Cryle and J. J. De Voss, Chem. Commun., 2004, 86–87, 10.1039/b311652b.
- M. J. Cryle, N. J. Matovic and J. J. De Voss, Org. Lett., 2003, 5, 3341–3344 CrossRef CAS PubMed.
- M. J. Cryle and I. Schlichting, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15696–15701 CrossRef CAS PubMed.
- M. J. Cryle, Metallomics, 2011, 3, 323–326 RSC.
- S. Huang, S. S. Elsayed, M. Lv, J. Tabudravu, M. E. Rateb, R. Gyampoh, K. Kyeremeh, R. Ebel, M. Jaspars, Z. Deng, Y. Yu and H. Deng, Chem. Biol., 2015, 22, 1633–1642 CrossRef CAS PubMed.
- S. K. P. Norah Alqahtani, E. D. James, D. M. Bis, J. A. Karty, A. L. Lane and R. Viswanathan, Org. Biomol. Chem., 2015, 13, 7177–7192 Search PubMed.
- R. Raju, A. M. Piggott, X.-C. Huang and R. J. Capon, Org. Lett., 2011, 13, 2770–2773 CrossRef CAS PubMed.
- W. Kitagawa, T. Ozaki, T. Nishioka, Y. Yasutake, M. Hata, M. Nishiyama, T. Kuzuyama and T. Tamura, ChemBioChem, 2013, 14, 1085–1093 CrossRef CAS PubMed.
- Y. Yasutake, W. Kitagawa, M. Hata, T. Nishioka, T. Ozaki, M. Nishiyama, T. Kuzuyama and T. Tamura, FEBS Lett., 2014, 588, 105–110 CrossRef CAS PubMed.
- A. Sandmann, J. Dickschat, H. Jenke-Kodama, B. Kunze, E. Dittmann and R. Müller, Angew. Chem., Int. Ed., 2007, 46, 2712–2716 CrossRef CAS PubMed.
- D. Pistorius, Y. Li, A. Sandmann and R. Müller, Mol. BioSyst., 2011, 7, 3308–3315 RSC.
- A. R. Howard-Jones and C. T. Walsh, Biochemistry, 2005, 44, 15652–15663 CrossRef CAS PubMed.
- A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 11016–11017 CrossRef CAS PubMed.
- A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 12289–12298 CrossRef CAS PubMed.
- M. Makino, H. Sugimoto, Y. Shiro, S. Asamizu, H. Onaka and S. Nagano, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11591–11596 CrossRef CAS PubMed.
- Y. Wang, H. Chen, M. Makino, Y. Shiro, S. Nagano, S. Asamizu, H. Onaka and S. Shaik, J. Am. Chem. Soc., 2009, 131, 6748–6762 CrossRef CAS PubMed.
- P. J. Goldman, K. S. Ryan, M. J. Hamill, A. R. Howard-Jones, C. T. Walsh, S. J. Elliott and C. L. Drennan, Chem. Biol., 2012, 19, 855–865 CrossRef CAS PubMed.
- K. S. Ryan, A. R. Howard-Jones, M. J. Hamill, S. J. Elliott, C. T. Walsh and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15311–15316 CrossRef CAS PubMed.
- A. P. Salas, L. Zhu, C. Sánchez, A. F. Braña, J. Rohr, C. Méndez and J. A. Salas, Mol. Microbiol., 2005, 58, 17–27 CrossRef CAS PubMed.
- H. Onaka, S. Asamizu, Y. Igarashi, R. Yoshida and T. Furumai, Biosci., Biotechnol., Biochem., 2005, 69, 1753–1759 CrossRef CAS PubMed.
- C. A. Jackson, M. B. Couger, M. Prabhakaran, K. D. Ramachandriya, P. Canaan and B. Z. Fathepure, J. Appl. Microbiol., 2017, 122, 940–952 CrossRef CAS PubMed.
