
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Roles of 2-oxoglutarate oxygenases and isopenicillin N synthase in β-lactam biosynthesis
Patrick
Rabe†
 ,
Jos J. A. G.
Kamps†
,
Christopher J.
Schofield†
* and
Christopher T.
Lohans†
*
,
Jos J. A. G.
Kamps†
,
Christopher J.
Schofield†
* and
Christopher T.
Lohans†
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk; christopher.lohans@chem.ox.ac.uk
First published on 29th May 2018
Abstract
Covering: up to 2017
2-Oxoglutarate (2OG) dependent oxygenases and the homologous oxidase isopenicillin N synthase (IPNS) play crucial roles in the biosynthesis of β-lactam ring containing natural products. IPNS catalyses formation of the bicyclic penicillin nucleus from a tripeptide. 2OG oxygenases catalyse reactions that diversify the chemistry of β-lactams formed by both IPNS and non-oxidative enzymes. Reactions catalysed by the 2OG oxygenases of β-lactam biosynthesis not only involve their typical hydroxylation reactions, but also desaturation, epimerisation, rearrangement, and ring-forming reactions. Some of the enzymes involved in β-lactam biosynthesis exhibit remarkable substrate and product selectivities. We review the roles of 2OG oxygenases and IPNS in β-lactam biosynthesis, highlighting opportunities for application of knowledge of their roles, structures, and mechanisms.
 Patrick Rabe | Patrick Rabe studied chemistry at the University of Braunschweig. He received the Fritz-Wagner Award of the Helmholtz Centre for Infection Research for his master's thesis in 2013. In 2016 he finished his PhD at the Kekulé-Institute of Organic Chemistry and Biochemistry at the University of Bonn on “Mechanistic Studies on Bacterial Terpene Synthases” with Prof. Jeroen S. Dickschat. For his doctoral thesis he received the 2017 Dechema doctoral award. Since 2017 he is working in the group of Prof. Chris Schofield on mutagenesis studies and time-resolved crystallography on isopenicillin N synthase with a scholarship funded by the Deutsche Akademie für Naturforscher Leopoldina. |
 Jos J. A. G. Kamps | Jos Kamps obtained his bachelor's degree in chemistry from the HAN University for Applied Sciences. He finished his master's degree at the Radboud University Nijmegen in organic chemistry in 2015. During this time he worked with Dr Jasmin Mecinović in the group of Prof. Floris Rutjes on the chemical recognition of methylated lysine in epigenetics. In 2015 he started his PhD as a student of the Synthesis for Biology and Medicine Centre for Doctoral Training with a Clarendon Award scholarship. Currently he is working in the groups of Prof. Chris Schofield and Prof. Timothy Claridge on the recognition of substrate analogues by isopenicillin N synthase. |
 Christopher J. Schofield | Chris Schofield is currently Head of Organic Chemistry, and a Fellow of Hertford College, University of Oxford. His current research interests include antibiotic biosynthesis and resistance, regulation of gene expression by oxygen, and epigenetics with a common theme being enzymes that catalyse chemically interesting reactions of biomedicinal importance. |
 Christopher T. Lohans | Christopher Lohans completed his PhD in 2014 in the group of Prof. John Vederas at the University of Alberta, Canada, studying the structure and biosynthesis of antimicrobial peptides. He then joined the group of Prof. Chris Schofield at the University of Oxford, UK, as a postdoctoral fellow supported by the Canadian Institutes of Health Research. His postdoctoral research has focused on investigating the mechanism and inhibition of the serine β-lactamases using NMR spectroscopy. |
1. Introduction
β-Lactam ring containing molecules remain amongst the most important small molecule medicines in use.1 Following the discovery of the penicillins in the 1940s, new generations of β-lactam antibacterials, based on natural products discovered in the 1970s and 1980s, were developed to improve their spectrum of activity and combat resistance (Fig. 1). β-Lactam antibiotics work by reacting with catalytically important nucleophilic serine residues in transpeptidases (penicillin binding proteins, PBPs) that are crucially involved in bacterial cell wall biosynthesis (Fig. 2). β-Lactam resistance is substantially, but not fully, mediated by β-lactamase enzymes which catalyse hydrolysis of the β-lactam ring (Fig. 2). The new generations of natural product-derived β-lactams include cephalosporins, carbapenems, clavams, and monocyclic β-lactams. Depending on the functionalisation patterns of the core β-lactam ring system, these compounds manifest different antibacterial activity profiles and susceptibilities to the constantly evolving β-lactamases. It should be noted that naturally occurring β-lactams have roles beyond inhibiting PBPs, sometimes with important biological consequences such as for the plant toxin tabtoxin (Fig. 1).2–4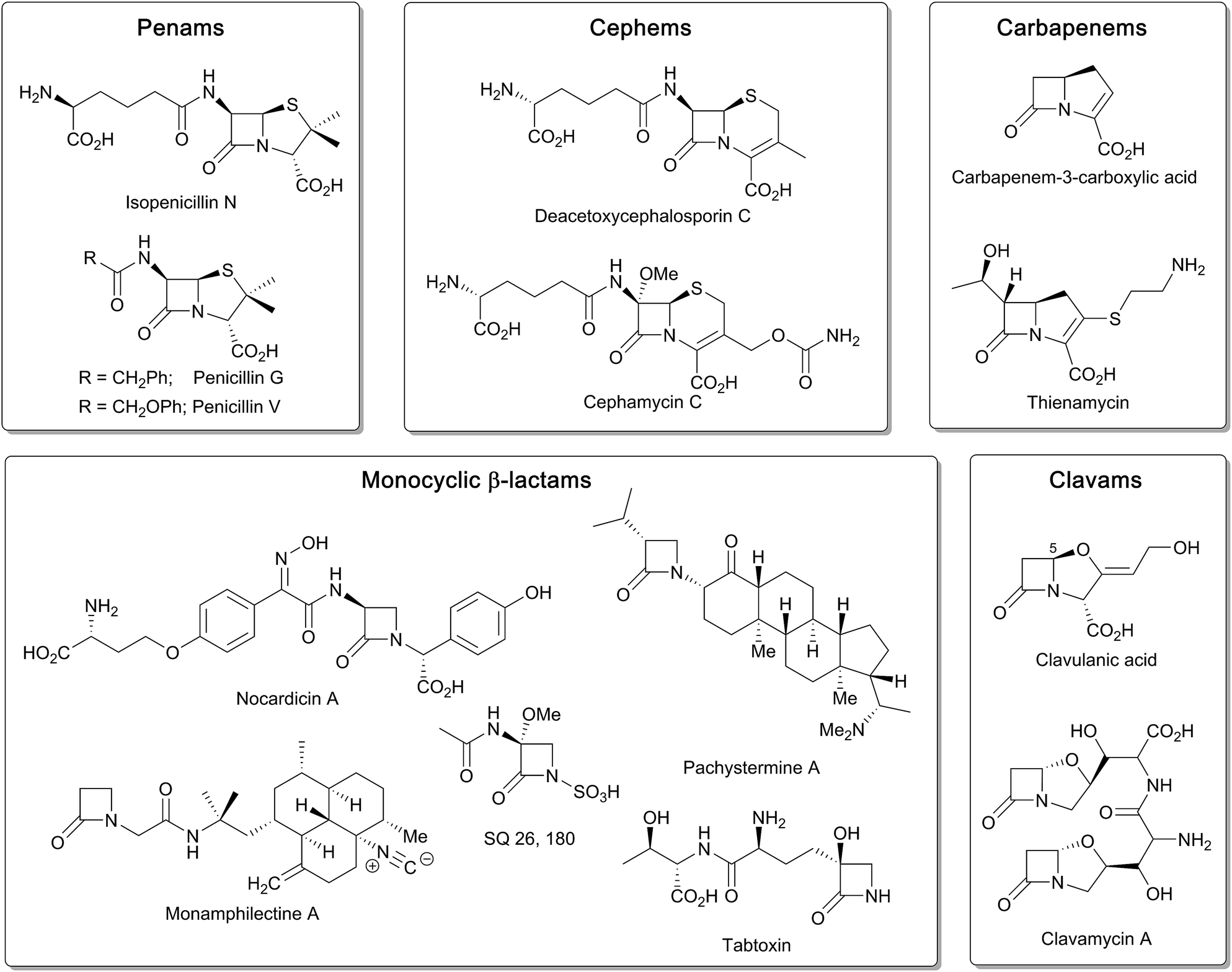 | ||
| Fig. 1 Examples of the different types of naturally occurring β-lactam ring containing molecules. | ||
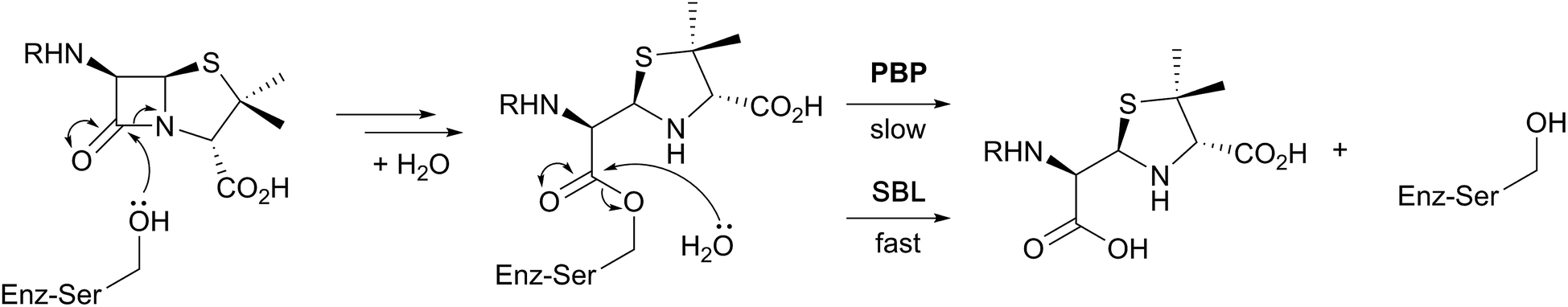 | ||
| Fig. 2 Outline interaction of β-lactams (exemplified by a penicillin) with penicillin binding proteins (PBPs) and serine β-lactamases (SBLs). A major difference between the PBPs and SBLs is the stability of the acyl-enzyme complex. | ||
Clavulanic acid, the sole clavam to be used clinically, has relatively weak antibacterial activity, but is a potent inhibitor of some nucleophilic serine β-lactamases. Hence, like the other clinically used β-lactamase inhibitors (sulbactam, tazobactam, and avibactam; the latter being the sole non-β-lactam approved for use as a β-lactamase inhibitor), it is used in combination with a more potent antibiotic (for example, amoxicillin and clavulanic acid are combined in the drug Augmentin).5 Synthetic β-lactams have found therapeutic applications other than as antibacterials.6 Despite extensive resistance, the longevity of β-lactams as antibacterials is remarkable in the history of anti-infective medicine.7 This has led to the proposal that the combination of β-lactam inhibitors of PBPs and their PBP targets is special from a chemical perspective.7 Thus, some naturally occurring β-lactams, which likely have ancient origins in bacteria, may well be highly evolved to their antibacterial roles. If this is the case it would seem likely that their biosynthesis may also be highly evolved – at least for secondary metabolites.
Whether or not β-lactams are chemically unique as antibacterials, at least from an enzymology perspective, some of the reactions occurring during β-lactam biosynthesis are spectacular (for prior relevant reviews on β-lactam biosynthesis, see ref. 4 and 8–25). Although non-oxidative reactions play crucial roles in β-lactam biosynthesis, reactions catalysed by ferrous iron and 2-oxoglutarate (2OG) dependent oxygenases and the related oxidase isopenicillin N synthase (IPNS) are of particular interest (Fig. 3). The 2OG oxygenases were first discovered during work on collagen biosynthesis and their involvement in β-lactam biosynthesis was unexpected. They typically catalyse two electron oxidations, most commonly hydroxylations, in which substrate oxidation is coupled to conversion of 2OG and dioxygen to succinate and carbon dioxide.22
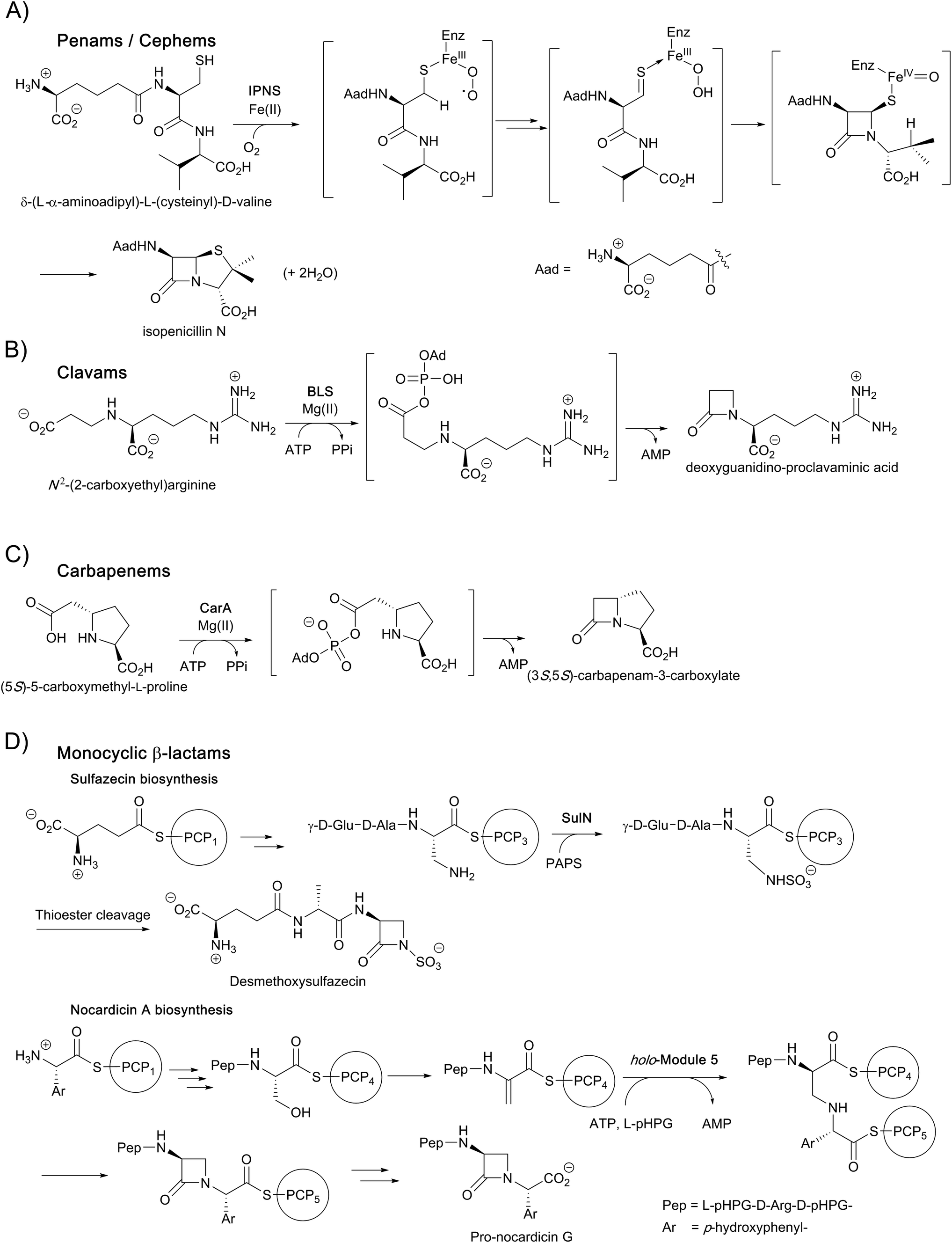 | ||
| Fig. 3 Enzymes catalysing β-lactam formation during antibiotic biosynthesis, including: (A) isopenicillin N synthase (IPNS), (B) β-lactam synthetase (BLS), (C) CarA, and (D) non-ribosomal peptide synthetases (NRPS) SulN and NocB. Outline mechanisms/steps are shown. Note that not all cofactors, co-substrates, and products are shown. PAPS: 3'-phosphoadenosine-5'-phosphosulfate; PCP: peptidyl-carrier protein. | ||
In the case of the non-penicillin β-lactam biosynthetic pathways, 2OG oxygenases play roles in diversifying the chemistry, and hence the activities, of the β-lactams formed by non-oxygen dependent chemistry. During biosynthesis of the clavam and the carbapenem bicyclic ring systems, β-lactam formation is catalysed by asparagine synthetase related enzymes (Fig. 3).4,8,13,16,22 These β-lactam synthetases catalyse formation of the β-lactam ring from appropriate β-amino acid precursors, i.e. in effect they catalyse the reverse of β-lactamase catalysis (Fig. 2), but use ATP to activate the carboxylic acid. In the case of the clavams, a monocyclic β-lactam ring is formed (Fig. 3); this acts as a precursor for the formation of the bicyclic clavam ring in reactions catalysed by the 2OG oxygenase clavaminic acid synthase (CAS). In the carbapenem pathways, β-lactam synthetases catalyse formation of the (3S,5S)-carbapenam ring system which, at least in the pathway to carbapen-2-em-3-carboxylate, is epimerised and desaturated by CarC, a 2OG oxygenase, to give a (5R)-carbapenem (Fig. 3). Thus, in these pathways, 2OG oxygenases play roles both in forming a bicyclic β-lactam ring system (clavams) and in modifying a bicyclic β-lactam ring system (carbapenems) to create antibacterials.4,8,13,16,18–22
Some monocyclic β-lactams, for example the nocardicins and monobactams (Fig. 3), are formed by the action of non-ribosomal peptide synthetases (NRPSs) which catalyse formation of the N1–C4 β-lactam bond from an appropriate synthetase-bound precursor peptide.11 In some cases, for example the monobactams and tabtoxin, 2OG oxygenases (likely) are involved in modifications subsequent to β-lactam formation.4
Notably, both the clavam and carbapenem biosynthetic pathways involve multiple steps en route to the clinically useful bicyclic antibiotics/β-lactamase inhibitors, for example, thienamycin and clavulanic acid (Fig. 1). Further, these pathways can result in multiple coproducts. Although it was possible to optimise clavulanic acid fermentation for commercial use,10 this has not yet been possible in the case of clinically useful carbapenems, which are instead produced by total synthesis.4 By contrast to the, at least superficially, relatively complex carbapenem and clavam biosynthesis pathways, the pathway leading to the penicillins and subsequently formed cephalosporins appears to be a paradigm of biosynthetic efficiency (Fig. 3).