- N. Funa, M. Funabashi, Y. Ohnishi and S. Horinouchi, J. Bacteriol., 2005, 187, 8149–8155 CrossRef CAS PubMed.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- R. H. Lambalot, D. E. Cane, J. J. Aparicio and L. Katz, Biochemistry, 1995, 34, 1858–1866 CrossRef CAS PubMed.
- D. Stassi, S. Donadio, M. J. Staver and L. Katz, J. Bacteriol., 1993, 175, 182–189 CrossRef CAS PubMed.
- J. F. Andersen and C. R. Hutchinson, J. Bacteriol., 1992, 174, 725–735 CrossRef CAS PubMed.
- J. M. Weber, J. O. Leung, S. J. Swanson, K. B. Idler and J. B. McAlpine, Science, 1991, 252, 114–117 CAS.
- Y. Xue, D. Wilson, L. Zhao, H. Liu and D. H. Sherman, Chem. Biol., 1998, 5, 661–667 CrossRef CAS PubMed.
- M. V. Mendes, N. Antón, J. F. Martín and J. F. Aparicio, Biochem. J., 2005, 386, 57–62 CrossRef CAS PubMed.
- M. V. Mendes, E. Recio, R. Fouces, R. Luiten, J. F. Martín and J. F. Aparicio, Cell Chem. Biol., 2001, 8, 635–644 CAS.
- H. Ogura, C. R. Nishida, U. R. Hoch, R. Perera, J. H. Dawson and P. R. Ortiz de Montellano, Biochemistry, 2004, 43, 14712–14721 CrossRef CAS PubMed.
- N. Moebius, C. Ross, K. Scherlach, B. Rohm, M. Roth and C. Hertweck, Chem. Biol., 2012, 19, 1164–1174 CrossRef CAS PubMed.
- M. Carmody, B. Murphy, B. Byrne, P. Power, D. Rai, B. Rawlings and P. Caffrey, J. Biol. Chem., 2005, 280, 34420–34426 CrossRef CAS PubMed.
- S. Chen, X. Mao, Y. Shen, Y. Zhou, J. Li, L. Wang, X. Tao, L. Yang, Y. Wang, X. Zhou, Z. Deng and D. Wei, Appl. Environ. Microbiol., 2009, 75, 1778–1781 CrossRef CAS PubMed.
- S.-P. Liu, P.-H. Yuan, Y.-Y. Wang, X.-F. Liu, Z.-X. Zhou, Q.-t. Bu, P. Yu, H. Jiang and Y.-Q. Li, Microbiol. Res., 2015, 173, 25–33 CrossRef CAS PubMed.
- E. M. Seco, S. Fotso, H. Laatsch and F. Malpartida, Chem. Biol., 2005, 12, 1093–1101 CrossRef CAS PubMed.
- C. Olano, B. Wilkinson, C. Sánchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Braña, C. J. Martin, M. Oliynyk, C. Méndez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87–97 CAS.
- M. T. Nelp and V. Bandarian, Angew. Chem., Int. Ed., 2015, 54, 10627–10629 CrossRef CAS PubMed.
- C. J. Schulze, W. M. Bray, F. Loganzo, M.-H. Lam, T. Szal, A. Villalobos, F. E. Koehn and R. G. Linington, J. Nat. Prod., 2014, 77, 2570–2574 CrossRef CAS PubMed.
- J. Li, X. Tang, T. Awakawa and B. S. Moore, Angew. Chem., Int. Ed., 2017, 56, 12234–12239 CrossRef CAS PubMed.
- M. L. Dickens and W. R. Strohl, J. Bacteriol., 1996, 178, 3389–3395 CrossRef CAS PubMed.
- M. L. Dickens, N. D. Priestley and W. R. Strohl, J. Bacteriol., 1997, 179, 2641–2650 CrossRef CAS PubMed.
- R. J. Walczak, M. L. Dickens, N. D. Priestley and W. R. Strohl, J. Bacteriol., 1999, 181, 298–304 CAS.
- F. Kudo, K. Kawamura, T. Furuya, H. Yamanishi, A. Motegi, A. Komatsubara, M. Numakura, A. Miyanaga and T. Eguchi, ChemBioChem, 2016, 17, 233–238 CrossRef CAS PubMed.