Following pioneering work identifying the tripeptide δ-(L-α-aminoadipoyl)-L-(cysteinyl)-D-valine (ACV) as a precursor of the penicillins,26,27 the ferrous iron dependent oxidase IPNS was isolated and shown to catalyse the four electron oxidation of ACV to form, in a single step, the bicyclic nucleus of isopenicillin N (IPN), the first formed β-lactam in the pathway. IPN is a branchpoint in some organisms; its δ-(L-α-aminoadipoyl)-side chain can be exchanged (by the action of an amidohydrolase/acyltransferase) for a more hydrophobic side chain to give a readily extractable clinically useful penicillin, e.g. penicillins G or V (Fig. 1). These penicillins with hydrophobic sidechains can be used as starting points for the production of penicillins with clinically better sidechains, via enzymatically produced 6-aminopenicillanic acid (6-APA), or for the synthesis of cephalosporins, via the oxidative chemical ring expansion of the penam nucleus to that of the cephems. This chemical ring expansion of penam sulphoxides to the cephems has echoes in the biosynthetic formation of the cephalosporins (see below).
During cephalosporin biosynthesis, the side chain of IPN is epimerised to give penicillin N, which is the substrate for deacetoxycephalosporin C synthase (DAOCS), a 2OG oxygenase catalysing an unusual oxidative rearrangement to give deacetoxycephalosporin C (DAOC).4,8,16,18–22 Studies on the chemically interesting mechanisms and structures of IPNS and DAOCS were pioneering for the 2OG oxygenase superfamily.28–31 The procollagen prolyl hydroxylases were the first 2OG oxygenases to be identified; however, structural studies on the (animal) procollagen prolyl hydroxylases have been difficult. Unexpectedly, crystallographic studies on IPNS and 2OG oxygenases acting on small molecules22 led to the discovery that 2OG oxygenases are very widely distributed in aerobic biology (archaea being a likely exception), including in humans where there are approximately 60 such enzymes.22 Work over the last two to three decades has shown that 2OG oxygenases in humans and other animals have diverse roles. In addition to collagen biosynthesis,32 these enzymes are involved in epigenetics, hypoxia sensing, chromatin regulation, DNA repair, ribosome/RNA modifications, and lipid biosynthesis.33–36 By contrast with some of their reactions in β-lactam biosynthesis, to date, the reactions assigned for 2OG oxygenases in animals involve chemically simple hydroxylations and N-methyl demethylations (also proceeding via initial hydroxylation).21
This brief review focuses on the roles of IPNS and 2OG oxygenases in β-lactam biosynthesis, describing current mechanistic knowledge and highlighting opportunities for the generation of new antibiotics and for the improved production procedures for existing antibiotics.
2. Isopenicillin N synthase
The discovery of isopenicillin N synthase,27,37 and its marvellous reaction that enables oxidative ring closure to the densely functionalised heterocyclic penicillin ring system, illustrates the power of biosynthesis. IPNS catalyses formation of the fused β-lactam and thiazolidine core of the penicillins via the four electron oxidation of a peptide precursor with concomitant reduction of dioxygen to two water molecules.22,38,39 The IPNS substrate, i.e. the tripeptide δ-(L-α-aminoadipoyl)-L-(cysteinyl)-D-valine (ACV) was identified more than 50 years ago.40 ACV is biosynthesised by action of the non-ribosomal peptide synthetase ACV synthetase from the precursors L-α-aminoadipic acid, L-cysteine, and L-valine, the stereochemistry of the latter being inverted during peptide formation.41–43The current detailed understanding of the mechanism of IPNS is based on extensive spectroscopic,44–46 computational,47–50 crystallographic,28,29,51–58 and substrate analogue studies (Fig. 4). Here we summarise these results – see earlier reviews for detailed descriptions of IPNS.4,6,15,17,21–24,39 The first crystal structure of IPNS (from Aspergillus nidulans) was obtained in complex with manganese substituting for iron; this structure revealed that in the resting enzyme state, the single active site metal is coordinated by two histidine (His214 and His270) and one aspartate residue (Asp216) as well as by the side-chain of Gln330 (Fig. 5A).28 The overall fold of IPNS is based on a distorted double-stranded β-helix (DSBH or ‘jelly roll’) core; this scaffold supports the triad of Fe(II) binding residues, and was subsequently shown to be conserved in 2OG oxygenases, including DAOCS.53,59 As shown in subsequent anaerobic structures obtained in the presence of ACV and Fe(II), Gln330 (ref. 28 and 60) (and one iron coordinating water) is displaced from the active site on binding of ACV, triggering conformational rearrangement of the C-terminal region (Asn326-Thr331).29 Together with other conformational changes, this arrangement means that the iron-ligated ACV is enclosed at the active site (Fig. 5B).29,57,61 Conformational changes at the active site upon binding of ACV are shown in Fig. 5D. Recently reported results for a C-terminally truncated IPNS variant support the proposal that the C-terminal α-helix shields the active site and hinders (potential) side reactions during catalysis.61
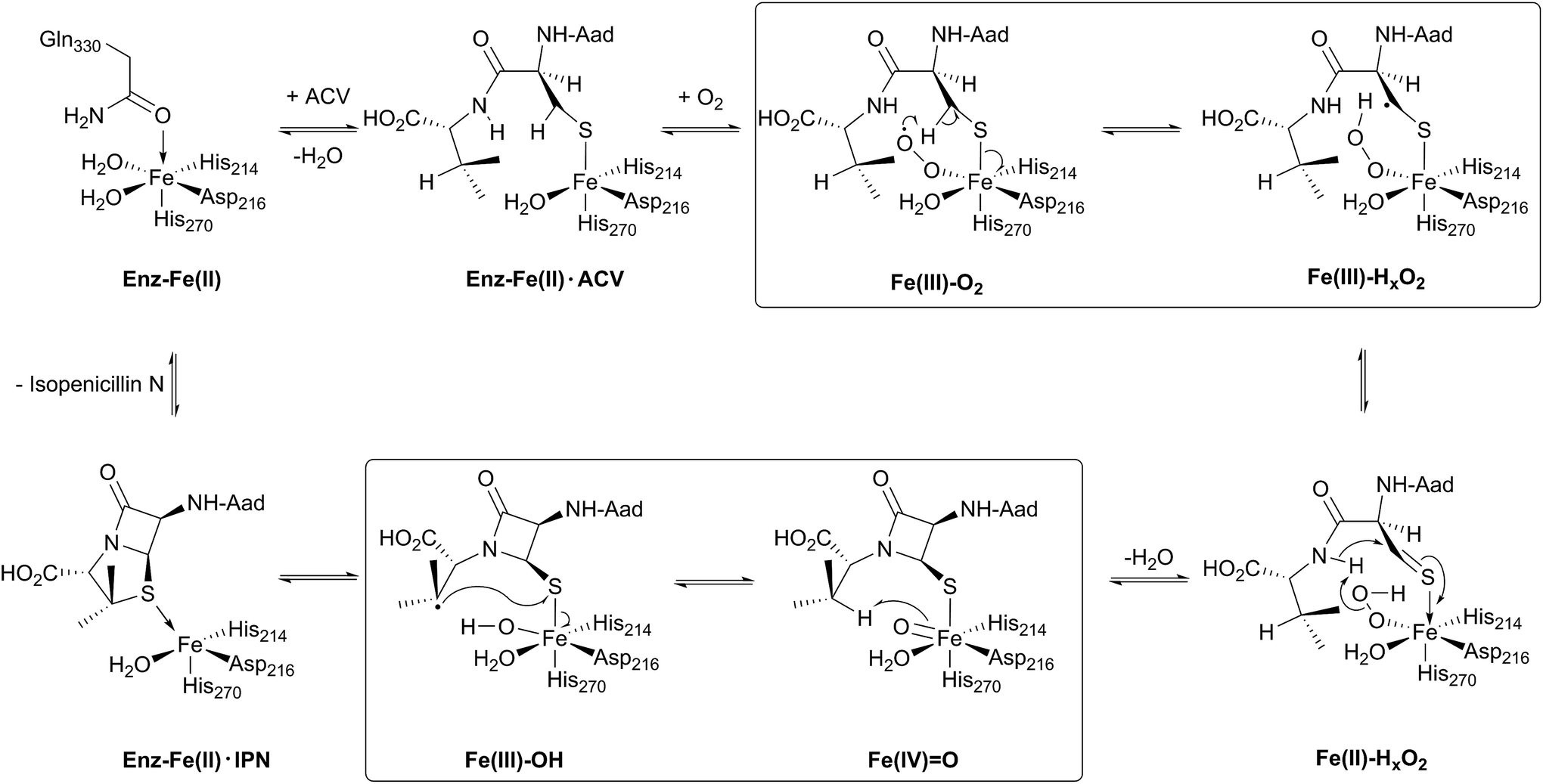 | ||
Fig. 4 Schematic overview of the currently proposed mechanism for isopenicillin N synthase (IPNS). Mechanism adapted from ref. 38. The high-spin Fe(III)–superoxo [Fe(III)–O2−] and high-spin Fe(IV)–oxo [Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] intermediates have been observed spectroscopically. O] intermediates have been observed spectroscopically. | ||
 | ||
| Fig. 5 Views from crystal structures of isopenicillin N synthase. (A) An IPNS–Mn complex (PDB 1IPS)28 showing coordination of metal in resting state by His214, His270, Asp216, as well as by the side-chain of Gln330 (Mn, substituting for Fe, in purple); (B) the IPNS–Fe–ACV–NO complex (PDB 1BLZ),29 showing binding of ACV by Arg87, the thiolate–Fe interaction, and enclosure of ACV at the active site, including by the extended helix; Thr331 and Lys98 form a salt bridge to constrain the conformation of the complex and to shield the active site/substrate (Fe in orange, extended helix in blue); (C) the IPNS–Fe–IPN complex (PDB 1QJE);55 (D) expanded view of the superposition of the IPNS–Fe–ACV complex active site [PDB 1BLZ; showing enclosure of ACV by the extended helix (in blue)] with the substrate-free IPNS–Mn complex (PDB 1IPS; α-helix indicated in red);29 (E) hydrophobic residues bind the ACV valine isopropyl group;29,61 (F) the IPNS–Fe–monocyclic β-lactam complex (PDB 1QJF)55 formed after incubation with δ-(L-α-aminoadipoyl)-L-(cysteinyl)-S-methyl-D-cysteine (ACMC) and O2, supporting initial β-lactam ring formation during stepwise bicyclisation to isopenicillin N. | ||
In the IPNS·ACV·Fe(II) complex, ACV coordinates to the Fe(II) via its cysteinyl thiolate and appears conformationally constrained in a manner appropriate for β-lactam ring formation. In addition to metal complexation, ACV is bound by both hydrophobic and electrostatic interactions, the latter including the interaction of the L-α-aminoadipoyl carboxylate group with Arg-87.29,51,61 The valine isopropyl group is bound in a hydrophobic pocket formed by the side-chains of Pro283, Leu223, Leu231, Val272, and Thr221 (Fig. 5E). Recent mutational studies on the role of these residues highlights their importance in substrate recognition and catalysis.61
Crystallographic analysis of the IPNS·ACV·Fe(II)·NO complex29 showed that NO (acting as a dioxygen analogue) binds adjacent to the cysteinyl thiol with the oxygen atom of the complexed NO directed toward the cysteinyl β-carbon. These observations support formation of the β-lactam ring prior to the thiazolidine ring (Fig. 5B) and are consistent with prior kinetic studies, in particular those employing deuterium labelled ACV substrates (with [3,3-2H2]-Cys or [3-2H1]-Val labelled ACV residues).22,57 Further support for initial β-lactam ring formation came from crystallographic analyses of IPNS with the substrate analogue δ-(L-α-aminoadipoyl)-L-(cysteinyl)-S-methyl-D-cysteine (ACMC), which was crystallised under anaerobic conditions, then exposed to a high pressure of oxygen. The resultant structure showed formation of a monocyclic β-lactam (with oxidation of the S-methyl group),55,56 providing strong experimental support for initial β-lactam formation (Fig. 5F and 6A).
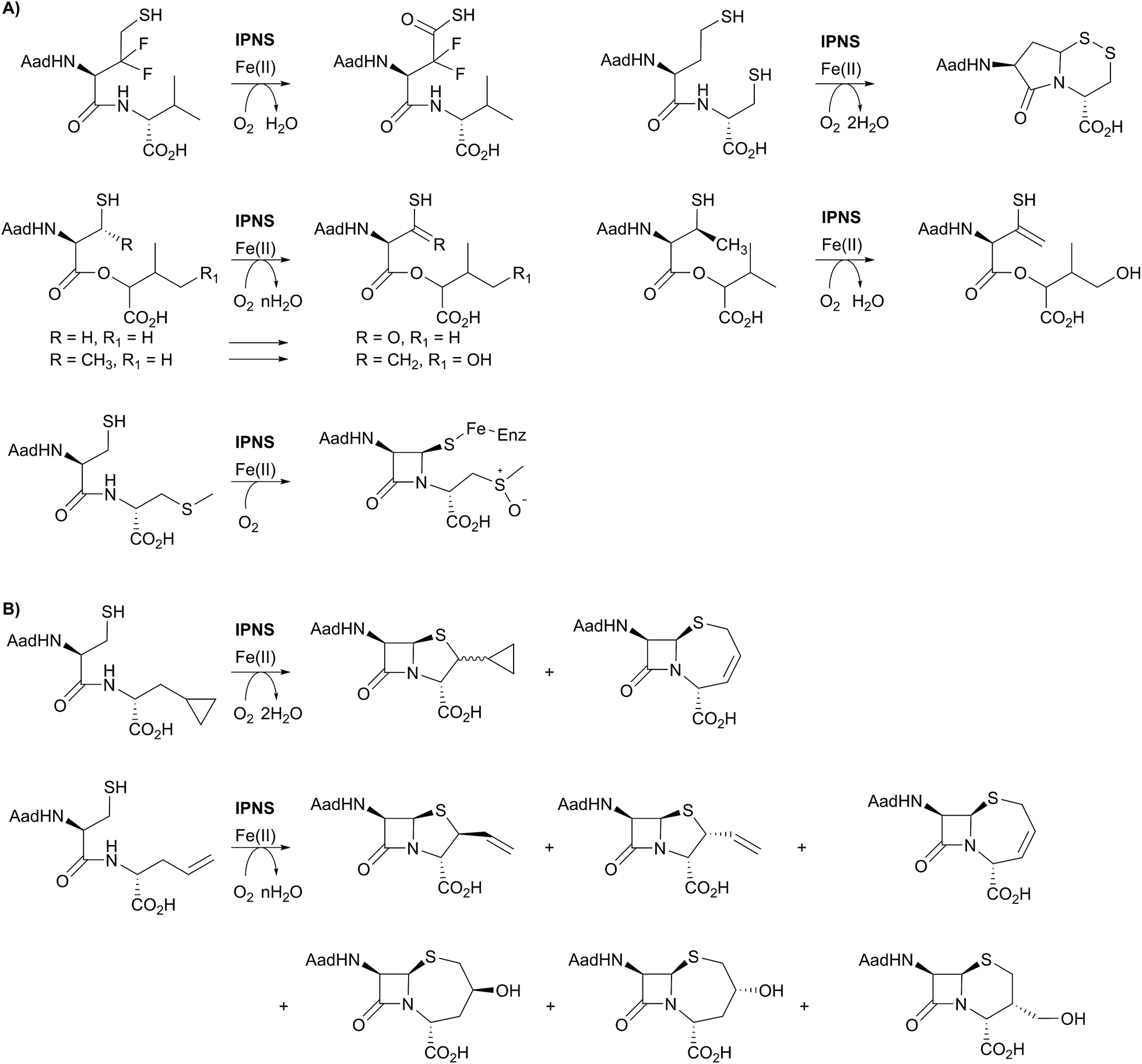 | ||
| Fig. 6 (A) Substrate analogues exemplifying the catalytic promiscuity of IPNS with respect to the type of oxidation reaction catalysed. The products derived from the depsipeptide analogues and the S-methyl cysteine analogue were observed crystallographically. (B) Examples showing the ability of IPNS to make different β-lactam ring systems by substituting the C-terminal valine of the natural (δ-(L-α-aminoadipoyl)-L-(cysteinyl)-D-valine) substrate. | ||
Mechanistic proposals for IPNS arising from early kinetics, substrate analogue, and crystallographic studies are supported by recent stopped flow, modelling, and spectroscopic (including by Mossbauer) studies.38 Following ACV binding/ligation to the active site Fe(II) (involving displacement of one metal ligated water and incompletely defined induced fit processes), dioxygen binds and reacts to form an (unusually stable) iron(III) superoxo species.38 The first step in the ACV-catalysed reaction involves abstraction of the pro-(S) cysteinyl C-3 hydrogen to give a thioalkyl radical. After inner-sphere electron transfer, a ferrous hydroperoxo intermediate [Fe(II)–OOH] and a thioaldehyde are formed. Heterolysis of the peroxo bond to form a water molecule, concomitant with abstraction of the cysteinyl-valine amido hydrogen by the terminal peroxide oxygen, and nucleophilic attack of the amide nitrogen onto the thioaldehyde forms a monocyclic β-lactam intermediate linked to a ferryl species [Fe(IV)O] (Fig. 4).55,57 This second key intermediate, which has been characterised by spectroscopy,38 mediates radical type abstraction of the ACV valine C-3 hydrogen resulting in a ferric hydroxyl [Fe(III)–OH] species. Reaction of the valinyl radical on the sulphur enables isopenicillin N product formation and restoration of the metal to the Fe(II) state (Fig. 4 and 5C).55
A feature of IPNS is its ability to accept substrate analogues, with a few selected examples shown in Fig. 6.4,15,22,62–78 The substrate analogue work has provided further insight into the mechanism of IPNS, and highlights the potential of non-heme oxygenases for production of synthetically challenging molecules. In terms of biocatalysis, the observation that ACV analogues lacking the amino group in the δ-(L-α-aminoadipoyl)-side chain are IPNS substrates has potential utility in pathway engineering.4,15 Some ACV analogues with hydrophobic groups substituting for the δ-(L-α-aminoadipoyl) residue of ACV are IPNS substrates, raising the possibility of engineering both ACV synthetase and IPNS to produce clinically useful penicillins in two enzyme catalysed steps.4,15
One mechanistic insight arising from the substrate analogue work is the delicate nature of the balance in regiochemistry of IPNS catalysed peptide oxidations. By way of example, δ-(L-α-aminoadipoyl)-L-(3,3-difluorohomocysteinyl)-D-valine is oxidised by IPNS giving neither bicyclic nor monocyclic products; instead the 3,3-difluorohomocysteinyl residue undergoes four electron oxidation resulting in a thiocarboxylic acid (Fig. 6A).62 Use of δ-(L-α-aminoadipoyl)-L-(homocysteinyl)-D-cysteine enabled trapping of a bicyclic γ-lactam as a disulfide (Fig. 6A).63,78 More recent work in the crystalline state has employed substrate analogues in which the cysteinyl-valine amide of ACV is replaced with an ester (i.e., depsipeptide analogues), with particularly interesting results being obtained with β-methyl cysteinyl analogues; the depsipeptide with a cysteinyl (3S)-methyl group yielded a thioenol/thioketone, whereas one depsipeptide without the methyl group yielded a thiocarboxylic acid.68,69
ACV analogues employing valine substitutes strikingly reveal the potential for promiscuity in the reactions of the monocyclic β-lactam ferryl intermediate, at least with unnatural substrates (Fig. 6).4,15 Multiple products have been isolated with some unsaturated or cyclopropane ring containing valine analogues, with the balance between products being manipulated by use of deuterium labels.15,22,63–66 This promiscuity, including the ability to form ‘expanded’ ring products, is clearly manifested with replacement of the D-valine of ACV by D-allylglycine or by D-cyclopropylalanine (Fig. 6B). At least some of the observed products are formed via radical intermediates.63–66
3. 2OG oxygenases in cephalosporin biosynthesis – deacetoxycephalosporin C and deacetylcephalosporin C synthase
The committed step in cephalosporin biosynthesis is catalysed by deacetoxycephalosporin C synthase (DAOCS).4 Eukaryotic microorganisms have a single bifunctional 2OG oxygenase DAOCS/DACS,79 which has both deacetoxycephalosporin C synthase (DAOCS) and deacetylcephalosporin C synthase (DACS) activities. By contrast, in prokaryotes two distinct enzymes are highly, but incompletely, selective for the penam expansion (DAOCS)80 and the DAOC hydroxylation steps (DACS).81–83 Isopenicillin N is not a substrate for DAOCS, though note that δ-(D-α-aminoadipoyl)-L-(cysteinyl)-D-valine is an IPNS substrate.4,84,85 In microorganisms producing cephalosporins, the δ-(L-α-aminoadipoyl) side chain of isopenicillin N undergoes epimerisation to give the δ-(D-α-aminoadipoyl) side chain of penicillin N, which is the DAOCS substrate. In prokaryotes, this reaction is catalysed by a pyridoxal dependent epimerase.86–90 In eukaryotes, a different epimerisation system is present, encoded for by genes cefD1 and cefD2; the encoded protein CefD1 is proposed to catalyse CoA ester formation, while CefD2 is proposed to catalyse epimerisation.91,92At least one 2OG oxygenase is also involved in the 7α-functionalisation of cephalosporins. Two types of 7α-functionalised cephalosporins with potent antibiotic activity are known: the cephamycins with a 7α-methoxy group, and the cephabacins with a 7α-formylamino group. Whilst the biosynthesis of the latter has not been defined, the methyl group and the oxygen of the 7α-methoxy group are derived from methionine and dioxygen, respectively.93 Studies with cell-free extracts of Streptomyces clavuligerus, using O-carbamoyl DAC as a substrate, showed the process is dependent on Fe(II), 2OG, and S-adenosylmethionine. Evidence for sequential hydroxylation then methylation came from the isolation of the 7α-hydroxy-O-carbamoyl DAC. Sequencing of the cephamycin biosynthesis gene cluster suggests cmcI and cmcJ encode for a methyltransferase and a 7α-hydroxylase, respectively.94 The assignment of CmcI as a methyltransferase was supported by structural studies;95 as yet no biochemical studies on recombinant CmcJ have been reported. The improvement of 7α-methoxycephalosporins production by overexpression of cmcJ and cmcI in S. clavuligerus has been recently reported.96
Insight into the DAOCS/DACS mechanisms has come from isotopic labelling experiments, spectroscopy, and crystallography.4,8,16,18,22,24,31 The DACS catalysed hydroxylation reaction is typical of 2OG-dependent oxygenase type reactions, albeit occurring at an allylic position (Fig. 7B); it likely proceeds via hydrogen abstraction involving a ferryl intermediate, generated by reaction of 2OG and dioxygen with consequent production of carbon dioxide and succinate, as proposed for other 2OG oxygenases.4,8,22 Consistent with this, the reaction proceeds with, at least, partial retention of stereochemistry.97 By contrast, the DAOCS catalysed oxidative ring expansion reaction (Fig. 7A) is unique to date in enzymology. It does, however, have precedent in the non-enzymatic oxidative rearrangement of penicillins to cephalosporins, in which a penicillin β-sulfoxide is converted into a cephalosporin where the (pro-S) β-methyl group of a penicillin is incorporated into the dihydrothiazine ring.98 Studies in cells and with isolated enzyme have shown that the β-methyl group of penicillin N forms the endocyclic C-3 carbon of DAOC (Fig. 7).98–100 By contrast with the results for the DACS reaction, cellular studies employing (3-pro-R)-valine, with a 1H/2H/3H labelled chiral methyl group at the position that forms the endocyclic C-3 of cephalosporin C, imply loss of stereochemistry during the DAOCS reaction (Fig. 7A), suggesting the involvement of a radical intermediate, as supported by biomimetic studies.101–103
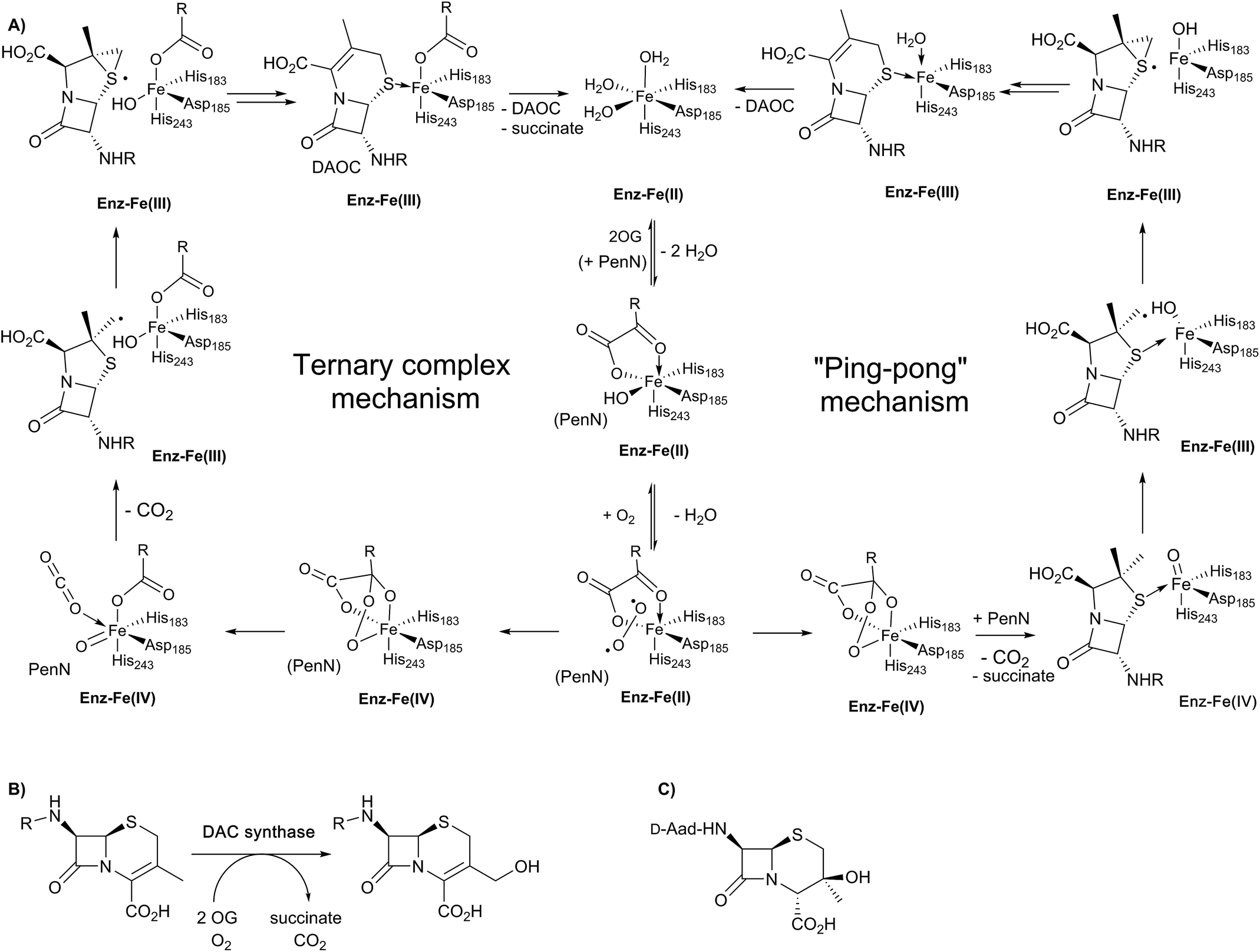 | ||
| Fig. 7 Schematic overview of proposed mechanisms for deacetoxycephalosporin C synthase (DAOCS). (A) The ternary complex mechanism shown employs the consensus process for formation of the ferryl intermediate in the presence of the substrate (penicillin N) as supported by stopped-flow kinetic assays and binding studies by non-denaturing mass spectrometry and NMR.106 An alternative “ping-pong” mechanism has been proposed based on crystallographic information – see text. Note that penicillin N (PenN) binds to the enzyme-Fe(II)-2OG complex in the case of the ternary complex mechanism. (B) The allylic oxidation of deacetoxycephalosporin C (DAOC) to give deacetylcephalosporin C (DAC) as catalysed by deacetylcephalosporin C synthase (DACS). (C) Structure of the byproduct observed in DAOCS catalysis. | ||
Work with deuterium labelled penicillin N has shown that during the DAOCS reaction, a hydrogen is removed from the (pro-S) β-methyl group prior to loss of the C-3 hydrogen.104,105 Further, incubation with (3-2H)penicillin N resulted in a substantial increase in the observed amount of 3β-hydroxy-3α-methylcepham (Fig. 7C), which is only otherwise observed as a minor by-product.104,105 Coupled with studies on other 2OG oxygenases, these and other kinetic observations led to a proposal for DAOCS catalysis in which 2OG binding is followed by that of penicillin N. Recent spectroscopic studies support this mechanism and the formation of a ternary DAOCS·2OG·penicillin N complex.106 Following binding of dioxygen, oxidative decarboxylation of 2OG can occur to form carbon dioxide, succinate, and a ferryl intermediate as proposed in DACS catalysis. The ferryl intermediate can abstract a hydrogen to give a β-methyl radical which ring expands to give a cepham radical, which then loses a hydrogen atom to give DAOC (i.e., the ternary complex mechanism in Fig. 7A). Deuteration at the penicillin N C-3 position favours hydroxylation to give 3β-hydroxy-3α-methylcepham (Fig. 7C), due to operation of a primary isotope effect.104
In 1998, an X-ray crystal structure of DAOCS from S. clavuligerus, the first of a 2OG oxygenase, was reported.30 The structure revealed the distorted double-stranded β-helix also present in IPNS28,29 and other 2OG oxygenases.4,8,22,107 As for IPNS,28,29 DAOCS (and by implication the other 2OG oxygenases involved in cephalosporin biosynthesis) has the conserved Fe(II) binding triad 2-His-1-Asp motif (His183, Asp185, and His243 in DAOCS). Structures were obtained for DAOCS·Fe(II)·2OG and DAOCS·Fe(II)·succinate complexes. These are consistent with operation of the consensus ordered sequential mechanism for 2OG oxygenases,107–111 in which 2OG binds prior to penicillin N, and the DAOC product leaves prior to succinate (Fig. 8A and B). 2OG was observed to bind in a bidentate manner to the active site metal via its oxalate group with displacement of two water molecules.30 A second critical interaction for 2OG binding is via its 5-carboxylate with Arg258 which is part of a conserved RXS 2OG binding motif present in DAOCS subfamily 2OG oxygenases (including biomedicinally important human enzymes)22 (Fig. 8A). Mutagenesis of Arg258 (to Gln258) severely reduces DAOCS activity.112
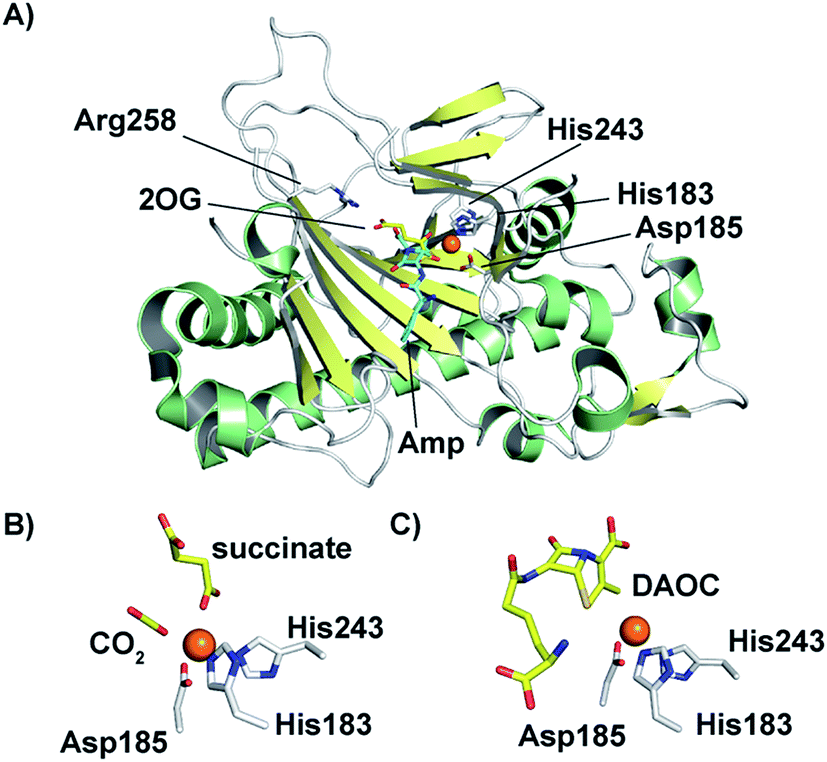 | ||
| Fig. 8 Structural views for deacetoxycephalosporin C synthase (DAOCS). (A) The overall fold (PDB 1UNB)114 and selected active site elements of DAOCS (including the Fe binding residues) are conserved with IPNS. (B) Active site view from a DAOCS–Fe–succinate–CO2 complex structure (DAOCS R307A mutant, PDB 1E5H);114 note the similar arrangement of Fe-binding ligands to that in IPNS (Fig. 5); (C) active site view from a DAOCS–Fe–DAOC product structure (PDB 1W2O);114 note the apparent overlap of the DAOC and 2OG/succinate binding sites – with other structures this led to the proposal of an unusual mechanism for DAOCS – see text and Fig. 7. | ||
Although it is predominantly monomeric in solution, DAOCS was initially crystallised as a trimer in which the C terminus of one protein monomer inserts into the active site of its neighbour monomer;30 this crystal form is likely not amenable to obtaining substrate complexes. Mutagenesis/truncation of the C-terminus yielded a catalytically active form of DAOCS which crystallises as a monomer.113,114 Structures were obtained of complexes with the (poor) substrate analogue penicillin G and ampicillin as well as one with the product DAOC (Fig. 8C).114 Structures of DAOCS-Fe(II)·penicillin G·2OG and DAOCS·Fe(II)·ampicillin·2OG revealed apparent overlapping of the penicillin substrate analogue with the co-substrate (2OG) binding sites. These results were interpreted as supporting an unprecedented “ping-pong” mechanism in which 2OG and dioxygen react to give a ferryl intermediate, succinate leaves, and only then does penicillin N bind with subsequent ring expansion as proposed above (Fig. 7A).114 However, more recent studies on DAOCS pre-steady-state kinetics and binding studies with different substrate analogues106 do not support such a mechanism, instead suggesting that the formation of a ternary DAOCS·Fe(II)·2OG·penicillin substrate complex occurs after 2OG binding. Future studies on DAOCS could focus on characterising details of the proposed radical mechanism of ring expansion.