- Y. Anzai, S. Li, M. R. Chaulagain, K. Kinoshita, F. Kato, J. Montgomery and D. H. Sherman, Chem. Biol., 2008, 15, 950–959 CrossRef CAS PubMed.
- J. C. Carlson, S. Li, S. S. Gunatilleke, Y. Anzai, D. A. Burr, L. M. Podust and D. H. Sherman, Nat. Chem., 2011, 3, 628–633 CrossRef CAS PubMed.
- R. A. Butcher, F. C. Schroeder, M. A. Fischbach, P. D. Straight, R. Kolter, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1506–1509 CrossRef CAS PubMed.
- J. J. Reddick, S. A. Antolak and G. M. Raner, Biochem. Biophys. Res. Commun., 2007, 358, 363–367 CrossRef CAS PubMed.
- S. H. Wong, S. G. Bell and J. J. De Voss, Pure Appl. Chem., 2017, 89, 841–852 CrossRef CAS.
- B. Zhao, F. P. Guengerich, A. Bellamine, D. C. Lamb, M. Izumikawa, L. Lei, L. M. Podust, M. Sundaramoorthy, J. A. Kalaitzis, L. M. Reddy, S. L. Kelly, B. S. Moore, D. Stec, M. Voehler, J. R. Falck, T. Shimada and M. R. Waterman, J. Biol. Chem., 2005, 280, 11599–11607 CrossRef CAS PubMed.
- B. Zhao, D. C. Lamb, L. Lei, S. L. Kelly, H. Yuan, D. L. Hachey and M. R. Waterman, Biochemistry, 2007, 46, 8725–8733 CrossRef CAS PubMed.
- K. Dornevil, I. Davis, A. J. Fielding, J. R. Terrell, L. Ma and A. Liu, J. Biol. Chem., 2017, 292, 13645–13657 CrossRef CAS PubMed.
- M. E. A. Richter, N. Traitcheva, U. Knüpfer and C. Hertweck, Angew. Chem., Int. Ed., 2008, 47, 8872–8875 CrossRef CAS PubMed.
- J. He, M. Müller and C. Hertweck, J. Am. Chem. Soc., 2004, 126, 16742–16743 CrossRef CAS PubMed.
- H. Ikeda, T. Nonomiya, M. Usami, T. Ohta and S. Ōmura, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9509–9514 CrossRef CAS.
- M. J. Smanski, R. M. Peterson and B. Shen, in Methods in Enzymology, ed. D. A. Hopwood, Academic Press, 2012, vol. 515, pp. 163–186 Search PubMed.
- G. Zocher, M. E. A. Richter, U. Mueller and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 2292–2302 CrossRef CAS PubMed.
- M. Richter, B. Busch, K. Ishida, B. S. Moore and C. Hertweck, ChemBioChem, 2012, 13, 2196–2199 CrossRef CAS PubMed.
- Y. Xie, B. Wang, J. Liu, J. Zhou, J. Ma, H. Huang and J. Ju, ChemBioChem, 2012, 13, 2745–2757 CrossRef CAS PubMed.
- Y. Xie, Q. Li, Y. Song, J. Ma and J. Ju, ChemBioChem, 2014, 15, 1183–1189 CrossRef CAS PubMed.
- Q. Li, Y. Chen, G. Zhang and H. Zhang, FEBS Lett., 2017, 591, 1295–1304 CrossRef CAS PubMed.
- M. E. Yurkovich, R. Jenkins, Y. Sun, M. Tosin and P. F. Leadlay, Chem. Commun., 2017, 53, 2182–2185 RSC.
- X. Tang, J. Li and B. S. Moore, ChemBioChem, 2017, 18, 1072–1076 CrossRef CAS PubMed.
- W. Tao, M. E. Yurkovich, S. Wen, K. E. Lebe, M. Samborskyy, Y. Liu, A. Yang, Y. Liu, Y. Ju, Z. Deng, M. Tosin, Y. Sun and P. F. Leadlay, Chem. Sci., 2016, 7, 376–385 RSC.