The polar δ-(D-α-aminoadipoyl)-side chain of naturally occurring cephalosporins is not clinically useful and makes them difficult to extract. In order to enable production of cephalosporins with hydrophobic (or, potentially clinically useful side-chains) which are easier to purify, approaches guided by structural studies or directed evolution to engineer the natural cephalosporin biosynthesis pathway are being employed.112,115–131 One approach involves using mutagenesis to alter the selectivity of DAOCS such that it efficiently accepts penicillins with hydrophobic side-chain (e.g. penicillin G) rather than the natural δ-(D-α-aminoadipoyl) side chain. The main focus of recent studies has been on modifications in the C-terminal region of DAOCS, which is involved in substrate recognition.118,123,131
4. The role of clavaminic acid synthase (CAS) in clavam biosynthesis
The biosynthetic pathways leading to clavams contrast with those for the penicillins and cephalosporins, both with respect to β-lactam ring formation and functionalisation processes. The initial steps of clavam biosynthesis are believed to occur via a conserved pathway leading to (3S,5S)-clavaminic acid, which acts as a branchpoint for diverging pathways yielding (3R,5R)-clavulanic acid and the (5S)-clavams.132 The formation of clavaminic acid involves the remarkable 2OG oxygenase clavaminic acid synthase (CAS), which catalyses three distinct oxidations, i.e., hydroxylation, cyclisation, and desaturation reactions.The committed step in clavam biosynthesis comprises reaction of L-arginine with glyceraldehyde-3-phosphate, yielding the β-amino acid N2-(2-carboxyethyl)arginine (CEA), as catalysed by the thiamine pyrophosphate enzyme, carboxyethylarginine synthase (CEAS). The (3S,5S) β-lactam core of the clavams is next formed via an ATP-dependent reaction catalysed by a β-lactam synthetase (BLS). CAS then catalyses the hydroxylation of deoxyguanidino-proclavaminic acid, yielding guanidino-proclavaminic acid, in a reaction typical of 2OG oxygenases.133 Prior to further oxidation by CAS, proclavaminate amidino hydrolase (PAH) catalyses hydrolysis of the guanidino group of guanidino-proclavaminic acid, forming proclavaminic acid.134–136 CAS then catalyses two further reactions: first the unusual bicyclisation of proclavaminic acid to form dihydroclavaminic acid, then a desaturation reaction to form clavaminic acid.137 The subsequent steps leading to clavulanic acid, in which the amino group of (3S,5S)-clavaminic acid is replaced with a hydroxyl group and the two stereocenters are epimerised, have not been fully elucidated.138 The final step in the pathway involves NADPH/NADH dependent reduction of (3R,5R)-clavaldehyde to give clavulanic acid.139–141 Another oxygenase, potentially a P450 type enzyme, may be involved in the installation of the allylic hydroxyl oxygen of clavulanic acid, as growth of the producing organism in an 18O2 atmosphere resulted in isotopic labeling at this position.142
Whilst investigating the cell-free production of clavulanic acid, Elson et al. discovered the production of clavaminic acid and proclavaminic acid by a clavulanate-producing strain of S. clavuligerus.134 They also described the isolation of native CAS from this strain, which catalysed the conversion of proclavaminic acid into clavaminic acid (Fig. 9). CAS was shown to be a 2OG oxygenase, with conversion of proclavaminic acid into clavaminic acid requiring two equivalents of 2OG. Incubation of 13C-labeled proclavaminic acid or clavaminic acid with the clavulanic acid producer resulted in isotopic incorporation into clavulanic acid, indicating their roles as intermediates in clavulanic acid biosynthesis.143
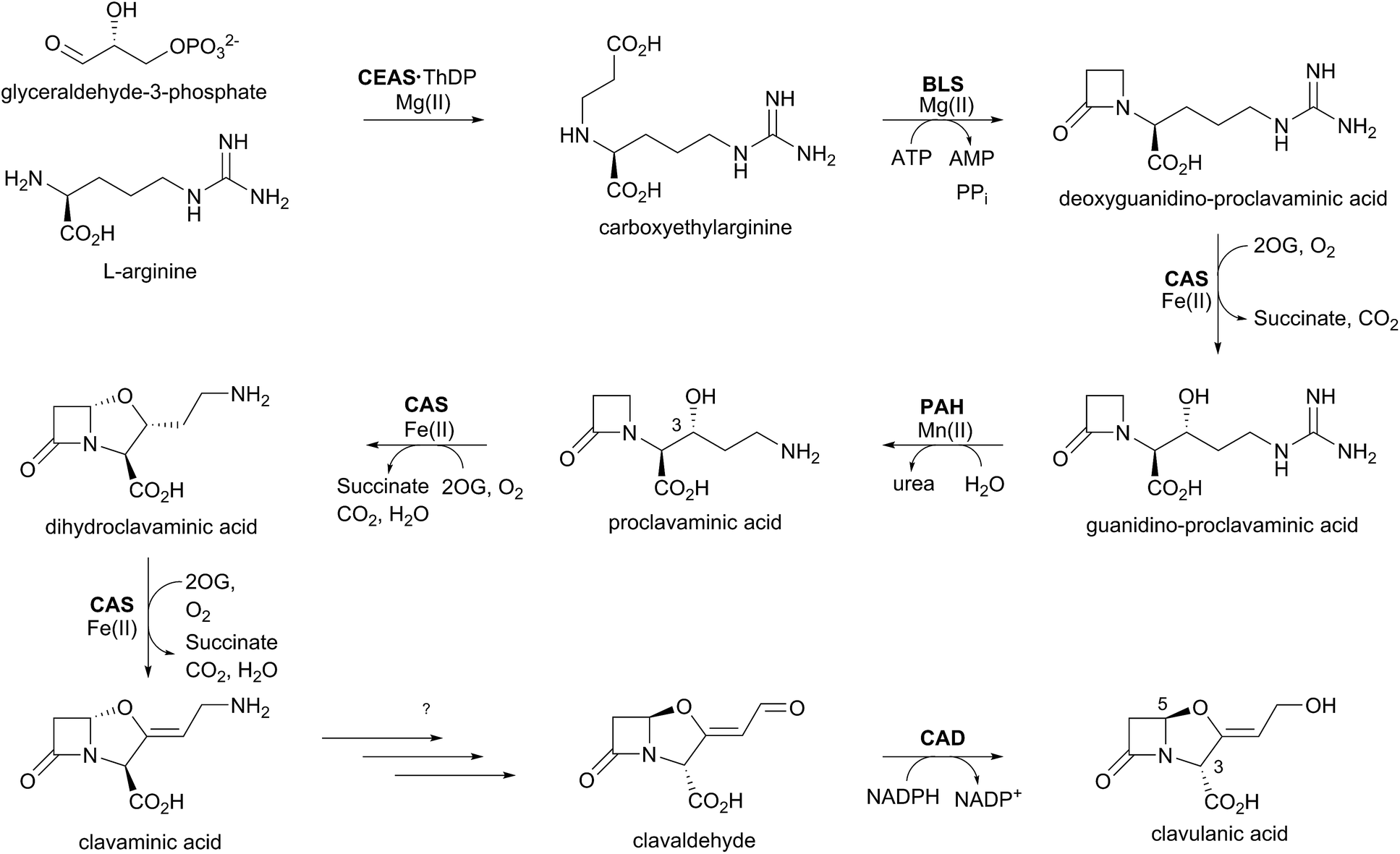 | ||
| Fig. 9 Biosynthetic scheme for clavulanic acid formation showing the trifunctional role of clavaminic acid synthase (CAS). Note that not all co-factors, co-substrates, and products are shown. CEAS: carboxyethylarginine synthase; BLS: β-lactam synthetase; PAH: proclavaminate amidino hydrolase; CAD: clavaldehyde dehydrogenase; ThDP: thiamine diphosphate. | ||
While monitoring the conversion of proclavaminic acid into clavaminic acid by CAS, the presence of a minor species was observed by 1H NMR;135 incubation of CAS with C-3 deuterium-labeled proclavaminic acid resulted in accumulation of this species, apparently due to a primary isotope effect, allowing for its isolation and characterisation as dihydroclavaminic acid (Fig. 9). This indicated that the cyclisation reaction catalysed by CAS, wherein the oxazolidine ring is formed, precedes the CAS-catalysed desaturation reaction. It was subsequently demonstrated that CAS catalyses the efficient hydroxylation of deoxyguanidino-proclavaminic acid to give guanidino-proclavaminic acid,133 which is the substrate for PAH. Studies employing an affinity label targeting CAS implied that all three reactions were catalysed by a single active site and required only a single iron center.141,142 Although the ability of a single active site to catalyse multiple reactions is precedented with other 2OG oxygenases,22 CAS is remarkable because of the three distinct types of reactions it catalyses (Fig. 10). Further, one of these – the first hydroxylation – occurs with a guanidine side chain, whereas the latter two – bicyclisation and desaturation – occur with an amino side chain. Thus, the role of PAH appears to be to substitute the substrate side chain in order to enable CAS to catalyse two additional reactions. Note the amino analogue of proclavaminic acid is neither an efficient inhibitor, nor a substrate of CAS.134
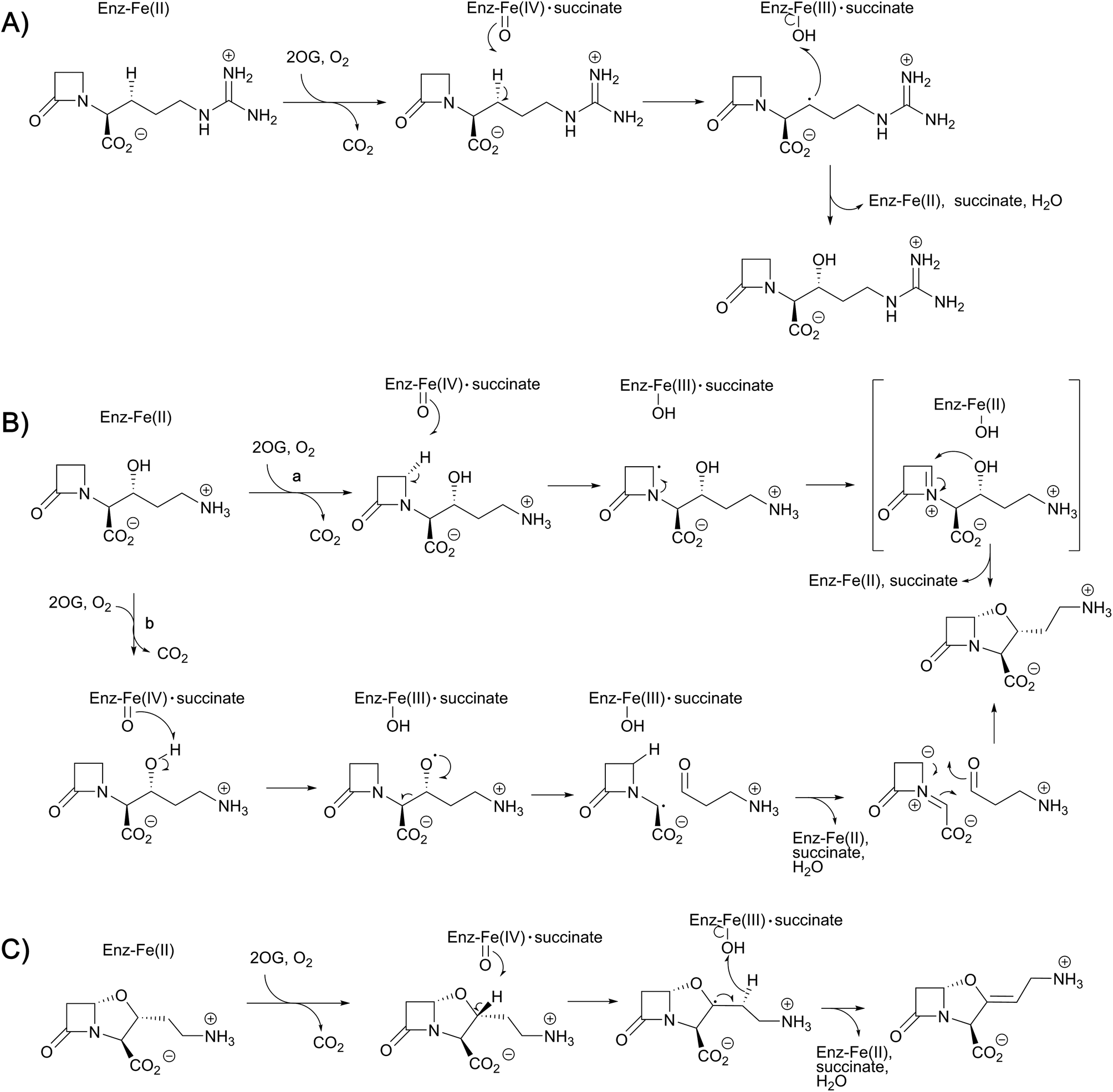 | ||
| Fig. 10 Proposed outline mechanisms for the three different reactions of clavaminic acid synthase (CAS). (A) Stereoselective hydroxylation of deoxyguanidino-proclavaminic acid – a typical reaction for the 2OG oxygenases; (B) two possible mechanisms (a and b) for the oxidative cyclisation of proclavaminic acid; and (C) the desaturation of dihydroclavaminic acid. | ||
The combined crystallographic and solution studies have yielded insight into the mechanisms of CAS. A crystal structure of the complex of CAS1 (one of two CAS isozymes in Streptomyces spp.136) with Fe(II) and 2-OG reveals the characteristic 2OG oxygenase core fold based on a distorted double stranded β-helix fold, consisting of eight β-strands, enveloped by two α-helical regions (Fig. 11).144 While there is little overall sequence similarity between CAS and the penicillin/cephalosporin β-lactam biosynthesis enzymes, DAOCS/DACS and IPNS, the similar overall structure and arrangement of active site motifs implies that, as with all characterised 2OG oxygenases, they ultimately arise from divergent evolution. There is more sequence similarity between CAS and the 2OG oxygenases of carbapenem biosynthesis, including CarC,145 suggesting a closer evolutionary relationship between these two sets of 2OG oxygenases.
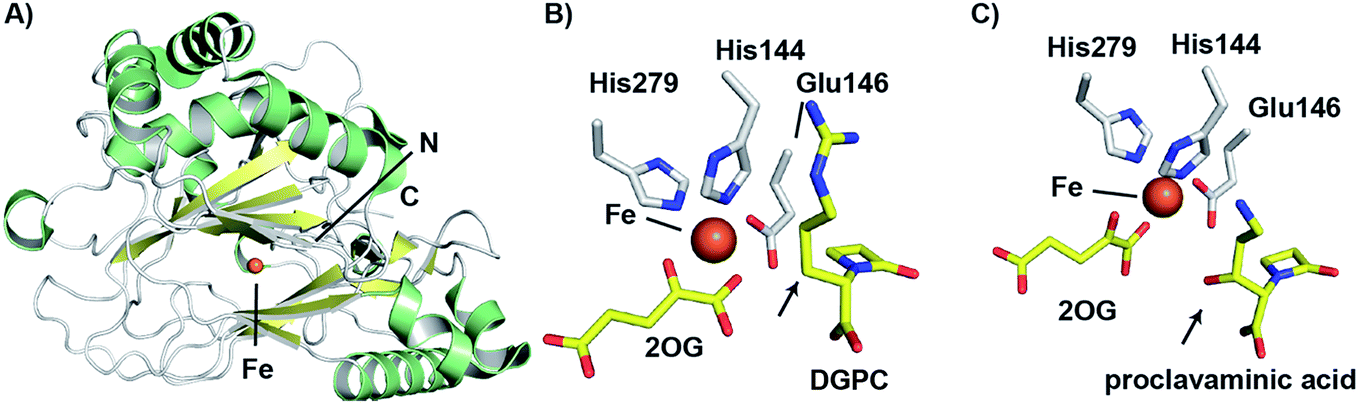 | ||
| Fig. 11 Views from structures of clavaminic acid synthase (CAS). (A) View from a crystal structure of CAS1 (PDB 1DS1)145 highlighting the core double-stranded β-helix fold (yellow). (B) View of the CAS1 active site with bound deoxyguanidinoproclavaminic acid (DGPC; PDB 1GVG),147 showing the orientation of the position to be hydroxylated (C-3) towards the metal (arrowed). Use of a Glu (rather than the more typical Asp) to bind to the iron may contribute to the catalytic flexibility of CAS. Note this structure was obtained using NO in place of O2. (C) View from the active site of a crystal structure of CAS1 with proclavaminic acid (PDB 1DRT).145 Note that the hydroxyl group is oriented towards the iron (arrowed), consistent with a mechanism in which the hydroxyl group hydrogen atom is abstracted by a ferryl species. | ||
The CAS structures revealed that iron binding is mediated by the side chains of His144, Glu146, and His279; the presence of an HXE⋯H motif in CAS rather than an HXD⋯H motif, as in IPNS and DAOCS is notable. The presence of a glutamyl residue in this motif, in place of the more commonly observed aspartyl residue is proposed to be related to the ability of CAS to catalyse multiple reactions in a single active site.145 As glutamyl residues are longer than aspartyl residues by a methylene unit, the position of the iron center may be relatively more flexible, which may permit the enzyme to catalyse different oxidation reactions with its three structurally distinct substrates.
As with other 2OG oxygenases (e.g. DAOCS – see above),106 bidentate binding of 2OG to the metal center is proposed to be followed by that of substrate proximate to the metal, an event which weakens binding of a metal-ligated water, so promoting ligation of dioxygen to the metal. An ordered sequential mechanism is supported by spectroscopic studies with CAS.146 Notably, the orientations of water and the 2OG 1-carboxylate around the metal center were noted to be reversed relative to what was observed in the DAOCS crystal structure. Studies using nitric oxide, as a mimic of dioxygen, revealed loss of a metal ligated water and rearrangement of the 2OG C-1 carboxylate on nitric oxide complexation.147 The precise mechanistic significance of this for CAS catalysis has yet to be defined, but analogous rearrangements are proposed with some other 2OG oxygenases, but in other cases (e.g. DAOCS) appear not to operate.24
Crystal structures have also been solved for CAS complexed with its substrates proclavaminic acid and deoxyguanidinoproclavaminate (DGPC), as well as the DGPC mimic N-acetylarginine.145,147 The binding positions observed for DGPC and N-acetylarginine are such that the 3-pro-R hydrogen atom is oriented towards the metal center, apparently allowing for its abstraction by the ferryl intermediate (Fig. 11B).145,147 The guanidino group of DGPC (and N-acetylarginine) appears to be snugly bound at the active site, being oriented to interact with the side chains of two aspartyl residues, while the β-lactam ring (or N-acetyl group) is positioned in a hydrophobic pocket.145,147 Thus, the CAS catalysed hydroxylation reaction appears to be enabled by precise enzyme-substrate interactions, consistent with solution studies on the high degree of stereoselectivity for the CAS catalysed hydroxylation of DGPC and NAA.148
While Arg297 is positioned so as to interact with the carboxylate of DGPC, the position of this residue is shifted somewhat in the structure with proclavaminic acid (Fig. 11C) so as to hydrogen bond with the hydroxyl group of the substrate for bicyclisation.145,147 Further, the amino group of proclavaminic acid does not appear positioned to interact directly with the aspartyl residues which are involved in binding to the guanidino group of the DGPC, but may interact with them through water molecules.145,147 Overall, the available structures imply proclavaminic acid is bound less precisely in the active site – possibly reflecting conformational changes during dihydroclavaminic acid formation. Studies using labelled proclavaminic acid have shown selective loss of the 4-pro-R β-lactam hydrogen during oxazolidine ring formation, i.e. reaction occurs with retention of stereochemistry.149 Modelling studies have led to the proposal that the ferryl intermediate first abstracts a hydrogen from the alcohol of proclavaminic acid; 1,5-hydrogen abstraction may then remove the β-lactam C-4 hydrogen so enabling subsequent oxazolidine ring formation, potentially via a radical retro aldol fragmentation followed by a 1,3-dipolar reaction (Fig. 10).150 Information on the CAS-catalysed desaturation is hindered by lack of a dihydroclavaminic acid complex structure, but solution studies reveal the CAS reaction proceeds with retention of stereochemistry141 and a stepwise process seem most probable (Fig. 10).
Given the clinical importance of clavulanic acid as an inhibitor of serine β-lactamases, there is interest in exploiting the clavam biosynthetic pathway to generate clavulanic acid analogues. Studies with CAS have demonstrated flexibility in terms of the substrates accepted by this enzyme, perhaps unsurprisingly given the catalytic flexibility of this enzyme. Notably, given the development of avibactam,7,151–154 CAS is able to generate a γ-lactam derivative of clavaminic acid from the γ-lactam variant of proclavaminic acid.155 Furthermore, CAS stereospecifically hydroxylates DGPC substrate analogues lacking a β-lactam ring, e.g. N-acetylarginine and N-acetylornithine, with alkene formation being observed in some cases, reflecting the flexibility in CAS catalysis.133,141,148 However, as the biosynthetic steps leading from clavaminic acid to clavulanic acid are unclear, it is also unclear whether the enzymes involved in these steps would tolerate such substitutions at an early stage in the pathway. Hence, engineering the end of the biosynthesis pathway to add functional groups to clavulanic acid may be more productive.
5. 2OG oxygenases in carbapenem biosynthesis
Biosynthetic studies of the carbapenems have primarily focused on the simplest carbapenem, (5R)-carbapen-2-em-3-carboxylic acid, and the more structurally complex and potent antibiotic thienamycin.4,11,13 The biosynthetic pathways to these two carbapenems may initially follow a common pathway, but diverge in later steps involving the desaturation and epimerisation of the five-membered ring, as well as the functionalisation at C-2 and C-6 as present in thienamycin.4,11,13,156,157The committed step of both pathways likely involves a decarboxylative aldol type reaction of malonyl-CoA with pyrroline-5-carboxylate, as catalysed by the crotonase-fold enzyme CarB/ThnE.158,159 CarB/ThnE have been shown to accept multiple substrate analogues and are amenable to engineering to produce multiple β-amino acids, many of which can be converted to β-lactams.158,160 In an analogous manner as occurring in the biosynthesis of clavulanic acid, but to give a bicycle, the carbapenem ring is formed via an ATP-dependent condensation catalysed by CarA/ThnM, forming (3S,5S)-carbapenam-3-carboxylic acid. The final steps in the biosynthesis of (5R)-carbapen-2-em-3-carboxylic acid are apparently catalysed by the 2OG-dependent oxygenase CarC. CarC catalyses the unusual epimerisation at the C-5 position, then desaturates the resultant (3S,5R)-carbapenam-3-carboxylic acid between C-2 and C-3 to give (5R)-carbapen-2-em-3-carboxylic acid (Fig. 12).4,11,13
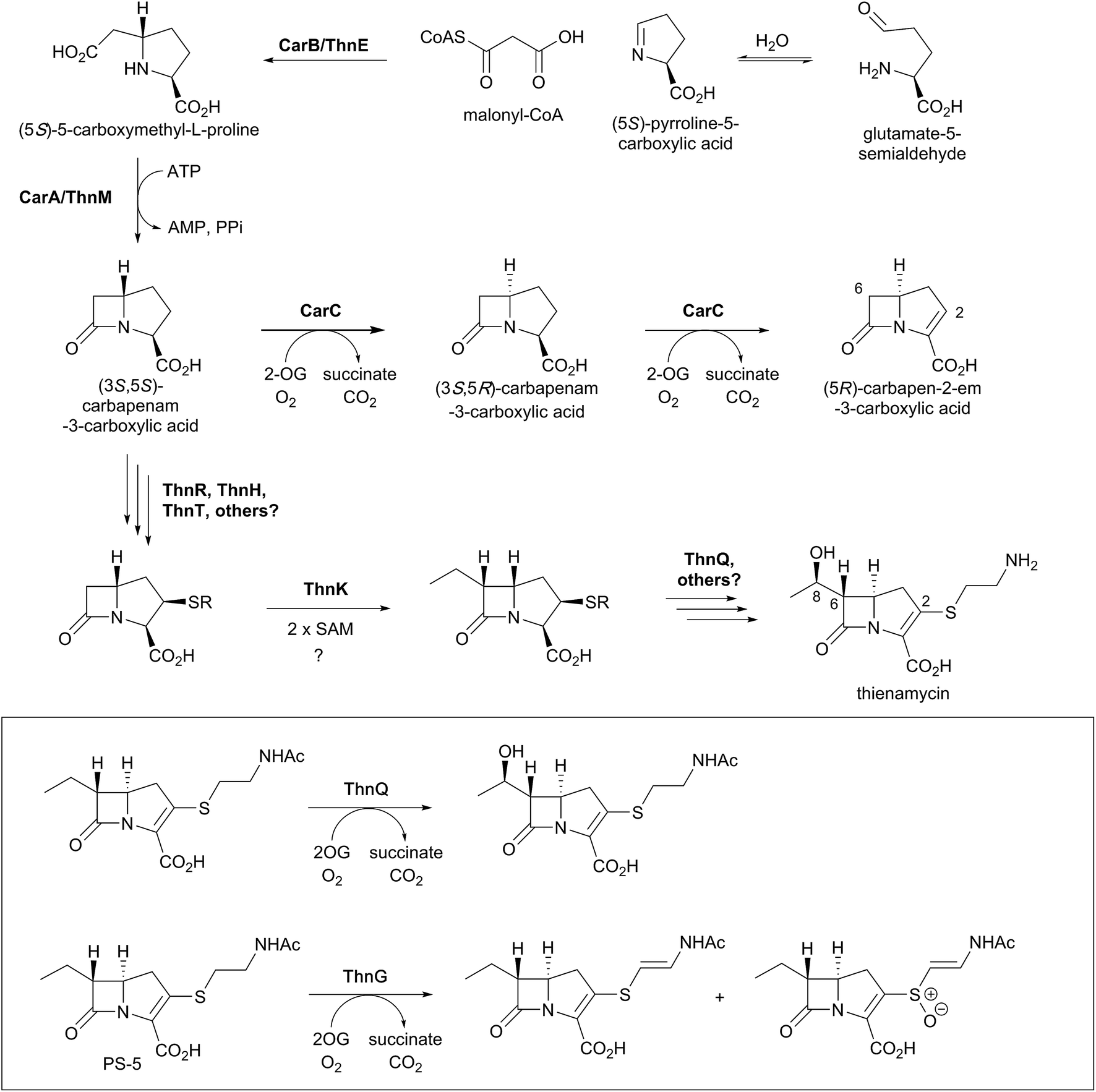 | ||
| Fig. 12 Biosynthetic scheme for carbapenem formation showing the roles of 2OG oxygenases. Carbapenem synthase (CarC) catalyses both epimerisation and desaturation steps in the biosynthesis of (5R)-carbapen-2-em-3-carboxylic acid. ThnQ and ThnG have (at least) roles in carbapenem side chain modification during thienamycin biosynthesis. Note that not all co-factors, co-substrates, and products are shown. | ||
Although recent studies have clarified key aspects of thienamycin biosynthesis, the overall thienamycin biosynthetic pathway is not fully elucidated. The biosynthetic gene cluster for thienamycin does not encode a close CarC homologue,161,162 and the mechanisms underlying carbapenam C-5 epimerisation and double bond formation in thienamycin biosynthesis are unclear. However, the pathways by which the C-2 and C-6 side chains are formed and functionalised have been elucidated, uncovering interesting chemistry involving 2OG oxygenases.156
Freeman et al. demonstrated that the C-2 cysteamine substituent of thienamycin is derived from coenzyme A via the action of hydrolytic enzymes ThnR, ThnH, and ThnT (Fig. 12).157 Whilst ThnR and ThnH sequentially degrade coenzyme A to pantetheine, ThnT degrades the pantetheinyl group to cysteamine, but only once it is covalently attached to the carbapenem core.157N-Acetylated thienamycin derivatives have been described; these are proposed to be formed through the action of the acetyltransferase ThnF.157 Further derivatives have been isolated which show desaturation of the C-2 cysteamine ethyl group and/or oxidation of the sulfur. Incubation of predicted substrates with the 2OG oxygenase ThnG showed that it catalyses sequential desaturation and sulfoxidation of the cysteamine side chain of deshydroxy N-acetylthienamycin (also referred to as PS-5; Fig. 12).156
The radical S-adenosylmethionine (SAM) enzyme ThnK is responsible for incorporation of the ethyl substituent at C-6 of thienamycin, in a mechanism involving two sequential radical-mediated methylation reactions.163 The 2OG oxygenase ThnQ has been shown to modify the C-6 ethyl group, by catalysing (8R)-hydroxylation (Fig. 12).156 Disruption of the ThnQ homolog involved in the biosynthesis of MM 4550 resulted in accumulation of an unhydroxylated product.164 Some carbapenems have different C-6 side chains, such as an (8S)-hydroxyl group or an (8S)-sulfate group (as seen in MM 4550).4 The epimerisation of the 8-hydroxyl group in MM 4550 is proposed to be catalysed by Cmm17, an enzyme homologous to the enoyl-CoA hydratases.157 The sulfotransferase CmmSu is proposed to catalyse sulfate formation, as supported by gene disruption studies.164
While these recent reports have greatly enhanced the understanding of the biosynthesis of the complex carbapenems, questions remain regarding some of the carbapenem biosynthetic steps. The mechanisms of desaturation and epimerisation of the five-membered ring in thienamycin biosynthesis are of particular interest. Further questions remain regarding the overall order in which modifications occur to the five-membered ring, the C-2, and the C-6 side chains. The enzymatic steps involved in the modifications seen in some other carbapenems (e.g., the C-6 hydroxyisopropylidene group of the asparenomycins)4,165,166 also remain to be elucidated.