- J. Havemann, M. E. Yurkovich, R. Jenkins, S. Harringer, W. Tao, S. Wen, Y. Sun, P. F. Leadlay and M. Tosin, Chem. Commun., 2017, 53, 1912–1915 RSC.
- J. G. McCoy, H. D. Johnson, S. Singh, C. A. Bingman, I.-K. Lei, J. S. Thorson and G. N. Phillips, Proteins: Struct., Funct., Bioinf., 2009, 74, 50–60 CrossRef CAS PubMed.
- Q. Zhao, Q. He, W. Ding, M. Tang, Q. Kang, Y. Yu, W. Deng, Q. Zhang, J. Fang, G. Tang and W. Liu, Chem. Biol., 2008, 15, 693–705 CrossRef CAS PubMed.
- W. Ding, W. Deng, M. Tang, Q. Zhang, G. Tang, Y. Bi and W. Liu, Mol. BioSyst., 2010, 6, 1071–1081 RSC.
- L. Song, L. Laureti, C. Corre, P. Leblond, B. Aigle and G. L. Challis, J. Antibiot., 2014, 67, 71–76 CrossRef CAS PubMed.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef CAS PubMed.
- D. M. Bis, Y. H. Ban, E. D. James, N. Alqahtani, R. Viswanathan and A. L. Lane, ChemBioChem, 2015, 16, 990–997 CrossRef CAS PubMed.
- B. Frank, S. C. Wenzel, H. B. Bode, M. Scharfe, H. Blöcker and R. Müller, J. Mol. Biol., 2007, 374, 24–38 CrossRef CAS PubMed.
- S. A. Borisova, L. Zhao, C. E. Melançon, C.-L. Kao and H.-w. Liu, J. Am. Chem. Soc., 2004, 126, 6534–6535 CrossRef CAS PubMed.
- M. C. Moncrieffe, M.-J. Fernandez, D. Spiteller, H. Matsumura, N. J. Gay, B. F. Luisi and P. F. Leadlay, J. Mol. Biol., 2012, 415, 92–101 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- Q. Zheng, H. Fang and W. Liu, Org. Biomol. Chem., 2017, 15, 3376–3390 CAS.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS PubMed.
- L. C. Foulston and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13461–13466 CrossRef CAS PubMed.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2016, 138, 15511–15514 CrossRef CAS PubMed.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 16012–16015 CrossRef CAS PubMed.
- T. S. Young and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13053–13058 CrossRef CAS PubMed.
- Q. Zheng, S. Wang, R. Liao and W. Liu, ACS Chem. Biol., 2016, 11, 2673–2678 CrossRef CAS PubMed.
- W. J. K. Crone, N. M. Vior, J. Santos-Aberturas, L. G. Schmitz, F. J. Leeper and A. W. Truman, Angew. Chem., Int. Ed., 2016, 55, 9639–9643 CrossRef CAS PubMed.
- W. J. K. Crone, F. J. Leeper and A. W. Truman, Chem. Sci., 2012, 3, 3516–3521 RSC.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 3522–3525 RSC.
- L. Huo, S. Rachid, M. Stadler, S. C. Wenzel and R. Müller, Chem. Biol., 2012, 19, 1278–1287 CrossRef CAS PubMed.
- J. G. Gober, S. V. Ghodge, J. W. Bogart, W. J. Wever, R. R. Watkins, E. M. Brustad and A. A. Bowers, ACS Chem. Biol., 2017, 12, 1726–1731 CrossRef CAS PubMed.
- J. A. E. Payne, M. Schoppet, M. H. Hansen and M. J. Cryle, Mol. BioSyst., 2017, 13, 9–22 RSC.
- G. H. Hur, C. R. Vickery and M. D. Burkart, Nat. Prod. Rep., 2012, 29, 1074–1098 RSC.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Sussmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100 CrossRef CAS PubMed.
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105–1118 CrossRef CAS PubMed.