5.1. CarC – a carbapenem synthase
In 1996, McGowan et al. described the gene cluster responsible for the biosynthesis of the simplest carbapenem (5R)-carbapen-2-em-3-carboxylic acid from Erwinia carotovora (now Pectobacterium carotovorum).167 In particular, homologies between CarA and CarC and the clavulanic acid biosynthetic enzymes β-lactam synthetase and CAS were noted.167 The role of CarC in carbapenem biosynthesis was supported the following year by mutational analysis.168 CarC is proposed to catalyse both epimerisation and desaturation of (3S,5S)-carbapenam-3-carboxylic acid, forming (5R)-carbapen-2-em-3-carboxylic acid [as well as (3S,5R)-carbapenam-3-carboxylic acid].169 Substrate studies demonstrated that (3S,5R)-carbapenam-3-carboxylic acid could also be converted to (5R)-carbapen-2-em-3-carboxylic acid by CarC, suggesting that the epimerisation activity of CarC precedes the desaturation activity (Fig. 13).170,171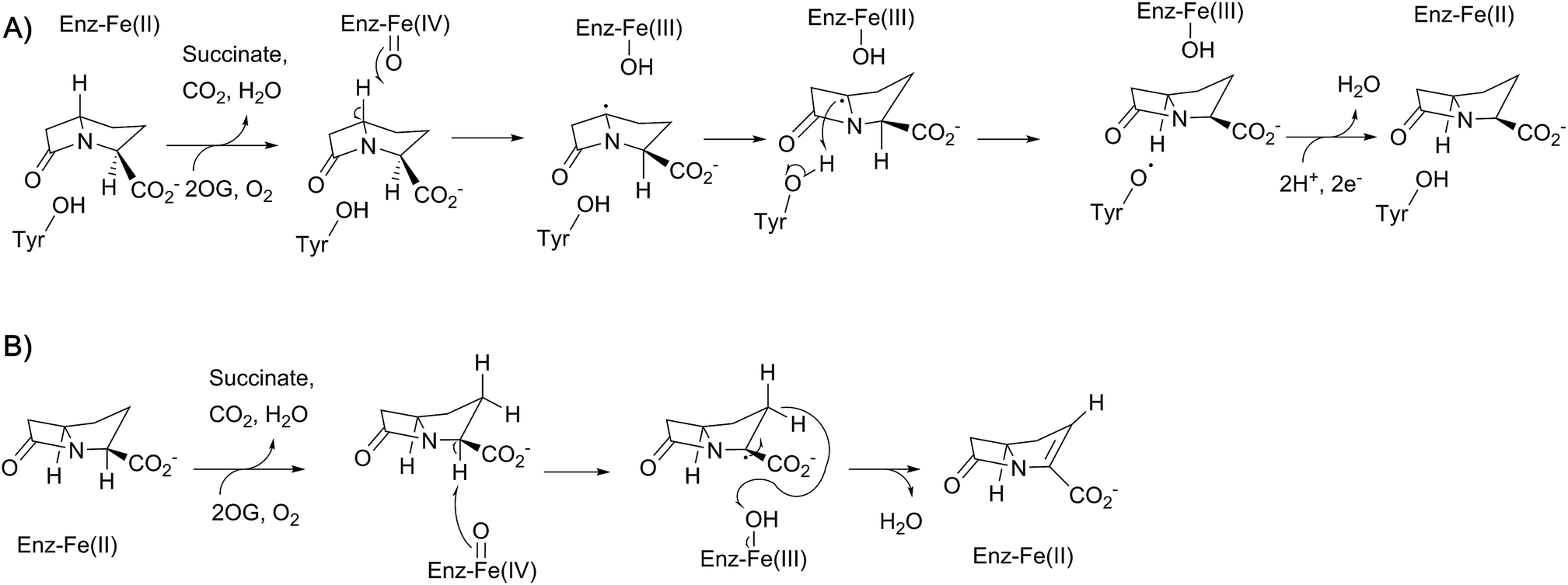 | ||
| Fig. 13 Proposed outline mechanisms for the two reactions involving CarC. (A) C5 inversion of stereochemistry; (B) oxidative C2–C3 desaturation. | ||
Crystallographic studies of CarC have revealed the double-stranded β-helix core typical of the 2OG oxygenases.172 The active site Fe(II) species was bound by a conserved 2-His-1-carboxylate triad as in IPNS/DAOCS, involving the side chains of His101, Asp103, and His251 (Fig. 14). 2OG was bound to the iron via the 2-keto and 1-carboxylate groups, while the C-5 carboxylate of 2OG forms an electrostatic interaction with the side chain of Arg263.172 Given the poor stability of (3S,5S)-carbapenam-3-carboxylic acid, a structure of CarC was first solved with bound L-N-acetylproline acting as a substrate analogue.172 While it was not possible to conclusively assign the orientation of L-N-acetylproline in this structure, modeling studies allowed the binding interactions of both (3S,5S)- and (3S,5R)-carbapenam-3-carboxylic acid with CarC to be studied.172 A later crystal structure of CarC was solved with bound (3S,5S)-carbapenam-3-carboxylic acid, in which two loops near the active site (which were disordered in previous structures) could be modeled.173 The orientation of (3S,5S)-carbapenam-3-carboxylic acid in this structure was such that the C-5 hydrogen atom was oriented towards the active site iron.173
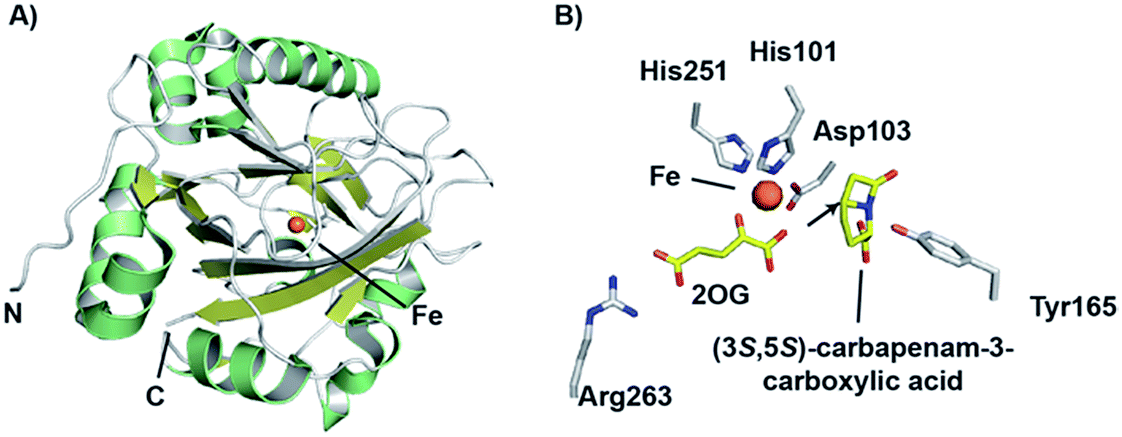 | ||
| Fig. 14 Views from structures of carbapenem synthase (CarC). (A) Overall view of a crystal structure of CarC (PDB 1NX4)172 showing the double-stranded β-helix fold. (B) View of the active site from a crystal structure of CarC with bound substrate (3S,5S)-carbapenam-3-carboxylic acid (PDB 4OJ8).173 Note that the C-5 hydrogen atom is likely abstracted by the ferryl iron species in the epimerisation step, with Tyr165 likely acting as the hydrogen atom donor on the other face. | ||
Labeling studies, in which Serratia marcescens was treated with deuterium- and tritium-labeled L-prolines, indicated that an isotopic label is retained at the C-5 position of (3S,5S)-carbapenam-3-carboxylic acid, but lost at the C-5 position of the diastereomer (and initial CarC product) (3S,5R)-carbapenam-3-carboxylic acid.174 This led to the proposal that the epimerisation step involves the abstraction of the hydrogen atom at the C-5 position; computational studies suggested that formation of a C-5 radical was energetically favourable, most likely through hydrogen abstraction by the ferryl radical (followed by stereochemical inversion).175 Further density functional theory calculations and in silico docking studies were consistent with such a mechanism, and suggested that the phenol group of a tyrosine residue may act as a hydrogen atom donor on the other face of the C-5 radical, thereby completing the epimerisation step.176,177 Based on a crystal structure of CarC with substrate (3S,5S)-carbapenam-3-carboxylic acid (as well as mutational analysis), Tyr165 was proposed to play this role.172,173 The epimerisation step catalysed by CarC is redox neutral, and so it is possible that the desaturation step may employ the same ferryl species as that involved in the epimerisation step. However, rapid-kinetic and spectroscopic studies indicate that the epimerisation step is likely stoichiometric, occurring with oxidation of Fe(II) to Fe(III).173 Substitution of Tyr165 with a phenylalanine residue yielded an enzyme that catalyses catalytic substrate oxidation.173 Therefore, desaturation may require the generation of a new ferryl species, thus probably requiring a second catalytic cycle involving 2OG. The desaturation step, a more typical reaction catalysed by a 2OG oxygenase, may involve initial abstraction of a hydrogen atom at C-2.173
6. Potential roles of 2OG oxygenases in the biosynthesis of monocyclic β-lactams
Pioneering reports informed on the amino acid origins, role of peptide synthetases, and stereochemistry of ring formation in monocyclic β-lactam biosynthesis.4,13,178,179 However, until recently, relatively little has been described regarding the enzymology of the biosynthesis of monocyclic β-lactams. At least in the case of some monocyclic β-lactams, i.e. the monobactams and tabtoxin, sequencing studies have implicated 2OG oxygenases in their biosynthetic pathways, though likely not in actual β-lactam formation.4,13,180–182 Recent reports have informed on biosynthesis of the monocyclic β-lactams, highlighting chemically interesting mechanisms of cyclisation.181,182 Notably, whilst the β-lactam rings of the nocardicins163 and sulfazecin182 are both formed in reactions catalysed by non-ribosomal peptide synthetases (NRPS), the precise mechanisms of β-lactam formation differ for these two β-lactam antibiotics as described below.The biosynthesis of nocardicin begins with NRPS proteins NocA and NocB.181,183 Based on the analysis of the five adenylation domains of these two proteins, a pentapeptide product/intermediate was predicted with a sequence of L-pHPG-L-Arg-D-pHPG-L-Ser-L-pHPG (pHPG, p-hydroxyphenylglycine).183 β-Lactam formation occurs while the nascent peptide chain is covalently attached to NocB (Fig. 3).181 β-Lactam formation was demonstrated to involve dehydration of the serine residue (as part of a NocB tetrapeptide peptide chain) bound to peptidyl carrier protein (PCP) domain PCP4, prior to the incorporation of the C-terminal pHPG residue. The α-amino group of this pHPG residue, while bound to PCP domain 5 as a thioester, can then undergo conjugate addition with the protein-linked dehydroalanine residue. The resultant secondary amino group of the PCP5-bound pHPG then attacks the thioester link to PCP4, forming the β-lactam and releasing the nascent peptide chain from PCP4. Finally, following the thioesterase-domain mediated hydrolysis, pro-nocardicin G is obtained; a further hydrolytic step and tailoring reactions lead to nocardicin A.181
While the β-lactam formation in nocardicin occurs on an NRPS PCP domain, the β-lactam ring of sulfazecin is proposed to occur via catalysis of an NRPS thioesterase domain.182 The NRPSs SulI and SulM, which contain three modules, generate an enzyme-bound tripeptide intermediate, consisting of a D-Glu residue (linked via its γ-carboxyl group), a D-Ala residue, and a L-2,3-diaminopropionate (Dap or β-aminoalanine) residue. The 3-amino group of Dap (in its SulM-bound tripeptide form) then undergoes an unusual N-sulfonation reaction, as catalysed by SulN, which uses 3′-phosphoadenosine 5′-phosphosulfate as a cosubstrate. The tripeptide is then transferred to a thioesterase domain, which catalyses nucleophilic attack of the sulfonated amino group onto the thioester carbonyl, thereby forming the β-lactam.182 This alternate strategy for β-lactam formation (cf. nocardicin) is possible as the β-lactam ring occurs at the C-terminus of the substrate, whereas that found in nocardicin occurs within the peptide sequence.
As seen with some cephalosporins (see above), the β-lactam rings of certain monocyclic β-lactams (such as sulfazecin and tabtoxin) manifest methoxylation at the C-3 position,4 a modification which can decrease susceptibility to some β-lactamases.184,185 The biosynthetic gene cluster associated with sulfazecin was noted to encode putative 2OG oxygenase, SulO, and methyltransferase, SulP.184,185 As indicated above, the NRPSs SulI and SulM and sulfotransferase SulN catalyse the formation of desmethoxysulfazecin. Addition of SulO and SulP to this in vitro reconstitution of the biosynthetic pathway (with added 2OG, ammonium ferrous sulfate, and ascorbate) yielded sulfazecin, confirming the role of SulO in monobactam C-3 hydroxylation (Fig. 15).182 The tabtoxin gene cluster encodes for putative 2OG oxygenase TblC, which likely plays an analogous role in tabtoxin biosynthesis.186
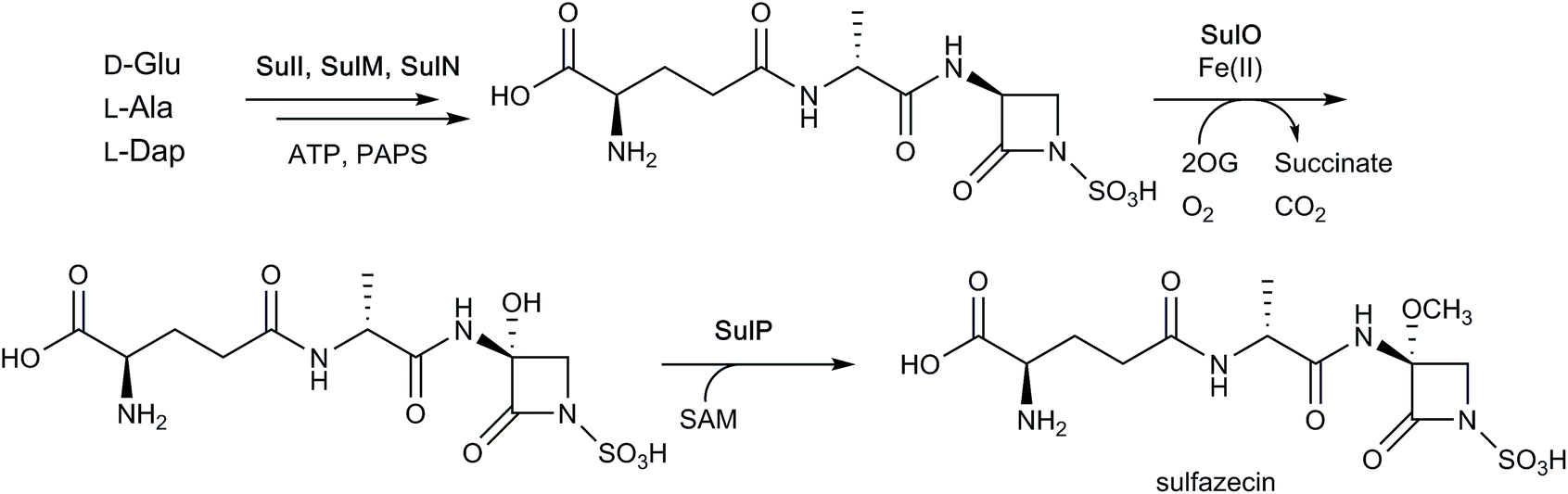 | ||
| Fig. 15 Final steps of sulfazecin biosynthesis. As shown in Fig. 3, the non-ribosomal peptide synthetases SulI and SulM are responsible for synthesising the sulfazecin peptide scaffold and β-lactam ring from D-glutamate, L-alanine, and L-2,3-diaminopropionate (L-Dap), while SulN acts as a sulfotransferase using 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as a co-factor. The 2OG-dependent oxygenase SulO then catalyses C-3 hydroxylation, followed by S-adenosylmethionine (SAM)-mediated methylation as catalysed by SulP.182 Note that not all co-factors, co-substrates, and products are shown. | ||
7. Conclusions
In the period following Arnstein's breakthrough proposal26 for the peptide origin of the penicillins and Abraham's direct evidence27 for the role of L,L,D-ACV (and by implication an oxidative mechanism for penicillin formation) very considerable advances have been made in our understanding of β-lactam biosynthesis. IPNS was isolated and shown to operate via a chemically remarkable mechanism.22 ATP dependent synthetases that catalyse β-lactam formation from β-amino acids have been identified and peptide synthetase templated β-lactam formation via thioester reactions180,181 has been shown to operate for at least two classes of monocyclic β-lactams. Aside from the central role of IPNS, 2OG oxygenases have been shown to play roles in the biosynthesis of bicyclic β-lactams (DAOCS) and in modifying both monocyclic and bicyclic β-lactams, sometimes by chemically interesting processes (especially the DAOCS, CAS, and CarC reactions).4 The biosynthetic work, including on the 2OG oxygenase superfamily members, has been justified in part on the basis that it may help enable improved routes to already used antibiotics (e.g. by enabling fermentation of carbapenems) and make synthetically challenging structures of interest for testing as antibiotics more accessible. Despite ongoing efforts to engineer cephalosporin biosynthesis, these ‘translational’ outcomes have yet to be realised. This is likely in part due to commercial and regulatory considerations associated with the introduction of new manufacturing procedures for (new) antibiotics, coupled with the success of classical empirical strain optimisation and lack of knowledge of ‘indirect’ limiting molecular factors for the highly efficient large scale fermentation and isolation of relatively labile small molecules. Recent work on metabolic engineering, e.g. TCA cycle glyoxylate metabolism (as well as deleting β-lactamases) holds promise for increasing β-lactam antibiotic production yields.119,187,188Combining these approaches with biosynthetic pathway engineering may be productive. The results to date on β-lactam biosynthesis do, however, mean that focused efforts to engineer β-lactam biosynthesis, e.g. by ‘rewiring’ the penicillin/cephalosporin pathway to produce new generation cephalosporins via modification of ACVS/IPNS/DAOCS, could at least proceed in the light of detailed knowledge of how the relevant enzymes work. Future biocatalytic research could also explore the use of modified enzymes in combination with non-enzymatic synthetic chemistry to access modified β-lactams of interest in the field of antibacterials and beyond. Engineering involving coupling the modular nature of peptide synthetase mediated β-lactam biosynthesis with the tailoring capabilities of 2OG oxygenases is one potential avenue.
It is, however, important to note that although the basic work on the enzymes of β-lactam biosynthesis has not (yet) delivered breakthroughs in the antibacterial field, the chemical interest driven work on enzyme mechanisms and structures has been enabling in other fields. The structural studies on IPNS and the β-lactam modifying 2OG oxygenases28–30,145 led to the identification of apparently homologous enzymes in most aerobic organisms. Some of these are used in biocatalysis, others are important in plant signalling (‘ethylene-forming-enzymes’) and as agrochemical targets (gibberellin biosynthesis oxygenases), whilst many others play crucial roles in animal biology, notably in post translational protein modifications and in the regulation of nucleic acid and protein biosynthesis.22 Inhibitors for human 2OG dependent prolyl-hydroxylases, which play a critical role in the cellular response to hypoxia, are presently in late stage clinical trials for the treatment of anaemia.189 Hence, at least, the work on the oxygenases of β-lactam biosynthesis is a nice example of how curiosity driven research on chemically interesting natural products can help open new fields of medicinal relevance.
8. Conflicts of interest
The authors declare no conflicts of interest.9. Acknowledgements
We thank the Biotechnology and Biological Sciences Research Council (BBSRC), the Engineering and Physical Sciences Research Council (EPSRC) (for a studentship to J. K. via the Synthesis for Biology and Medicine, Centre for Doctoral Training, and a Clarendon Scholarship), the Medical Research Council, the Innovative Medicines Initiative, and the Wellcome Trust, for funding our work on β-lactams. P. R. thanks the Deutsche Akademie für Naturforscher Leopoldina, Germany, for funding a postdoctoral fellowship. We also thank and acknowledge our coworkers and those in other groups for their work on β-lactam biosynthesis and apologise for incomplete citations in this brief review.10. References
- B. Hamad, Nat. Rev. Drug Discov., 2010, 9, 675 CrossRef PubMed.
- K. Tahlan and S. E. Jensen, J. Antibiot., 2013, 66, 401 CrossRef PubMed.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, a025247 CrossRef PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21 RSC.
- M. Babic, A. M. Hujer and R. A. Bonomo, Drug Resist. Updates, 2006, 9, 142 CrossRef PubMed.
- D. A. Burnett, Curr. Med. Chem., 2004, 11, 1873 CrossRef PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063 CrossRef PubMed.
- G. Ozcengiz and A. L. Demain, Biotechnol. Adv., 2013, 31, 287 CrossRef PubMed.
- C. C. Muniz, T. E. C. Zelaya, G. R. Esquivel and F. J. Fernandez, Rev. Latinoam. Microbiol., 2007, 49, 88 Search PubMed.
- K. H. Baggaley, A. G. Brown and C. J. Schofield, Nat. Prod. Rep., 1997, 14, 309 RSC.
- N. J. Kershaw, M. E. C. Caines, M. C. Sleeman and C. J. Schofield, Chem. Commun., 2005, 4251 RSC.
- C. J. Schofield, J. E. Baldwin, M. F. Byford, I. Clifton, J. Hajdu, C. Hensgens and P. Roach, Curr. Opin. Struct. Biol., 1997, 7, 857 CrossRef PubMed.
- C. A. Townsend, Curr. Opin. Chem. Biol., 2016, 35, 97 CrossRef PubMed.
- J. E. Baldwin and E. Abraham, Nat. Prod. Rep., 1988, 5, 129 RSC.
- J. E. Baldwin and M. Bradley, Chem. Rev., 1990, 90, 1079 CrossRef.
- S. S. Weber, R. A. L. Bovenberg and A. J. M. Driessen, Biotechnol. J., 2012, 7, 225 CrossRef PubMed.
- R. P. Elander, Appl. Microbiol. Biotechnol., 2003, 61, 385 CrossRef PubMed.
- T. R. M. Barends, H. Yoshida and B. W. Dijkstra, Curr. Opin. Biotechnol., 2004, 15, 356 CrossRef PubMed.
- A. A. Brakhage, Q. Al-Abdallah, A. Tüncher and P. Spröte, Phytochemistry, 2005, 66, 1200 CrossRef PubMed.
- R. D. G. Cooper, Bioorg. Med. Chem., 1993, 1, 1 CrossRef PubMed.
- Y. Aharonowitz and G. Cohen, Annu. Rev. Microbiol., 1992, 46, 461 CrossRef PubMed.
- P. J. Rutledge, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 1–487 Search PubMed.
- S. C. Peck and W. A. v. d. Donk, J. Biol. Inorg Chem., 2017, 22, 381 CrossRef PubMed.
- I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino and C. J. Schofield, J. Inorg. Biochem., 2006, 100, 644 CrossRef PubMed.
- J. E. Baldwin and C. J. Schofield, in The Chemistry of β-lactams, ed. M. I. Page, Springer, Berlin/Heidelberg, 1992, pp. 1–78 Search PubMed.
- H. R. V. Arnstein and D. Morris, Biochem. J., 1960, 76, 357 CrossRef PubMed.
- P. A. Fawcett, J. J. Usher, J. A. Huddleston, R. C. Bleaney, J. J. Nisbet and E. P. Abraham, Biochem. J., 1976, 157, 651 CrossRef PubMed.
- P. L. Roach, I. J. Clifton, V. Fulop, K. Harlos, G. J. Barton, J. Hajdu, I. Andersson, C. J. Schofield and J. E. Baldwin, Nature, 1995, 375, 700 CrossRef PubMed.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827 CrossRef PubMed.
- K. Valegard, A. C. V. Terwisscha Scheltinga, M. D. Lloyd, T. Hara, S. Ramaswamy, A. Perrakis, A. Thompson, H. J. Lee, J. E. Baldwin, C. J. Schofield, J. Hajdu and I. Andersson, Nature, 1998, 394, 805 CrossRef PubMed.
- I. Andersson and K. Valegard, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 385–400 Search PubMed.
- J. Myllyharju, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 149–168 Search PubMed.
- S. E. Wilkins, E. Flashman, J. S. Scotti, R. J. Hopkinson, R. Chowdhury and C. J. Schofield, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 169–209 Search PubMed.
- T. A. Müller and R. P. Hausinger, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 246–262 Search PubMed.
- P. Ø. Falnes and A. Y. Yen Ho, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 275–288 Search PubMed.
- R. J. A. Wanders, S. Ferdinandusse, M. S. Ebberink and H. R. Waterham, in 2-Oxoglutarate-Dependent Oxygenases, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 338–349 Search PubMed.
- C.-P. Pang, B. Chakravarti, R. M. Adlington, H.-H. Ting, R. L. White, G. S. Jayatilake, J. E. Baldwin and E. P. Abraham, Biochem. J., 1984, 222, 789 CrossRef PubMed.
- X. E. Tamanaha, B. Zhang, Y. Guo, W.-c. Chang, E. W. Barr, G. Xing, J. St. Clair, S. Ye, F. Neese, M. Bollinger Jr and C. Krebs, J. Am. Chem. Soc., 2016, 138, 8862 CrossRef PubMed.
- P. J. Rutledge, in 2-Oxoglutarate-Dependent Oxygenasesm, ed. R. P. Hausinger and C. J. Schofield, RSC, Cambridge, 2015, pp. 414–424 Search PubMed.
- H. R. V. Arnstein and M. E. Clubb, Biochem. J., 1958, 68, 528 CrossRef PubMed.
- K. Tahlan, M. A. Moore and S. E. Jensen, J. Ind. Microbiol. Biotechnol., 2017, 44, 517 CrossRef PubMed.
- M. F. Byford, J. E. Baldwin, C.-Y. Shiau and C. J. Schofield, Chem. Rev., 1997, 97, 2631 CrossRef PubMed.
- W. Kallow, J. Kennedy, B. Arezi, G. Turner and H. von Döhren, J. Mol. Biol., 2000, 297, 395 CrossRef PubMed.
- V. J. Chen, A. M. Orville, M. R. Harpe, C. A. Frolik, K. K. Surerus, E. Münck and J. D. Lipscomb, J. Biol. Chem., 1989, 264, 21677 Search PubMed.
- R. A. Scott, S. Wang and M. K. Eidsness, Biochemistry, 1992, 31, 4596 CrossRef PubMed.
- J. E. Baldwin, R. M. Adlington, S. E. Moroney, L. D. Field and H.-H. Ting, J. Chem. Soc., Chem. Commun., 1984, 984 RSC.
- M. Lundberg, P. E. M. Siegbahn and K. Morokuma, Biochemistry, 2008, 47, 1031 CrossRef PubMed.
- C. D. Brown-Marshall, A. R. Diebold and E. I. Solomon, Biochemistry, 2010, 49, 1176 CrossRef PubMed.
- M. Lundberg, T. Kawatsu, T. Vreven, M. J. Frisch and K. Morokuma, J. Chem. Theory Comput., 2009, 5, 222 CrossRef PubMed.
- M. Lundberg and K. Morokuma, J. Phys. Chem. B, 2007, 111, 9380 CrossRef PubMed.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, A. J. Long, R. W. Strange, S. S. Hasnain, C. J. Schofield, J. E. Baldwin and J. Hajdu, Eur. J. Biochem., 1996, 242, 736 Search PubMed.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, C. J. Schofield and P. J. Rutledge, Biochem. J., 2003, 372, 687 CrossRef PubMed.
- I. J. Clifton, W. Ge, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, FEBS Lett., 2013, 587, 2705 CrossRef PubMed.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, P. J. Rutledge and C. J. Schofield, Biochemistry, 2005, 44, 6619 CrossRef PubMed.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. Hensgens, M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721 CrossRef PubMed.
- P. J. Rutledge, N. I. Burzlaff, J. M. Elkins, M. Pickford, J. E. Baldwin and P. L. Roach, Anal. Biochem., 2002, 308, 265 CrossRef PubMed.
- J. E. Baldwin, R. M. Adlington, S. E. Moroney, L. D. Field and H. H. Ting, J. Chem. Soc., Chem. Commun., 1984, 15, 984 RSC.
- A. C. Stewart, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2007, 8, 2003 CrossRef PubMed.
- C. J. Schofield and Z. H. Zhang, Curr. Opin. Struct. Biol., 1999, 9, 722 CrossRef PubMed.
- M. Sami, T. J. N. Brown, P. L. Roach, C. J. Schofield and J. E. Baldwin, FEBS Lett., 1997, 405, 191 CrossRef PubMed.
- L. A. McNeill, T. J. N. Brown, M. Sami, I. J. Clifton, N. I. Burzlaff, T. D. W. Claridge, R. M. Adlington, J. E. Baldwin, P. J. Rutledge and C. J. Schofield, Chem.–Eur. J., 2017, 23, 12815 CrossRef PubMed.
- J. E. Baldwin, G. P. Lynch and C. J. Schofield, Tetrahedron, 1992, 48, 9085 CrossRef.
- J. M. Blackburn, J. D. Sutherland and J. E. Baldwin, Biochemistry, 1995, 34, 7548 CrossRef PubMed.
- J. E. Baldwin, M. Bradley, S. D. Abbott and R. M. Adlington, Tetrahedron, 1991, 47, 5309 CrossRef.
- J. E. Baldwin, R. M. Adlington, B. P. Domayne-Hayman, G. Knight and H.-H. Ting, J. Chem. Soc., Chem. Commun., 1987, 1661 RSC.
- A. R. Howard-Jones, J. M. Elkins, I. J. Clifton, P. L. Roach, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, Biochemistry, 2007, 46, 4755 CrossRef PubMed.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2011, 14, 1881 Search PubMed.
- W. Ge, I. J. Clifton, A. R. Howard-Jones, J. E. Stok, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2009, 10, 2025 CrossRef PubMed.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2006, 7, 351 CrossRef PubMed.
- J. E. Baldwin, J. M. Blackburn, M. Sako and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1989, 970 RSC.
- J. E. Baldwin, R. M. Adlington, A. E. Derome, H. H. Ting and N. J. Turner, J. Chem. Soc., Chem. Commun., 1984, 1211 RSC.
- J. E. Baldwin and M. Bradley, Chem. Rev., 1990, 90, 1079 CrossRef.
- J. E. Baldwin, R. M. Adlington, N. Moss and N. G. Robinson, J. Chem. Soc., Chem. Commun., 1987, 1664 RSC.
- J. E. Baldwin, G. P. Lynch and C. J. Schofield, Tetrahedron, 1992, 48, 9085 CrossRef.
- J. E. Baldwin, W. J. Norris, R. T. Freeman, M. Bradley, R. M. Adlington, S. Long-Fox and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1988, 1128 RSC.
- G. A. Bahadur, J. E. Baldwin and J. J. Usher, J. Am. Chem. Soc., 1981, 103, 7650 CrossRef.
- G. W. Huffman, P. D. Gesellchen, J. R. Turner, R. B. Rothenberger, H. E. Osborne, F. D. Miller, J. L. Chapman and S. W. Queener, J. Med. Chem., 1992, 35, 1897 CrossRef PubMed.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2013, 16, 599 CrossRef PubMed.
- S. M. Samson, J. E. Dotzlaf, M. L. Slisz, G. W. Becker, R. M. van Frank, L. E. Veal, W.-K. Yeh, J. R. Miller, S. W. Queener and T. D. Ingolia, Nat. Biotechnol., 1987, 5, 1207 CrossRef.
- S. Kovacevic, B. J. Weigel, M. B. Tobin, T. D. Ingolia and J. R. Miller, J. Bacteriol., 1989, 171, 754 CrossRef PubMed.
- S. Kovacevic and J. R. Miller, J. Bacteriol., 1991, 173, 398 CrossRef PubMed.
- J. J. R. Coque, F. J. Enguita, R. E. Cardoza, J. F. Martin and P. Liras, Appl. Microbiol. Biotechnol., 1996, 44, 605 CrossRef PubMed.
- B. J. Baker, J. E. Dotzlaf and W.-K. Yeh, J. Biol. Chem., 1991, 266, 5087 Search PubMed.
- J. E. Baldwin, E. P. Abraham, R. M. Adlington, G. A. Bahadur, B. Chakravarti, B. P. Domayne-Hayman, L. D. Field, S. L. Flitsch, G. S. Jayatilake, A. Špakovaskis, H.-H. Ting, N. J. Turner, R. L. White and J. J. Usher, J. Chem. Soc., Chem. Commun., 1984, 1225 RSC.
- Y.-Q. Shen, S. Wolfe and A. L. Demain, J. Antibiot., 1984, 37, 1044 CrossRef PubMed.
- S. Kovacevic, M. B. Tobin and J. R. Miller, J. Bacteriol., 1990, 172, 3952 CrossRef PubMed.
- J. J. R. Coque, J. F. Martin and P. Liras, Mol. Gen. Genet., 1993, 236, 453 CrossRef PubMed.
- S. E. Jensen, D. W. S. Westlake and S. Wolfe, Can. J. Microbiol., 1983, 29, 1526 CrossRef PubMed.
- F. J. Enguita, J. J. R. Coque, P. Liras and J. F. Martin, J. Bacteriol., 1998, 180, 5489 Search PubMed.
- Y. Martínez-Burgoa, R. Álvarez-Álvareza, R. Pérez-Redondob and P. Lirasa, J. Biotechnol., 2014, 186, 21 CrossRef PubMed.
- R. V. Ullán, J. Casqueiro, O. Banûelos, F. J. Fernández, S. Gutiérrez and J. F. Martin, J. Biol. Chem., 2002, 277, 46216 CrossRef PubMed.
- R. V. Ullan, J. Casqueiro, L. Naranjo, I. Vaca and J. F. Martin, Mol. Genet. Genomics, 2004, 272, 562 CrossRef PubMed.
- J. O'Sullivan, R. Aplin, C. Stevens and E. Abraham, Biochem. J., 1979, 179, 47 CrossRef.
- L. M. Iyer, S. Abhiman, R. F. de Souza and L. Aravind, Nucleic Acids Res., 2010, 38, 5261 CrossRef PubMed.