- O. Puk, P. Huber, D. Bischoff, J. Recktenwald, G. Jung, R. D. Sussmuth, K.-H. van Pee, W. Wohlleben and S. Pelzer, Chem. Biol., 2002, 9, 225–235 CrossRef CAS PubMed.
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol. Chem., 2010, 285, 24562–24574 CrossRef CAS PubMed.
- H. Chen and C. T. Walsh, Chem. Biol., 2001, 8, 301–312 CrossRef CAS PubMed.
- Y. Hirose, K. Watanabe, A. Minami, T. Nakamura, H. Oguri and H. Oikawa, J. Antibiot., 2011, 64, 117–122 CrossRef CAS PubMed.
- C. Zhang, L. Kong, Q. Liu, X. Lei, T. Zhu, J. Yin, B. Lin, Z. Deng and D. You, PLoS One, 2013, 8, e56772 CAS.
- K. Koketsu, H. Oguri, K. Watanabe and H. Oikawa, Org. Lett., 2006, 8, 4719–4722 CrossRef CAS PubMed.
- U. Galm, E. Wendt-Pienkowski, L. Wang, N. P. George, T.-J. Oh, F. Yi, M. Tao, J. M. Coughlin and B. Shen, Mol. BioSyst., 2009, 5, 77–90 RSC.
- H. Chen, B. K. Hubbard, S. E. O'Connor and C. T. Walsh, Chem. Biol., 2002, 9, 103–112 CrossRef CAS PubMed.
- C. E. Wise and T. M. Makris, ACS Chem. Biol., 2017, 12, 1316–1326 CrossRef CAS PubMed.
- S. Mulyani, E. Egel, C. Kittel, S. Turkanovic, W. Wohlleben, R. D. Süssmuth and K.-H. van Pée, ChemBioChem, 2010, 11, 266–271 CrossRef CAS PubMed.
- M. Pacholec, N. J. Hillson and C. T. Walsh, Biochemistry, 2005, 44, 12819–12826 CrossRef CAS PubMed.
- A. V. Ramaswamy, C. M. Sorrels and W. H. Gerwick, J. Nat. Prod., 2007, 70, 1977–1986 CrossRef CAS PubMed.
- A. Muliandi, Y. Katsuyama, K. Sone, M. Izumikawa, T. Moriya, J. Hashimoto, I. Kozone, M. Takagi, K. Shin-ya and Y. Ohnishi, Chem. Biol., 2014, 21, 923–934 CrossRef CAS PubMed.
- F. G. Healy, S. B. Krasnoff, M. Wach, D. M. Gibson and R. Loria, J. Bacteriol., 2002, 184, 2019–2029 CrossRef CAS PubMed.
- S. Pohle, C. Appelt, M. Roux, H.-P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 CrossRef CAS PubMed.
- V. Schubert, F. Di Meo, P.-L. Saaidi, S. Bartoschek, H.-P. Fiedler, P. Trouillas and R. D. Süssmuth, Chem.–Eur. J., 2014, 20, 4948–4955 CrossRef CAS PubMed.
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef CAS PubMed.
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Süssmuth and M. J. Cryle, Angew. Chem., Int. Ed., 2014, 53, 8518–8522 CrossRef CAS PubMed.
- B. Kokona, E. S. Winesett, A. Nikolai von Krusenstiern, M. J. Cryle, R. Fairman and L. K. Charkoudian, Anal. Biochem., 2016, 495, 42–51 CrossRef CAS PubMed.
- C. Fu, L. Keller, A. Bauer, M. Brönstrup, A. Froidbise, P. Hammann, J. Herrmann, G. Mondesert, M. Kurz, M. Schiell, D. Schummer, L. Toti, J. Wink and R. Müller, J. Am. Chem. Soc., 2015, 137, 7692–7705 CrossRef CAS PubMed.
- X. Wang, J. Tabudravu, M. E. Rateb, K. J. Annand, Z. Qin, M. Jaspars, Z. Deng, Y. Yu and H. Deng, Mol. BioSyst., 2013, 9, 1286–1289 RSC.
- D. Zabala, J. W. Cartwright, D. M. Roberts, B. J. C. Law, L. Song, M. Samborskyy, P. F. Leadlay, J. Micklefield and G. L. Challis, J. Am. Chem. Soc., 2016, 138, 4342–4345 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS PubMed.
- E. Stegmann, H.-J. Frasch and W. Wohlleben, Curr. Opin. Microbiol., 2010, 13, 595–602 CrossRef CAS PubMed.
- R. S. Al Toma, C. Brieke, M. J. Cryle and R. D. Suessmuth, Nat. Prod. Rep., 2015, 32, 1207–1235 RSC.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef CAS PubMed.
- B. Hadatsch, D. Butz, T. Schmiederer, J. Steudle, W. Wohlleben, R. Suessmuth and E. Stegmann, Chem. Biol., 2007, 14, 1078–1089 CrossRef CAS PubMed.
- D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann, G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben and R. D. Suessmuth, ChemBioChem, 2005, 6, 267–272 CrossRef CAS PubMed.
- D. Bischoff, S. Pelzer, A. Holtzel, G. J. Nicholson, S. Stockert, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew. Chem., Int. Ed., 2001, 40, 1693–1696 CrossRef CAS PubMed.
- D. Bischoff, S. Pelzer, B. Bister, G. J. Nicholson, S. Stockert, M. Schirle, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew. Chem., Int. Ed., 2001, 40, 4688–4691 CrossRef CAS PubMed.
- R. D. Sussmuth, S. Pelzer, G. Nicholson, T. Walk, W. Wohlleben and G. Jung, Angew. Chem., Int. Ed., 1999, 38, 1976–1979 CrossRef CAS.
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. Robinson, R. D. Suessmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640–653 CrossRef CAS PubMed.
- P. C. Schmartz, K. Wolfel, K. Zerbe, E. Gad, E. El Tamany, H. K. Ibrahim, K. Abou-Hadeed and J. A. Robinson, Angew. Chem., Int. Ed., 2012, 51, 11468–11472 CrossRef CAS PubMed.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 CrossRef CAS PubMed.
- K. Zerbe, K. Woithe, D. B. Li, F. Vitali, L. Bigler and J. A. Robinson, Angew. Chem., Int. Ed., 2004, 43, 6709–6713 CrossRef CAS PubMed.
- K. Haslinger, C. Redfield and M. J. Cryle, Proteins: Struct., Funct., Bioinf., 2015, 83, 711–721 CrossRef CAS PubMed.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 CrossRef CAS PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- C. Brieke, M. Peschke, K. Haslinger and M. J. Cryle, Angew. Chem., Int. Ed., 2015, 54, 15715–15719 CrossRef CAS PubMed.
- J. Tailhades, M. Schoppet, A. Greule, M. Peschke, C. Brieke and M. J. Cryle, Chem. Commun., 2018, 54(17), 2146–2149 RSC.
- M. Peschke, C. Brieke, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Biochemistry, 2017, 56, 1239–1247 CrossRef CAS PubMed.
- V. Ulrich, M. Peschke, C. Brieke and M. Cryle, Mol. BioSyst., 2016, 12, 2992–3004 RSC.
- V. Ulrich, C. Brieke and M. Cryle, Beilstein J. Org. Chem., 2016, 12, 2849–2864 CrossRef CAS PubMed.
- M. Peschke, C. Brieke and M. J. Cryle, Sci. Rep., 2016, 6, 35584 CrossRef CAS PubMed.
- C. Brieke, V. Kratzig, K. Haslinger, A. Winkler and M. J. Cryle, Org. Biomol. Chem., 2015, 13, 2012–2021 CAS.
- K. Haslinger, E. Maximowitsch, C. Brieke, A. Koch and M. J. Cryle, ChemBioChem, 2014, 15, 2719–2728 CrossRef CAS PubMed.
- H.-T. Chiu, B. K. Hubbard, A. N. Shah, J. Eide, R. A. Fredenburg, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8548–8553 CrossRef CAS PubMed.
- B. Nazari, C. C. Forneris, M. I. Gibson, K. Moon, K. R. Schramma and M. R. Seyedsayamdost, MedChemComm, 2017, 8, 780–788 RSC.
- O.-K. Park, H.-Y. Choi, G.-W. Kim and W.-G. Kim, ChemBioChem, 2016, 17, 1725–1731 CrossRef CAS PubMed.
- A. Mollo, A. N. von Krusenstiern, J. A. Bulos, V. Ulrich, K. S. Akerfeldt, M. J. Cryle and L. K. Charkoudian, RSC Adv., 2017, 7, 35376–35384 RSC.
- W.-T. Liu, R. D. Kersten, Y.-L. Yang, B. S. Moore and P. C. Dorrestein, J. Am. Chem. Soc., 2011, 133, 18010–18013 CrossRef CAS PubMed.
- X. Jin, M. Rao, W. Wei, M. Ge, J. Liu, D. Chen and Y. Liang, Biotechnol. Lett., 2012, 34, 2283–2289 CrossRef CAS PubMed.
- J. Ma, Z. Wang, H. Huang, M. Luo, D. Zuo, B. Wang, A. Sun, Y.-Q. Cheng, C. Zhang and J. Ju, Angew. Chem., Int. Ed., 2011, 50, 7797–7802 CrossRef CAS PubMed.
- H. Zhang, J. Chen, H. Wang, Y. Xie, J. Ju, Y. Yan and H. Zhang, FEBS Lett., 2013, 587, 1675–1680 CrossRef CAS PubMed.
- M. Schorn, J. Zettler, J. P. Noel, P. C. Dorrestein, B. S. Moore and L. Kaysser, ACS Chem. Biol., 2014, 9, 301–309 CrossRef CAS PubMed.
- S. Zhang, J. Zhu, D. L. Zechel, C. Jessen-Trefzer, R. T. Eastman, T. Paululat and A. Bechthold, ChemBioChem, 2018, 19, 272–279 CrossRef CAS PubMed.
- S. M. Barry, J. A. Kers, E. G. Johnson, L. Song, P. R. Aston, B. Patel, S. B. Krasnoff, B. R. Crane, D. M. Gibson, R. Loria and G. L. Challis, Nat. Chem. Biol., 2012, 8, 814–816 CrossRef CAS PubMed.
- H. Tomita, Y. Katsuyama, H. Minami and Y. Ohnishi, J. Biol. Chem., 2017, 292, 15859–15869 CrossRef CAS PubMed.
- M. Gunsior, S. D. Breazeale, A. J. Lind, J. Ravel, J. W. Janc and C. A. Townsend, Chem. Biol., 2004, 11, 927–938 CrossRef CAS PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed.
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 CrossRef CAS PubMed.
- A. M. Reeve, S. D. Breazeale and C. A. Townsend, J. Biol. Chem., 1998, 273, 30695–30703 CrossRef CAS PubMed.
- W. L. Kelly and C. A. Townsend, J. Am. Chem. Soc., 2002, 124, 8186–8187 CrossRef CAS PubMed.
- M. Sørensen, E. H. J. Neilson and B. L. Møller, Mol. Plant, 2018, 11, 95–117 CrossRef PubMed.
- L. Ray, K. Yamanaka and B. S. Moore, Angew. Chem., Int. Ed., 2015, 55, 364–367 CrossRef PubMed.
- D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRef CAS PubMed.
- M. U. Huynh, M. C. Elston, N. M. Hernandez, D. B. Ball, S.-i. Kajiyama, K. Irie, W. H. Gerwick and D. J. Edwards, J. Nat. Prod., 2010, 73, 71–74 CrossRef CAS PubMed.
- M. Moutiez, P. Belin and M. Gondry, Chem. Rev., 2017, 117, 5578–5618 CrossRef CAS PubMed.
- M. J. Cryle, S. G. Bell and I. Schlichting, Biochemistry, 2010, 49, 7282–7296 CrossRef CAS PubMed.
- P. Belin, M. H. Le Du, A. Fielding, O. Lequin, M. Jacquet, J.-B. Charbonnier, A. Lecoq, R. Thai, M. Courçon, C. Masson, C. Dugave, R. Genet, J.-L. Pernodet and M. Gondry, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7426–7431 CrossRef PubMed.
- K. J. McLean, P. Carroll, D. G. Lewis, A. J. Dunford, H. E. Seward, R. Neeli, M. R. Cheesman, L. Marsollier, P. Douglas, W. E. Smith, I. Rosenkrands, S. T. Cole, D. Leys, T. Parish and A. W. Munro, J. Biol. Chem., 2008, 283, 33406–33416 CrossRef CAS PubMed.
- M. Fonvielle, M.-H. Le Du, O. Lequin, A. Lecoq, M. Jacquet, R. Thai, S. Dubois, G. Grach, M. Gondry and P. Belin, J. Biol. Chem., 2013, 288, 17347–17359 CrossRef CAS PubMed.
- H. E. Seward, A. Roujeinikova, K. J. McLean, A. W. Munro and D. Leys, J. Biol. Chem., 2006, 281, 39437–39443 CrossRef CAS PubMed.
- D. Leys, C. G. Mowat, K. J. McLean, A. Richmond, S. K. Chapman, M. D. Walkinshaw and A. W. Munro, J. Biol. Chem., 2003, 278, 5141–5147 CrossRef CAS PubMed.
- V. G. Dumas, L. A. Defelipe, A. A. Petruk, A. G. Turjanski and M. A. Marti, Proteins: Struct., Funct., Bioinf., 2014, 82, 1004–1021 CrossRef CAS PubMed.
- J. B. Patteson, W. Cai, R. A. Johnson, K. C. Santa Maria and B. Li, Biochemistry, 2018, 57(1), 61–65 CrossRef CAS PubMed.
- S. Meng, W. Han, J. Zhao, X.-H. Jian, H.-X. Pan and G.-L. Tang, Angew. Chem., Int. Ed., 2018, 57, 719–723 CrossRef CAS PubMed.
- F. G. Healy, M. Wach, S. B. Krasnoff, D. M. Gibson and R. Loria, Mol. Microbiol., 2000, 38, 794–804 CrossRef CAS PubMed.
- B. Gu, S. He, X. Yan and L. Zhang, Appl. Microbiol. Biotechnol., 2013, 97, 8439–8453 CrossRef CAS PubMed.
- H. Kries, D. L. Niquille and D. Hilvert, Chem. Biol., 2015, 22, 640–648 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS PubMed.
- P. J. Belshaw, C. T. Walsh and T. Stachelhaus, Science, 1999, 284, 486–489 CrossRef CAS PubMed.
- F. Yu, M. Li, C. Xu, Z. Wang, H. Zhou, M. Yang, Y. Chen, L. Tang and J. He, PLoS One, 2013, 8, e81526 Search PubMed.
- S. C. Dodani, J. K. B. Cahn, T. Heinisch, S. Brinkmann-Chen, J. A. McIntosh and F. H. Arnold, ChemBioChem, 2014, 15, 2259–2267 CrossRef CAS PubMed.
Footnotes |
| † The authors would like to acknowledge the outstanding and inspirational contributions of Paul Ortiz de Montellano to the field of cytochrome P450 research and of Prof. Sir Alan R. Battersby (1925–2018) to the molecular understanding of biosynthesis over the course of their careers. |
| ‡ Epothilone biosynthesis is performed by hybrid PKS/NRPS machinery, although the majority of the machinery and the region modified by EpoK is the result of PKS mediated biosynthesis. |
| § The intermediate postulated to be the substrate of NsnF is strictly bound to a PCP-domain, as the terminal module of the nocardiopsin mixed PKS/NRPS is an NRPS module; however, as the substrate of the P450 is a PKS-portion of the molecule, this compound is included in the PKS section. |
| This journal is © The Royal Society of Chemistry 2018 |