- L. M. Öster, D. R. Lester, A. Terwisscha van Scheltinga, M. Svenda, M. van Lun, C. Généreux and I. Andersson, J. Mol. Biol., 2006, 358, 546 CrossRef PubMed.
- L. Shao, J. J. Huang, Y. Yu, M. X. Li, T. Pu, S. D. Kan and D. J. Chen, J. Appl. Microbiol., 2014, 117, 1645 CrossRef PubMed.
- C. A. Townsend, A. B. Theis, A. S. Neese, E. B. Barrabee and D. Poland, J. Am. Chem. Soc., 1985, 107, 4760 CrossRef.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, N. J. Turner and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1989, 1141 RSC.
- H. Kluender, C. H. Bradley, C. J. Sih, P. Fawcett and E. P. Abraham, J. Am. Chem. Soc., 1973, 95, 6149 CrossRef.
- N. Neuss, C. H. Nash, J. E. Baldwin, P. A. Lemke and J. B. Grutzner, J. Am. Chem. Soc., 1973, 95, 3797 CrossRef PubMed.
- C. A. Townsend, J. Nat. Prod., 1985, 48, 708 CrossRef.
- C. P. Pang, R. L. White, E. P. Abraham, D. H. Crout, M. Lutstorf, P. J. Morgan and A. E. Derome, Biochem. J., 1984, 222, 777 CrossRef PubMed.
- J. E. Baldwin, R. M. Adlington, T. W. Kang, E. Lee and C. J. Schofield, Tetrahedron, 1988, 44, 5953 CrossRef.
- J. E. Baldwin, R. M. Adlington, R. T. Aplin, N. P. Crouch, G. Knight and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1987, 1651 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, C. J. Schofield, N. J. Turner and R. T. Aplin, Tetrahedron, 1991, 47, 9881 CrossRef.
- H. Tarhonskaya, A. Szöllössi, I. K. H. Leung, J. T. Bush, L. Henry, R. Chowdhury, A. Iqbal, T. D. W. Claridge, C. J. Schofield and E. Flashman, Biochemistry, 2014, 53, 2483 CrossRef PubMed.
- I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino and C. J. Schofield, J. Inorg. Biochem., 2006, 100, 644 CrossRef PubMed.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger and C. Krebs, Biochemistry, 2003, 42, 7497 CrossRef PubMed.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484 CrossRef PubMed.
- J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger, J. Am. Chem. Soc., 2003, 125, 13008 CrossRef PubMed.
- P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger and C. Krebs, J. Am. Chem. Soc., 2004, 126, 8108 CrossRef PubMed.
- L. M. Oester, A. C. T. van Scheltinga, K. Valegard, A. M. Hose, A. Dubus, J. Hajdu and I. Andersson, J. Mol. Biol., 2004, 343, 157 CrossRef PubMed.
- H. J. Lee, M. D. Lloyd, K. Harlos, I. J. Clifton, J. E. Baldwin and C. J. Schofield, J. Mol. Biol., 2001, 308, 937 CrossRef PubMed.
- K. Valegård, A. C. T. van Scheltinga, A. Dubus, G. Ranghino, L. M. Öster, J. Hajdu and I. Andersson, Nat. Struct. Mol. Biol., 2004, 11, 95 Search PubMed.
- M. D. Lloyd, H. J. Lee, K. Harlos, Z. H. Zhang, J. E. Baldwin, C. J. Schofield, J. M. Charnock, C. D. Garner, T. Hara, A. C. T. van Scheltinga, K. Valegard, J. A. C. Viklund, J. Hajdu, I. Andersson, A. Danielsson and R. Bhikhabhai, J. Mol. Biol., 1999, 287, 943 CrossRef PubMed.
- J. Ji, X. Tian, K. Fan and K. Yang, Appl. Microbiol. Biotechnol., 2012, 93, 23951 CrossRef PubMed.
- H. S. Chin, K. S. Goo and T. S. Sim, Appl. Environ. Microbiol., 2004, 70, 607 CrossRef PubMed.
- H. S. Chin and T. S. Sim, Biochem. Biophys. Res. Commun., 2002, 295, 55 CrossRef PubMed.
- K. Fan, B. Lin, Y. Tao and K. Yang, J. Ind. Microbiol. Biotechnol., 2017, 44, 705 CrossRef PubMed.
- K. S. Goo, C.-S. Chua and T.-S. Sim, Appl. Environ. Microbiol., 2008, 74, 1167 CrossRef PubMed.
- K. S. Goo, C.-S. Chua and T.-S. Sim, J. Ind. Microbiol. Biotechnol., 2009, 36, 619 CrossRef PubMed.
- J.-S. Hsu, Y.-B. Yang, C.-H. Deng, C.-L. Wei, S.-H. Liaw and Y.-C. Tsai, Appl. Environ. Microbiol., 2004, 70, 6257 CrossRef PubMed.
- J. Ji, K. Fan, X. Tian, X. Zhang, Y. Zhang and K. Yang, Appl. Environ. Microbiol., 2012, 78, 7809 CrossRef PubMed.
- H.-J. Lee, C. J. Schofield and M. D. Lloyd, Biochem. Biophys. Res. Commun., 2002, 292, 66 CrossRef PubMed.
- H.-J. Lee, M. D. Lloyd, K. Harlos, I. J. Clifton, J. E. Baldwin and C. J. Schofield, J. Mol. Biol., 2001, 308, 937 CrossRef PubMed.
- M. D. Lloyd, S. J. Lipscomb, K. S. Hewitson, C. M. H. Hensgens, J. E. Baldwin and C. J. Schofield, J. Biol. Chem., 2009, 279, 15420 CrossRef PubMed.
- C.-L. Wei, Y.-B. Yang, W.-C. Wang, W.-C. Liu, J.-S. Hsu and Y.-C. Tsai, Appl. Environ. Microbiol., 2003, 69, 2306 CrossRef PubMed.
- C.-L. Wei, Y.-B. Yang, C.-H. Deng, W.-C. Liu, J.-S. Hsu, Y.-C. Lin, S.-H. Liaw and Y.-C. Tsai, Appl. Environ. Microbiol., 2005, 71, 8673 Search PubMed.
- X.-B. Wu, X.-Y. Tian, J.-J. Ji, W.-B. Wu, K.-Q. Fan and K.-Q. Yang, Biotechnol. Lett., 2011, 33, 805 CrossRef PubMed.
- X.-B. Wu, K.-Q. Fan, Q.-H. Wang and K.-Q. Yang, FEMS Microbiol. Lett., 2005, 246, 103 CrossRef PubMed.
- N. Balakrishnan, S. Ganesan, P. Rajasekaran, L. Rajendran, S. Teddu and M. Durairaaj, Appl. Environ. Microbiol., 2016, 82, 3711 CrossRef PubMed.
- J. W. Janc, L. A. Egan and C. A. Townsend, J. Biol. Chem., 1995, 270, 5399 CrossRef PubMed.
- J. E. Baldwin, M. D. Lloyd, B. Wha-Son, C. J. Schofield, S. W. Elson, K. H. Baggaley and N. H. Nicholson, J. Chem. Soc., Chem. Commun., 1993, 500 RSC.
- S. W. Elson, K. H. Baggaley, J. Gillett, S. Holland, N. H. Nicholson, J. T. Sime and S. R. Woroniecki, J. Chem. Soc., Chem. Commun., 1987, 1736 RSC.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, A. O. Bringhen, J. B. Coates, N. P. Crouch, M. D. Lloyd, C. J. Schofield, S. W. Elson, K. H. Baggaley, R. Cassels and N. Nicholson, Tetrahedron, 1991, 47, 4089 CrossRef.
- S. P. Salowe, E. N. Marsh and C. A. Townsend, Biochemistry, 1990, 29, 6499 CrossRef PubMed.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, A. O. Bringhen, J. B. Coates, N. P. Crouch, M. D. Lloyd, C. J. Schofield, S. W. Elson and K. H. Baggaley, J. Chem. Soc., Chem. Commun., 1990, 617 RSC.
- S. Salowe, W. Krol, D. Iwata-Reuyl and C. Townsend, Biochemistry, 1991, 30, 2281 CrossRef PubMed.
- A. K. MacKenzie, N. J. Kershaw, H. Hernandez, C. V. Robinson, C. J. Schofield and I. Andersson, Biochemistry, 2007, 46, 1523 CrossRef PubMed.
- N. H. Nicholson, K. H. Baggaley, R. Cassels, M. Davison, S. W. Elson, M. Fulston, J. W. Tyler and S. R. Woroniecki, J. Chem. Soc., Chem. Commun., 1994, 1281 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, D. J. Drake, Y. Fujishima, S. W. Elson and K. H. Baggaley, J. Chem. Soc., Chem. Commun., 1994, 1133 RSC.
- C. A. Townsend and W. J. Krol, J. Chem. Soc., Chem. Commun., 1988, 1234 RSC.
- B. W. Bycroft, A. Penrose, J. Gillett and S. W. Elson, J. Chem. Soc., Chem. Commun., 1988, 980 RSC.
- L. A. Egan, R. W. Busby, D. Iwata-Reuyl and C. A. Townsend, J. Am. Chem. Soc., 1997, 119, 2348 CrossRef.
- Z. Zhang, J. Ren, D. K. Stammers, J. E. Baldwin, K. Marios and C. J. Schofield, Nat. Struct. Biol., 2000, 7, 127 CrossRef PubMed.
- J. Zhou, M. Gunsior, B. O. Bachmann, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 13539 CrossRef.
- Z. Zhang, J.-S. Ren, K. Harlos, C. H. McKinnon, I. J. Clifton and C. J. Schofield, FEBS Lett., 2002, 517, 7 CrossRef PubMed.
- M. D. Lloyd, K. D. Merritt, V. Lee, T. J. Sewell, B. Wha-Son, J. E. Baldwin, C. J. Schofield, S. W. Elson, K. H. Baggaley and N. H. Nicholson, Tetrahedron, 1999, 55, 10201 CrossRef.
- A. Basak, S. P. Salowe and C. A. Townsend, J. Am. Chem. Soc., 1990, 112, 1654 CrossRef.
- T. Borowski, S. de Marothy, E. Broclawik, C. J. Schofield and P. E. M. Siegbahn, Biochemistry, 2007, 46, 3682 CrossRef PubMed.
- A. Endimiani, K. M. Hujer, A. M. Hujer, M. E. Pulse, W. J. Weiss and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 82 CrossRef PubMed.
- T. Stachyra, M.-C. Pechereau, J.-M. Bruneau, M. Claudon, J.-M. Frere, C. Miossec, K. Coleman and M. T. Black, Antimicrob. Agents Chemother., 2010, 54, 5132 CrossRef PubMed.
- T. Stachyra, P. Levasseur, M.-C. Péchereau, A.-M. Girard, M. Claudon, C. Miossec and M. T. Black, J. Antimicrob. Chemother., 2009, 64, 326 CrossRef PubMed.
- D. E. Ehmann, H. Jahić, P. L. Ross, R. F. Gu, J. Hu, G. Kern, G. K. Walkup and S. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11663 CrossRef PubMed.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, M. D. Lloyd, T. J. Sewell, C. J. Schofield, K. H. Baggaley and R. Cassels, Tetrahedron, 1997, 53, 7011 CrossRef.
- M. J. Bodner, R. M. Phelan, M. F. Freeman, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2010, 132, 12 CrossRef PubMed.
- M. F. Freeman, K. A. Moshos, M. J. Bodner, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11128 CrossRef PubMed.
- R. B. Hamed, L. Henry, J. R. Gomez-Castellanos, J. Mecinović, C. Ducho, J. L. Sorensen, T. D. W. Claridge and C. J. Schofield, J. Am. Chem. Soc., 2012, 134, 471 CrossRef PubMed.
- M. C. Sleeman and C. J. Schofield, J. Biol. Chem., 2004, 279, 6730 CrossRef PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, A. Thalhammer, D. Harding, C. Ducho, T. D. W. Claridge and C. J. Schofield, Nat. Chem., 2011, 3, 365 CrossRef PubMed.
- L. E. Nunez, C. Mendez, A. F. Brana, G. Blanco and J. A. Salas, Chem. Biol., 2003, 10, 301 CrossRef PubMed.
- R. B. Hamed, E. T. Batchelar, J. Mecinović, T. D. W. Claridge and C. J. Schofield, ChemBioChem, 2009, 10, 246 CrossRef PubMed.
- D. R. Marous, E. P. Lloyd, A. R. Buller, K. A. Moshos, T. L. Grove, A. J. Blaszczyk, S. J. Booker and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10354 CrossRef PubMed.
- R. Li, E. P. Lloyd, K. A. Moshos and C. A. Townsend, ChemBioChem, 2014, 15, 320 CrossRef PubMed.
- J. Shoji, H. Hinoo, R. Sakazaki, N. Tsuji, K. Nagashima, K. Matsumoto, Y. Takahashi, S. Kozuki, T. Hattori, E. Kondo and K. Tanaka, J. Antibiot., 1982, 35, 15 CrossRef PubMed.
- N. Tsuji, K. Nagashima, M. Kobayashi, J. Shoji, T. Kato, Y. Terui, H. Nakai and M. Shiro, J. Antibiot., 1982, 35, 24 CrossRef PubMed.
- S. J. McGowan, M. Sebaihia, L. E. Porter, G. S. A. B. Stewart, P. Williams, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1996, 22, 415 CrossRef PubMed.
- S. J. McGowan, M. Sebaihia, S. O'Leary, K. R. Hardie, P. Williams, G. S. A. B. Stewart, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1997, 26, 545 CrossRef PubMed.
- R. F. Li, A. Stapon, J. T. Blanchfield and C. A. Townsend, J. Am. Chem. Soc., 2000, 122, 9296 CrossRef.
- A. Stapon, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2003, 125, 15746 CrossRef PubMed.
- M. C. Sleeman, P. Smith, B. Kellam, S. R. Chhabra, B. W. Bycroft and C. J. Schofield, ChemBioChem, 2004, 5, 879 CrossRef PubMed.
- I. J. Clifton, L. X. Doan, M. C. Sleeman, M. Topf, H. Suzuki, R. C. Wilmouth and C. J. Schofield, J. Biol. Chem., 2003, 278, 20843 CrossRef PubMed.
- W.-C. Chang, Y. Guo, C. Wang, S. E. Butch, A. C. Rosenzweig, A. K. Boal, C. Krebs and J. M. Bollinger Jr, Science, 2014, 242, 1140 CrossRef PubMed.
- A. Stapon, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2003, 125, 8486 CrossRef PubMed.
- M. Topf, G. M. Sandala, D. M. Smith, C. J. Schofield, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2004, 126, 9932 CrossRef PubMed.
- T. Borowski, E. Broclawik, C. J. Schofield and P. E. M. Siegbahn, J. Comput. Chem., 2006, 27, 74 CrossRef PubMed.
- R. M. Phelan and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 7496 CrossRef PubMed.
- J. O'Sullivan, A. M. Gillum, C. A. Aklonis, M. L. Souser and R. B. Sykes, Antimicrob. Agents Chemother., 1982, 21, 558 CrossRef.
- J. O'Sullivan, M. L. Souser, C. C. Kao and C. A. Aklonis, Antimicrob. Agents Chemother., 1983, 23, 598 CrossRef.
- M. E. Horsman, D. R. Marous, R. Li, R. A. Oliver, B. Byun, S. J. Emrich, B. Boggess, C. A. Townsend and S. Mobashery, ACS Chem. Biol., 2017, 12, 2552 CrossRef PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383 CrossRef PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24 CrossRef PubMed.
- J. M. Davidsen, D. M. Bartley and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 1749 CrossRef PubMed.
- M. Asai, K. Haibara, M. Muroi, K. Kintaka and T. Kishi, J. Antibiot., 1981, 34, 621 CrossRef PubMed.
- W. Stephen Faraci and R. F. Pratt, Biochemistry, 1986, 25, 2934 CrossRef.
- T. G. Kinscherf and D. K. Willis, J. Antibiot., 2005, 58, 817 CrossRef PubMed.
- B. Lin, K. Fan, J. Zhao, J. Ji, L. Wu, K. Yang and Y. Tao, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9855 CrossRef PubMed.
- A. R. Awan, B. A. Blount, D. J. Bell, W. M. Shaw, J. C. H. Ho, R. M. McKiernan and T. Ellis, Nat. Commun., 2017, 8 DOI:10.1038/ncomms15202.
- T.-L. Yeh, T. M. Leissing, M. I. Abboud, C. C. Thinnes, O. Atasoylu, J. P. Holt-Martyn, D. Zhang, A. Tumber, K. Lippl, C. T. Lohans, I. K. H. Leung, H. Morcrette, I. J. Clifton, T. D. W. Claridge, A. Kawamura, E. Flashman, X. Lu, P. J. Ratcliffe, R. Chowdhury, C. W. Pugh and C. J. Schofield, Chem. Sci., 2017, 8, 7651 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |