
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A tendril perversion in a helical oligomer: trapping and characterizing a mobile screw-sense reversal†
Michael
Tomsett
a,
Irene
Maffucci
b,
Bryden A. F.
Le Bailly
a,
Liam
Byrne
c,
Stefan M.
Bijvoets
c,
M. Giovanna
Lizio
cd,
James
Raftery
c,
Craig P.
Butts
 a,
Simon J.
Webb
cd,
Alessandro
Contini
b and
Jonathan
Clayden
*a
a,
Simon J.
Webb
cd,
Alessandro
Contini
b and
Jonathan
Clayden
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: j.clayden@bristol.ac.uk
bDipartimento di Scienze Farmaceutiche − Sezione di Chimica Generale e Organica “Alessandro Marchesini”, Università degli Studi di Milano, Via Venezian, 21 20133 Milano, Italy
cSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
dManchester Institute of Biotechnology, University of Manchester, 131 Princess St, Manchester M1 7DN, UK
First published on 25th January 2017
Abstract
Helical oligomers of achiral monomers adopt domains of uniform screw sense, which are occasionally interrupted by screw-sense reversals. These rare, elusive, and fast-moving features have eluded detailed characterization. We now describe the structure and habits of a screw-sense reversal trapped within a fragment of a helical oligoamide foldamer of the achiral quaternary amino acid 2-aminoisobutyric acid (Aib). The reversal was enforced by compelling the amide oligomer to adopt a right-handed screw sense at one end and a left-handed screw sense at the other. The trapped reversal was characterized by X-ray crystallography, and its dynamic properties were monitored by NMR and circular dichroism, and modelled computationally. Raman spectroscopy indicated that a predominantly helical architecture was maintained despite the reversal. NMR and computational results indicated a stepwise shift from one screw sense to another on moving along the helical chain, indicating that in solution the reversal is not localised at a specific location, but is free to migrate across a number of residues. Analogous unconstrained screw-sense reversals that are free to move within a helical structure are likely to provide the mechanism by which comparable helical polymers and foldamers undergo screw-sense inversion.
Introduction
Rigid macroscopic helical structures are either entirely left handed (most ‘spiral’ staircases), or entirely right handed (most bolts and screws).1 However, macroscopic helices with some flexibility of structure,2 such as the tendrils of climbing plants or the cord of a telephone handset, may contain stretches of uniform helical screw sense interrupted by ‘tendril perversions’,3,4 in which the screw sense reverses from left to right or right to left (Fig. 1a and b). These perversions are structures of local mirror symmetry that impart a kink to the helical structure.5–7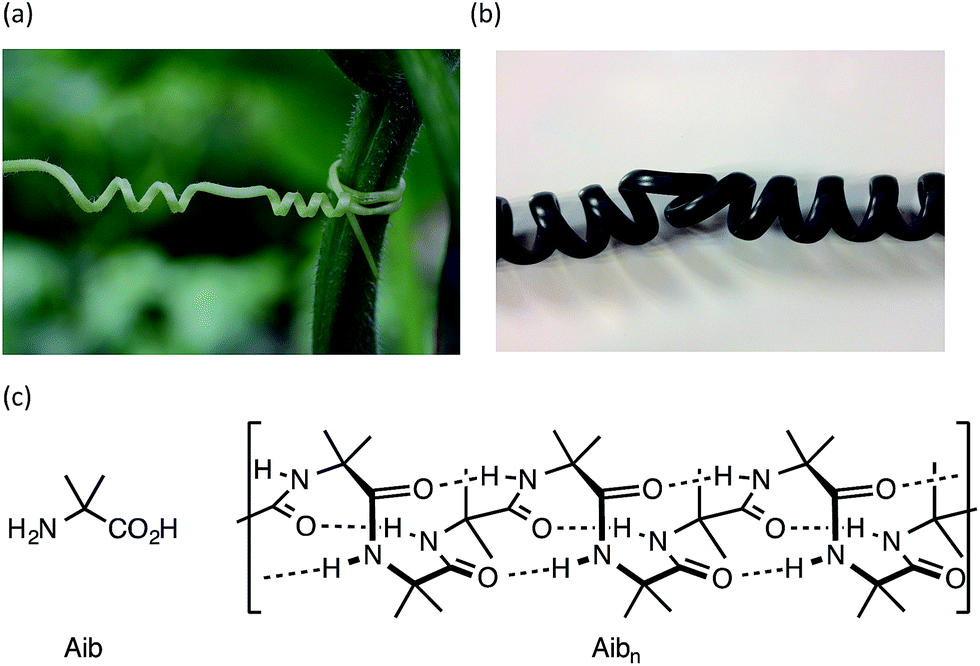 | ||
| Fig. 1 Helical perversions in (a) a tendril of the garden cucumber, Cucumis sativa, and (b) the cord of a telephone handset; (c) Aib and a portion of its 310 helical homo-oligomer, shown in its right-handed screw-sense conformation. | ||
On a molecular scale, helices are common features of polymeric structures.8–14 While polymers of chiral monomers typically adopt a single screw sense, polymers of achiral monomers may adopt helical structures in which the screw sense is left-handed or right-handed, or even both, with domains of opposite screw sense present within a single helical structure.15–20 The lengths of these domains of uniform screw-sense have been evaluated in certain classes of polymer and oligomer,16,21,22 but the structure and dynamics of the screw-sense reversals that separate them have not been explored. A screw-sense reversal provides a molecular analogy of the macroscopic tendril perversion, and in this paper we describe synthetic, spectroscopic, and computational work allowing us to trap and characterize these dynamic structural features.
Oligomers of the achiral quaternary amino acid Aib form hydrogen-bonded 310 helices23–30 (Fig. 1c) that invert rapidly (>1000 per second at room temperature31,32). In an achiral oligomer the two screw-sense conformers are necessarily equally populated, but covalent32–43 or non-covalent44–48 bonding to a chiral species may induce a local preference for either right-handed (P) or left-handed (M) screw sense. This conformational preference propagates through the oligomer, but decreases detectably with distance from the chiral inducer.49 By monitoring the decay of a terminally-induced screw-sense preference with increasing distance from the helix terminus, the intrusion of screw-sense reversals into the otherwise uniform helical polyamide structure can be accurately quantified.22 Screw-sense reversals are rare, but are more common in polar, hydrogen bonding solvents, and at higher temperatures. In THF, for example, any single Aib residue has 0.5% chance of hosting a screw-sense reversal; in MeOH this figure rises to 6%. Their scarcity, along with their rapid migration along the oligomer chain (which provides a plausible mechanism for the remarkably rapid kinetic screw-sense inversion of Aib oligomers50) has made structural characterization of this elusive motif51 particularly challenging.
Trapping a helical reversal within a screw-sense mismatched domain
Our method for quantifying the probability of screw-sense reversals was based on the fact that a helical structure with the same screw sense at each terminus must contain an even number of screw-sense reversals, while a helical structure with a left-handed screw sense at one terminus and a right-handed screw sense at the other must contain an odd number of screw-sense reversals.52 Thus, a helical structure in which the termini are forced to adopt opposing screw senses must contain at least one reversal, and such a molecule provides a scaffold for trapping the dynamic reversal motif within an oligo-Aib domain.Using solution-phase methods49,53 for synthesizing hindered peptides (see ESI†), we made the pairs of diastereoisomeric molecules 1a and 1b and 2a and 2b (Fig. 2). In each of 1a and 1b, a domain formed from five Aib residues is capped at each terminus by a homochiral pair of α-methylvaline (α-MeVal) residues. Like Aib, the hindered quaternary amino acid α-methylvaline favours 310 helical conformations,54,55 but its S enantiomer is accommodated preferentially by a right-handed helix, and its R enantiomer by a left-handed helix. Thus L-(α-MeVal)2 is a powerful inducer of local P screw-sense, and D-(α-MeVal)2 a powerful inducer of local M screw-sense, to the extent that L-(α-MeVal)2 induces quantitative adoption of a right-handed screw sense in an Aib oligomer.35,49
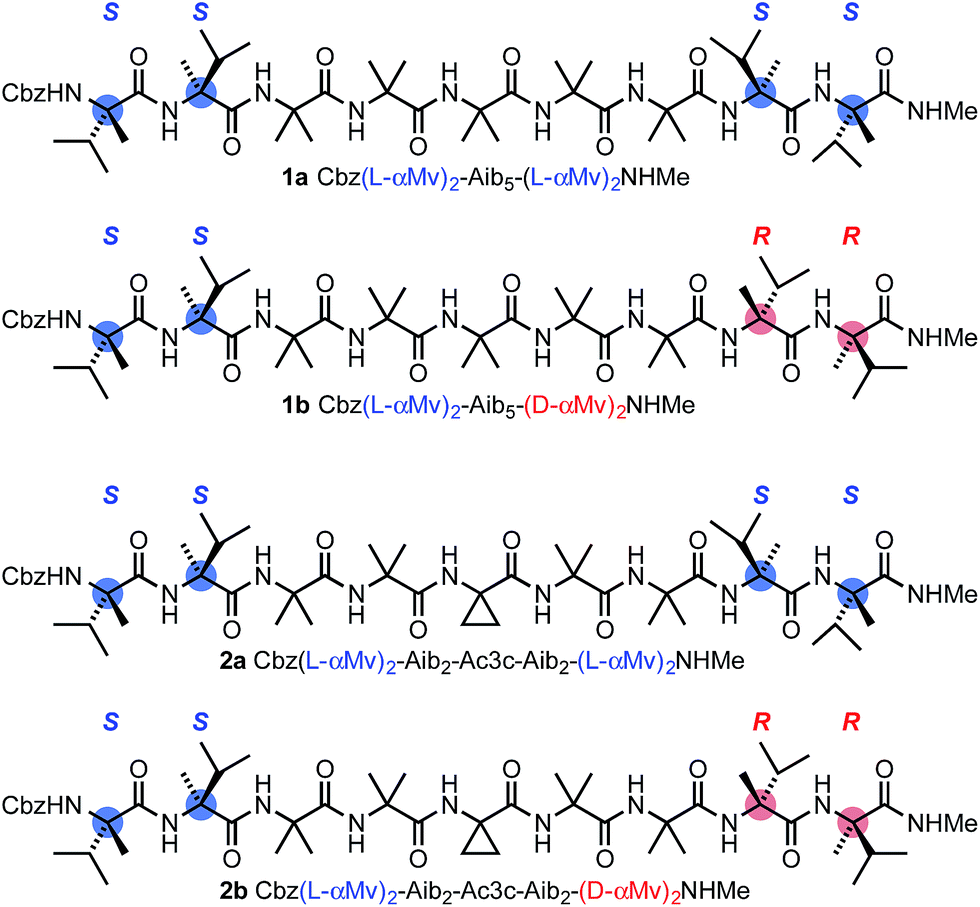 | ||
| Fig. 2 The diastereoisomeric pairs of compounds 1a and 1b and 2a and 2b. Cbz = BnOCO. | ||
As a consequence, we expected both termini of 1a to adopt the same screw sense, while in 1b we expected a right-handed screw sense to be enforced at the N-terminus and a left-handed screw sense to be enforced at the C-terminus. The screw-sense mismatch in 1b should ensure that the central Aib5 domain always contains a helical reversal, while the matched screw senses at the termini of 1a make the formation of a helical reversal unlikely. Diastereoisomers 2a and 2b present a stereochemically analogous pair, but with the central Aib residue replaced by aminocyclopropylcarboxylic acid Ac3c. Achiral quaternary residues other than Aib may also favour 310 helical conformations,23,56,57 but residues related to Ac3c have also been associated with the γ-turn screw-sense reversal motif.58 The contrast in structure between 1a and 1b and the analogous diastereoisomeric pairs was explored by a variety of analytical and computational techniques, with the aim of identifying the distinguishing features of 1b and 2b that could allow characterization of the screw-sense reversal.
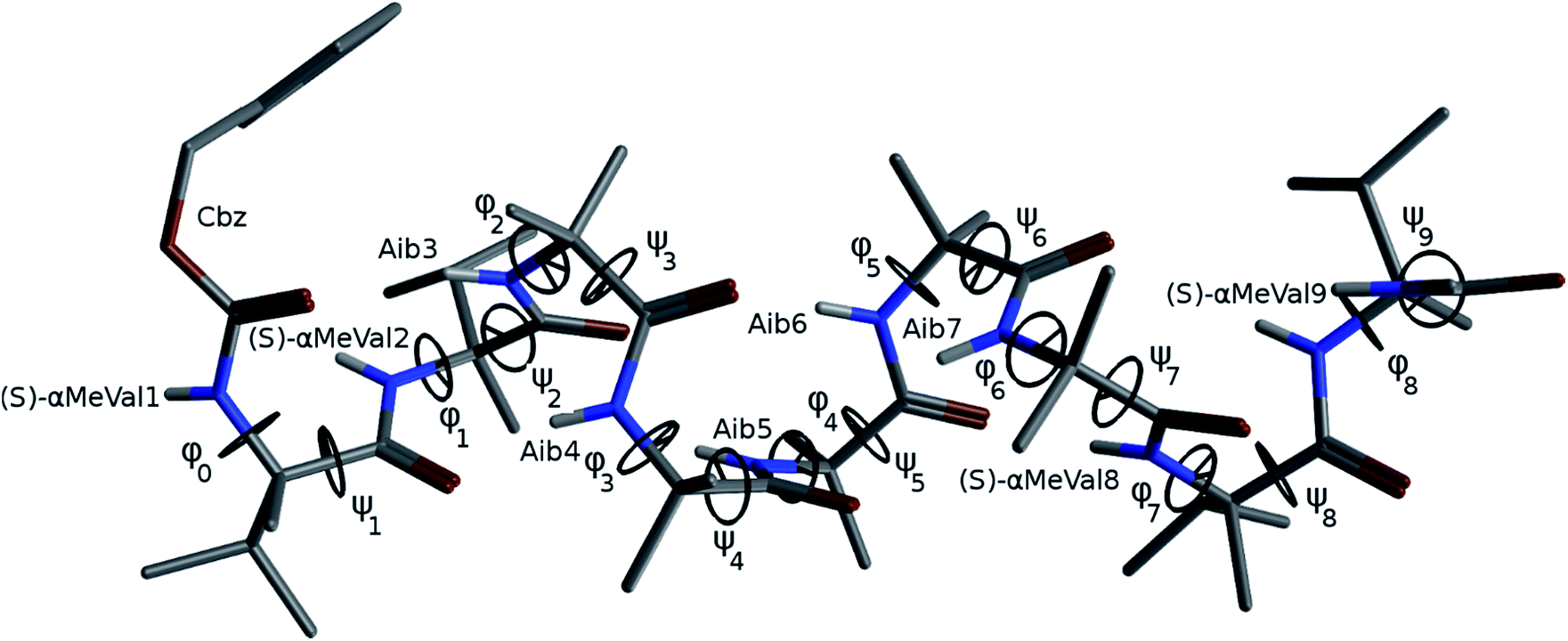
The X-ray crystal structure† of the ‘matched’ oligomer 1a (Fig. 3a) revealed a well-formed 310 helix with a right-handed screw sense, fully in accordance with structures reported in the literature.27–30,59,60 Dihedral angles ϕ and φ (see Table 1) closely approximate those of an idealized 310 helix (ϕ = −49°; φ = −26°). By contrast, the X-ray crystal structure† of the screw-sense mismatched 1b (Fig. 3b) showed, as expected, a right-handed helix at the N-terminus and a left-handed helix at the C-terminus, with a switch from right- to left-handed screw sense happening at residue 6. The change in the sign of the dihedral angles at this point in the structure (Table 1, rows indicated ‘X-ray’) confirms this localized screw-sense reversal in the solid state. The conformational consequence of the reversal becomes clear by viewing the helix end-on: the reversal takes a form that has the general appearance of an antisymmetric tendril perversion;7 the amide carbonyl groups present their Re face to the outside of the helix in the N-terminal P domain and their Si face to the outside of the helix in the C-terminal M domain. At the point of the helical reversal in 1b, there is one unsatisfied hydrogen bond donor (the NH of α-MeVal8) and one unsatisfied hydrogen bond acceptor (the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Aib5), as illustrated in Fig. 3b.
O of Aib5), as illustrated in Fig. 3b.
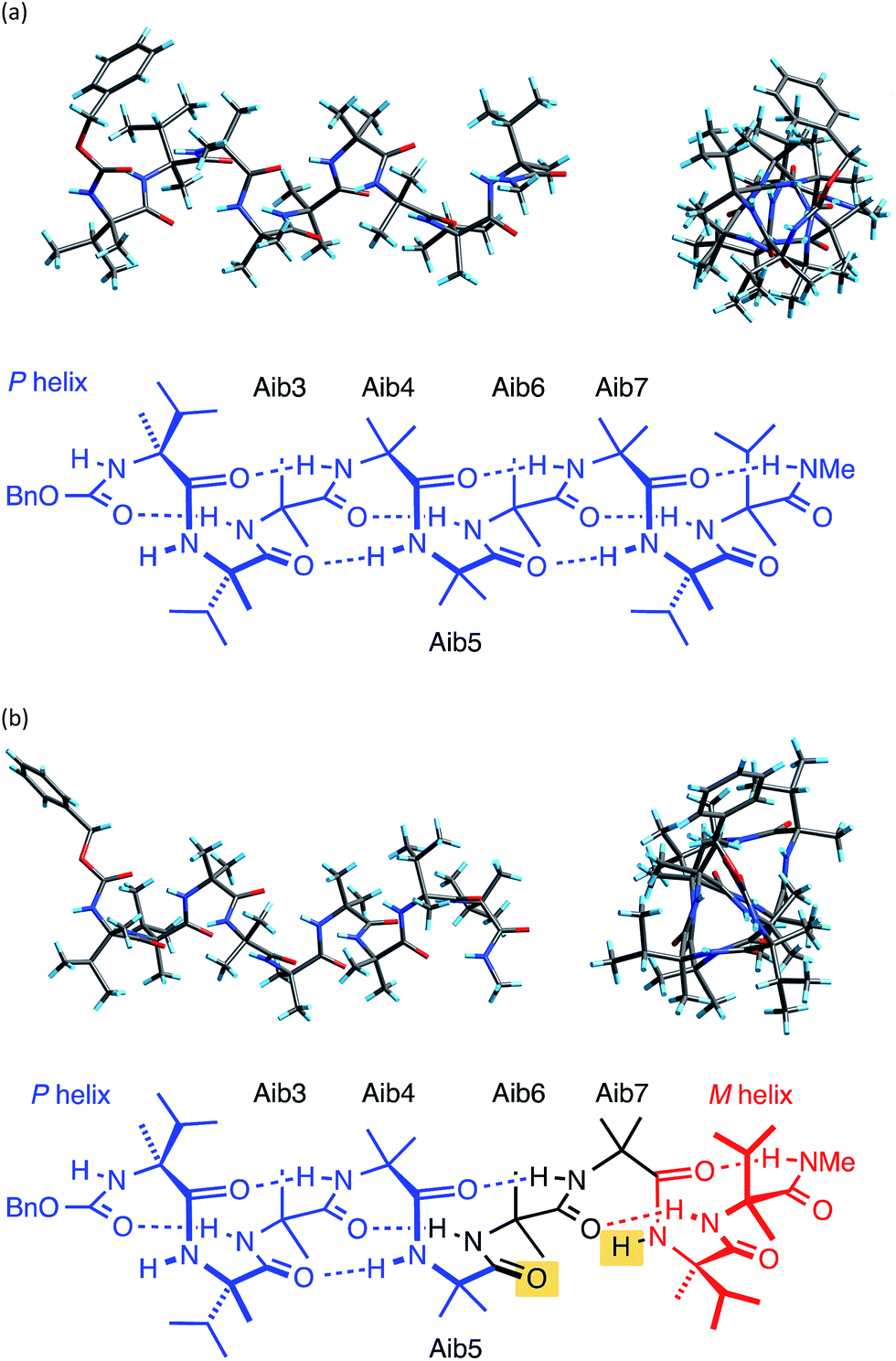 | ||
| Fig. 3 X-ray crystal structure viewed along, and perpendicular to, the helical axis, along with a schematic diagram of the hydrogen bonding in (a) 1a and (b) 1b. Non-hydrogen bonded CO and N–H are highlighted in yellow. | ||
O groups with unsatisfied hydrogen bonds. Dihedral angles are illustrated by the X-ray crystal structure of 1a
Solution state studies were carried out using circular dichroism (CD) and NMR. CD in methanol suggested that the solution state conformational preferences of 1a and 2a were consistent with the crystal structure of 1a: the CD spectra are characteristic of an Aib-containing 310 helix, showing a clear negative maximum at 208 nm (Fig. 4), consistent with the formation of a right-handed helix in solution.38,49 By contrast, the form of the CD spectra of 1b and 2b does not correspond closely to that expected for a 310 helix, suggesting that neither an M nor a P 310 helix predominates. The CD spectra are less intense than those of 1a and 2a, suggesting a less well-defined conformation in solution, and consistent with several possible situations: the adoption of stable M and P screw senses in separate domains within each molecule, the population of a dynamic, interconverting mixture of screw senses across an ensemble of molecules, or a population of conformations rich in non-310-helical motifs.
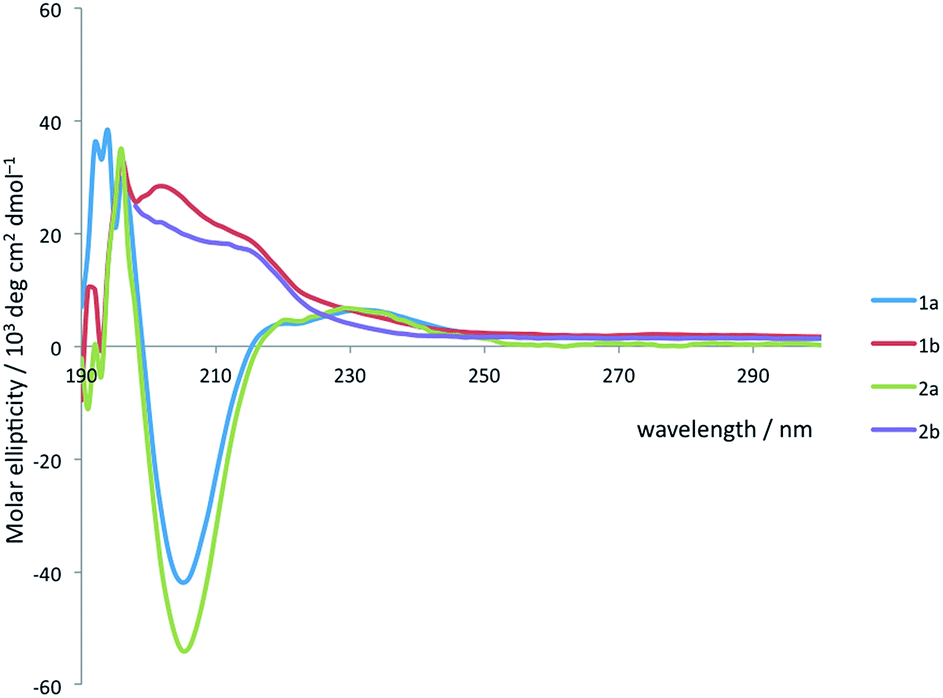 | ||
| Fig. 4 CD spectra of 1a and b and 2a and b recorded at 2.4 × 10−4 M in MeOH at 20 °C. | ||
To distinguish between these alternatives, Raman spectra of 1a and 1b were acquired in chloroform (see ESI, Fig. S1†). In the amide I region both spectra show a band at 1664 cm−1, and deconvolution of this band showed it to be composed of a single contributor for both 1a and 1b. Amide I bands at ∼1660 cm−1 have been ascribed to Aib oligomers in 310 helical conformations,61 so these Raman spectra are consistent with foldamer populations that have significant 310 helix content in either single or multiple domains.
A more detailed picture of the solution-state conformational populations of 1a and 1b was gained by NMR spectroscopy. The screw-sense preference (‘helical excess’, or h.e., defined as the excess population of one screw-sense conformer over the other35) at specific residues in an Aib helix may be quantified by measuring the chemical shift difference (anisochronicity, Δδ) between the 13C signals of the Aib residue's methyl groups.22,32,49 Using 1H–13C HMBC experiments to assign the paired signals of the diastereotopic methyl groups within each Aib residue of 1a and 1b, and a combination of 1H–15N and 1H–13C HSQC to confirm their location in the chain, we quantified the chemical shift separations Δδ in MeOH at 23 °C for the pairs of methyl groups corresponding to each of the residues Aib3–Aib7. Fig. 5 shows the variation of Δδ with position in the chain,62 along with a value for the helical excess calculated using the reported slow-exchange value for Δδ in related compounds.22,32,49
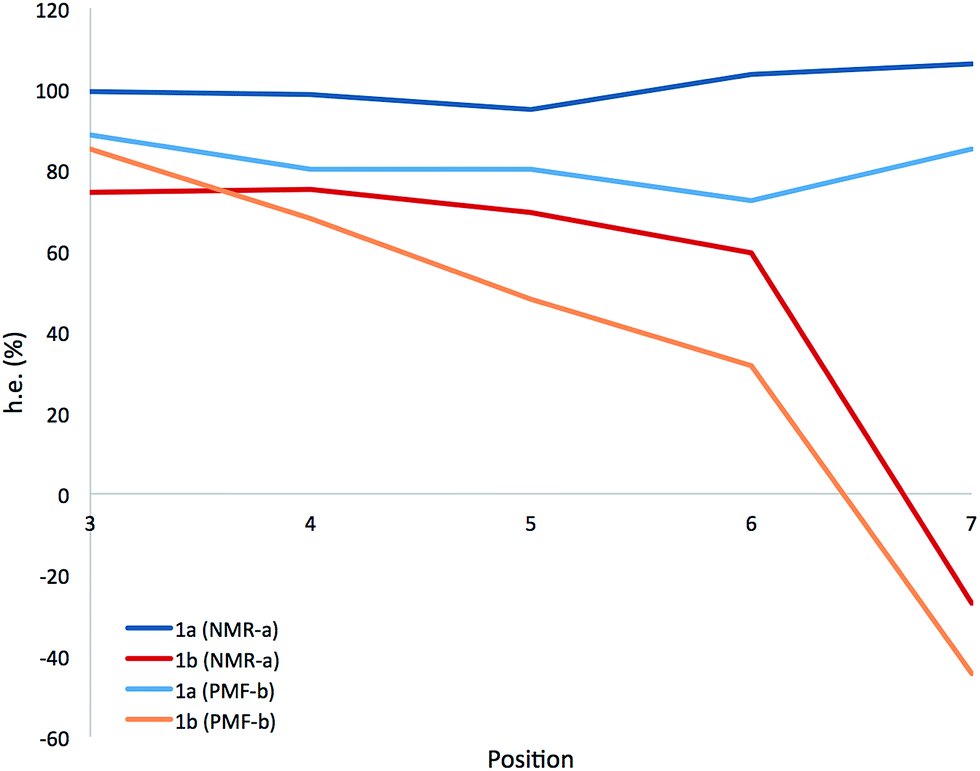 | ||
| Fig. 5 Variation of helical excess with position in the chain for the achiral Aib5 domain of 1a and 1b (a) calculated from the chemical shift difference Δδ in the 13C NMR spectrum; (b) calculated from the Boltzmann distributions resulting from PMF profiles in implicit solvent (see below). | ||
Helical excess in the ‘matched’ oligomer 1a was more or less consistent along the entire length of the chain, as a consequence of the uniform screw sense, with, intriguingly, a slight drop in the central portion of the chain that may indicate a small population of conformers in which a central left-handed domain is flanked by two screw-sense inversions. Values of h.e. that apparently exceed 100% are likely to be a result of the proximity of Aib7 to the terminal α-MeVal residues, which may perturb the simple dependence of chemical shift on helical excess.
The chemical shift differences of the Aib residues of the ‘mismatched’ oligomer 1b vary according to their position in the chain, being greatest at the N-terminus and falling towards the C-terminus. In the absence of enantioselective 13C labelling,38,49,53,63 the scalar value Δδ cannot distinguish positive and negative values of h.e., so (with support from computational work described below) we make the assumption that the gradual reduction in h.e. from Aib3 to Aib6 represents a decreasing preference for right-handed screw sense, crossing over to a left-handed screw sense at Aib7.
The progressive, rather than instantaneous, change in Δδ indicates that the screw-sense reversal located between Aib6 and Aib7 in the crystal structure of the mismatched oligomer 1b is a mobile, dynamic feature in solution, with the shift from a right-handed screw-sense preference at the N-terminus to a left-handed screw sense at the C-terminus distributed over several residues of the chain. The change in h.e. from one residue (A) to another (B) in a dynamic helical structure can be described by a value p(reversal)A–B for the likelihood of a screw-sense reversal being located between those two residues:
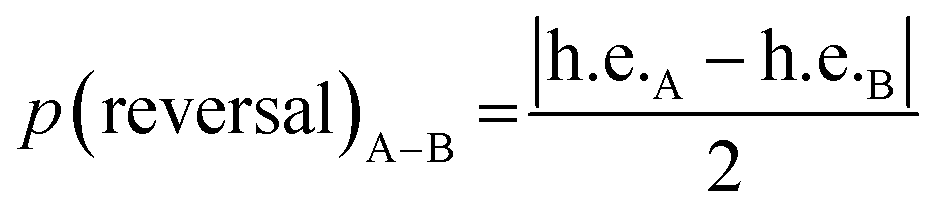
| p(reversal)A−B ∝ |ΔδA − ΔδB| |
By this reasoning, we deduce that the most likely location for a screw-sense reversal in 1b is between residues 6 and 7.
Given that the crystallographic screw-sense reversal entails the loss of an intramolecular hydrogen bond, we carried out 1H NMR studies of 1a and 1b with the aim of identifying NH protons not participating in intramolecular hydrogen bonding. Addition of increasing quantities of DMSO (0–10%) to a solution of either 1a or 1b in CDCl3 led to significant changes in chemical shift of two NH signals, suggesting that only two NH groups have the potential for intermolecular hydrogen bonding. Natural abundance 15N HSQC (see ESI†) experiments on 1b showed most of the 15N signals falling in a narrow band between −250 and −260 ppm, with the exception of the N-terminal carbamate 15N and the C-terminal amide 15N, which fell significantly upfield of the others, at around −280 ppm. These two upfield 15N signals also correspond to the two NH signals that experienced a large shift in the DMSO titrations, suggesting that in this case these NH shifts in DMSO merely show some fraying of the helix termini in polar solvent. The 15N spectrum of 1a shows a tighter grouping of most 15N signals, mostly between −240 and −246 ppm, with three signals at −260 ppm, none of which correspond to the NH signals that shift during the DMSO titrations. Earlier studies have shown that correlations between 15N chemical shift and hydrogen-bond strength are subject to complex and subtle effects,64–66 and this data does not allow us to assign with confidence the population of hydrogen bonds experimentally.
Computational analysis
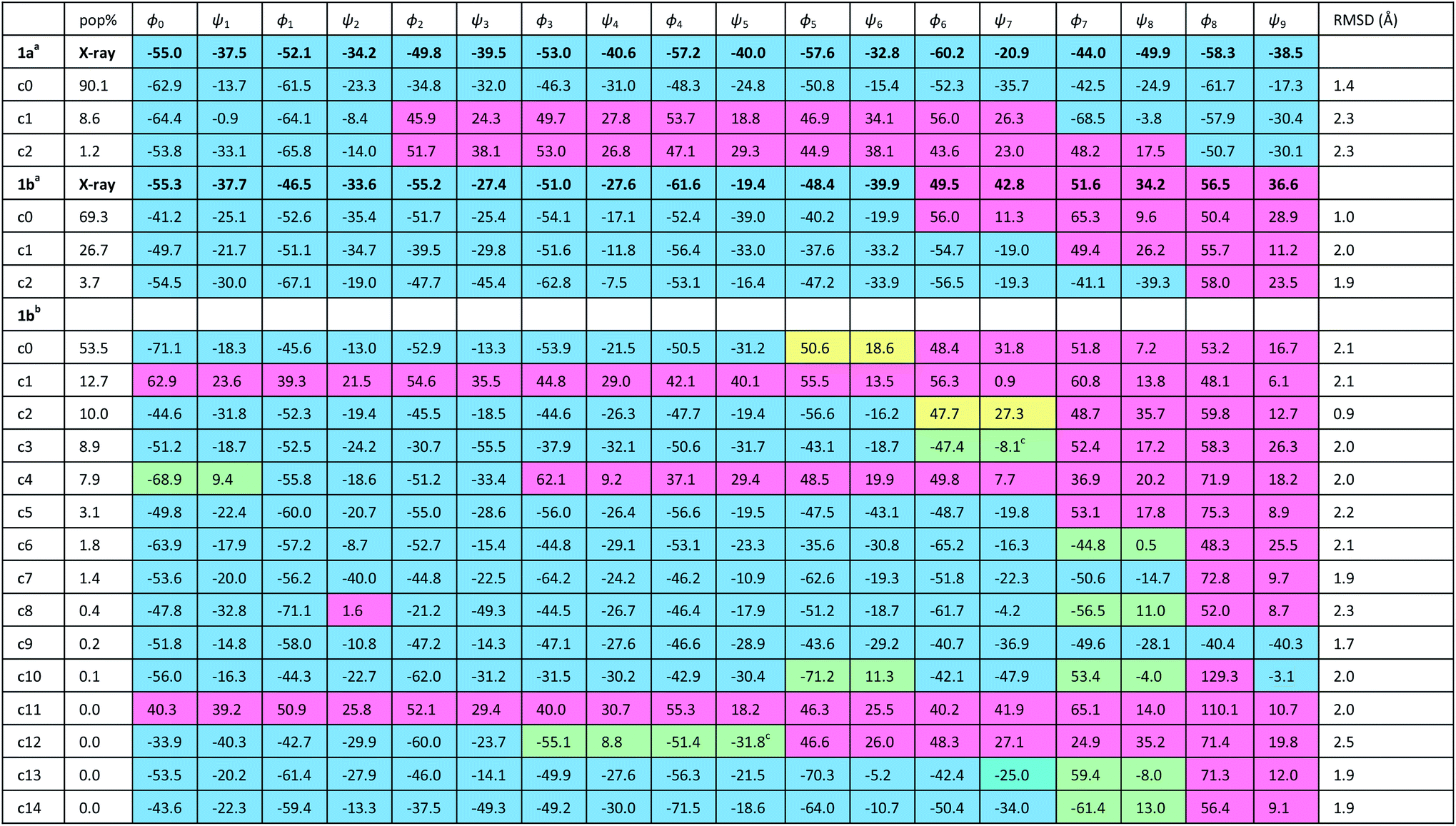
In order to gain insight into the conformations populated by oligomer 1b in the solution phase, we explored the structures of 1a and 1b computationally, using Replica Exchange Molecular Dynamics (REMD). REMD is a generalized-ensemble algorithm performing random walks in energy space, allowing exploration of the whole conformational space and statistical evaluation of the most energetically favoured conformations at a chosen temperature. It has been successfully applied to the study of conformational changes in biomolecules, including peptide folding.67Cluster analyses performed on the implicit solvent trajectories of oligomers 1a and 1b, reported in Table 1, show that in both cases the REMD simulations reproduce the crystallographic data (Fig. 6). The most populated clusters calculated for oligomers 1a (90.1%) and 1b (69.3%) have a root-mean-squared (RMS) deviation from the backbone of the corresponding X-ray structures of 1.4 Å and 1.0 Å respectively, and the computation and crystallographic structures are essentially superimposable (Fig. 6a and b). The most populated cluster of oligomer 1a is a continuous right-handed 310 helix, while that of oligomer 1b is a right-handed 310 helix from the N-terminus to Aib6 and a left-handed 310 helix from Aib7 to the C-terminus.
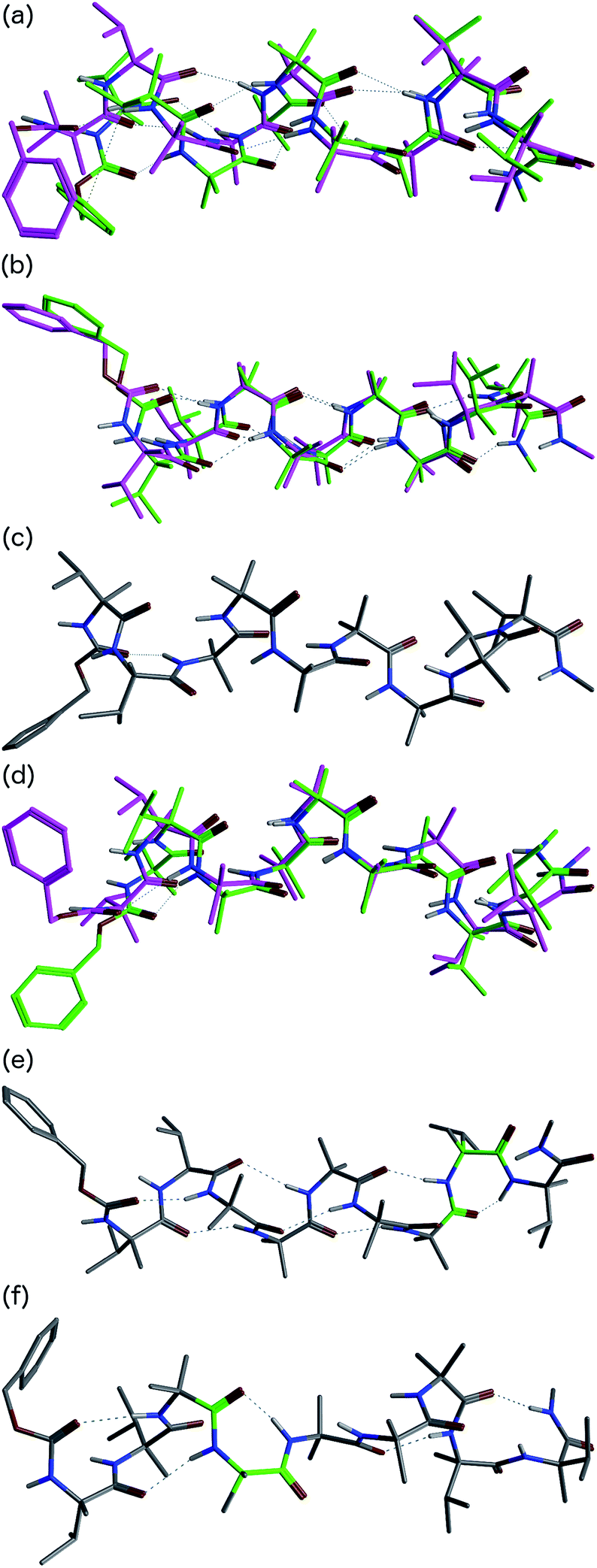 | ||
| Fig. 6 Superposition of the X-ray structures (green) and a representative structure of the most populated cluster (magenta) of (a) oligomer 1a and (b) oligomer 1b. (c) Representative structure of the most populated cluster of the REMD trajectory of 1b in explicit solvent at 303.60 K. (d) Superposition of the crystallographic structure of oligomer 1b (green) and the representative structure of cluster c2 (magenta) of the REMD trajectory in explicit solvent at 303.60 K. Representative structures of clusters (e) c6 and (f) c12 from the REMD trajectory of 1b in explicit methanol at 303.60 K. The γ-turns associated with i + 2 → i hydrogen bonds are highlighted in green. | ||
The most stable hydrogen bonds (occupancies >50%: see Table S1, ESI†) involve i + 3 and i residues and form β-turns that build up the 310 helix identifiable in the crystal structures (Fig. 6a and b and Table S1, ESI†). However, the occupancies of the hydrogen bonds between D-αMeVal8 and Aib5 and between D-αMeVal9 and Aib6 of peptide 1b are about 20% lower than the equivalent hydrogen-bonded interactions between L-αMeVal8 and Aib5 and between L-αMeVal9 and Aib6 of oligomer 1a, as a result of the screw-sense reversal. The reversal also manifests itself in the population distribution among the clusters. The most populated conformational cluster of oligomer 1a, corresponding to the right-handed 310 helix, comprises >90% of the total. Simulation of oligomer 1b suggests a less uniform conformational preference, with two major clusters, each having a screw-sense reversal at a different point along the chain, between residues 6 and 7, or 7 and 8 (Table 1).
Given the importance of hydrogen bonding in the detailed solution phase structures of 1a and 1b, and the possible association of the screw-sense reversal with the loss of an intramolecular hydrogen bond,15,26 a second REMD simulation of oligomer 1b was performed with methanol as an explicit solvent (Table 1). A few differences from the earlier simulation in implicit solvent were evident. The representative structure of the most populated cluster (53.5%) now showed a helical screw-sense reversal between Aib5 and Aib6, with an RMS deviation from the crystallographic structure of 2.06 Å (Fig. 6c). Nonetheless, the representative structure of cluster c2 (10.0%) has a conformation that is superimposable on the X-ray structure with a RMSD of 0.90 Å (Fig. 6d).
The screw-sense reversal in clusters c0 and c2 is accompanied by an unsatisfied hydrogen bond, as seen in the X-ray crystal structure (Fig. 3b). Additionally, some minor clusters (e.g. c3, c4, c6, c8, c10, c12–14: Table 1 and Fig. 6e and f) clearly display γ-turns, which involve i + 2 → i hydrogen bonds at various points along the chain. The γ-turns are all associated with screw-sense reversals. Hydrogen-bond analysis of the REMD trajectories in explicit methanol (Table S2, ESI†) indicate that while these γ-turns can involve any residue, the highest occupancies of i + 2 → i hydrogen bonds are those between Aib7 and Aib5, D-αMeVal8 and Aib6 and D-αMeVal9 and Aib7.
From these data, we deduce that explicit methanol lowers the barriers for the reversal of screw sense at any point along the chain, possibly through the stabilization of γ-turn intermediates. The near-planar γ-turns occur more frequently at the boundary between domains of opposite helical screw sense. A γ-turn has been observed centred on a quaternary derivative of Ac3c,58 and we are currently exploring the hypothesis that γ-turns may play a role in the kinetic mechanism of screw-sense inversion.50
The different behaviour of the two oligomers 1a and 1b is confirmed by potential of mean force (PMF) profiles as a function of φ and ψ dihedral angles (Fig. 7), initially obtained from the implicit solvent trajectories. Two minima are evident for each dihedral angle, with the values of φ = ±50° and ψ = ±30° corresponding to the right- and left-handed 310 helical conformations. The barriers to interconversion between these enantiomeric conformations of φ are significantly higher than the barriers to interconversion between the enantiomeric conformations of ψ. PMF profiles of oligomer 1a show a consistent preference for a right-handed helical conformation (φ = −50°; ψ = −30°) at every residue in the chain, with the preference being strongest (ca. 2.5 kcal mol−1) near the termini and weakening (to about 1 kcal mol−1) in the middle of the chain (residues 4–6). For oligomer 1b, the PMF profiles show a right-handed conformational preference at the N-terminus (2.5 kcal mol−1 at residues 1, 2) which steadily decreases through residues 3–6. Dihedral angles φ3–5 and ψ4–6 display a progressive reduction in the energy difference between the two minima, culminating in a reversal of the screw-sense preference on passing φ6 and ψ7 at Aib7. Through the remaining dihedral angles φ6–8 and ψ7–9, the global minimum corresponding to the left-handed helix becomes progressively more stable. The change in distribution as a function of position in the chain suggests that the inversion from right-handed to left-handed screw sense in 1b occurs most commonly between Aib6 and Aib7, but that conformers with the reversal between Aib5 and Aib6 or between Aib7 and αMeVal8 are not greatly disfavoured.
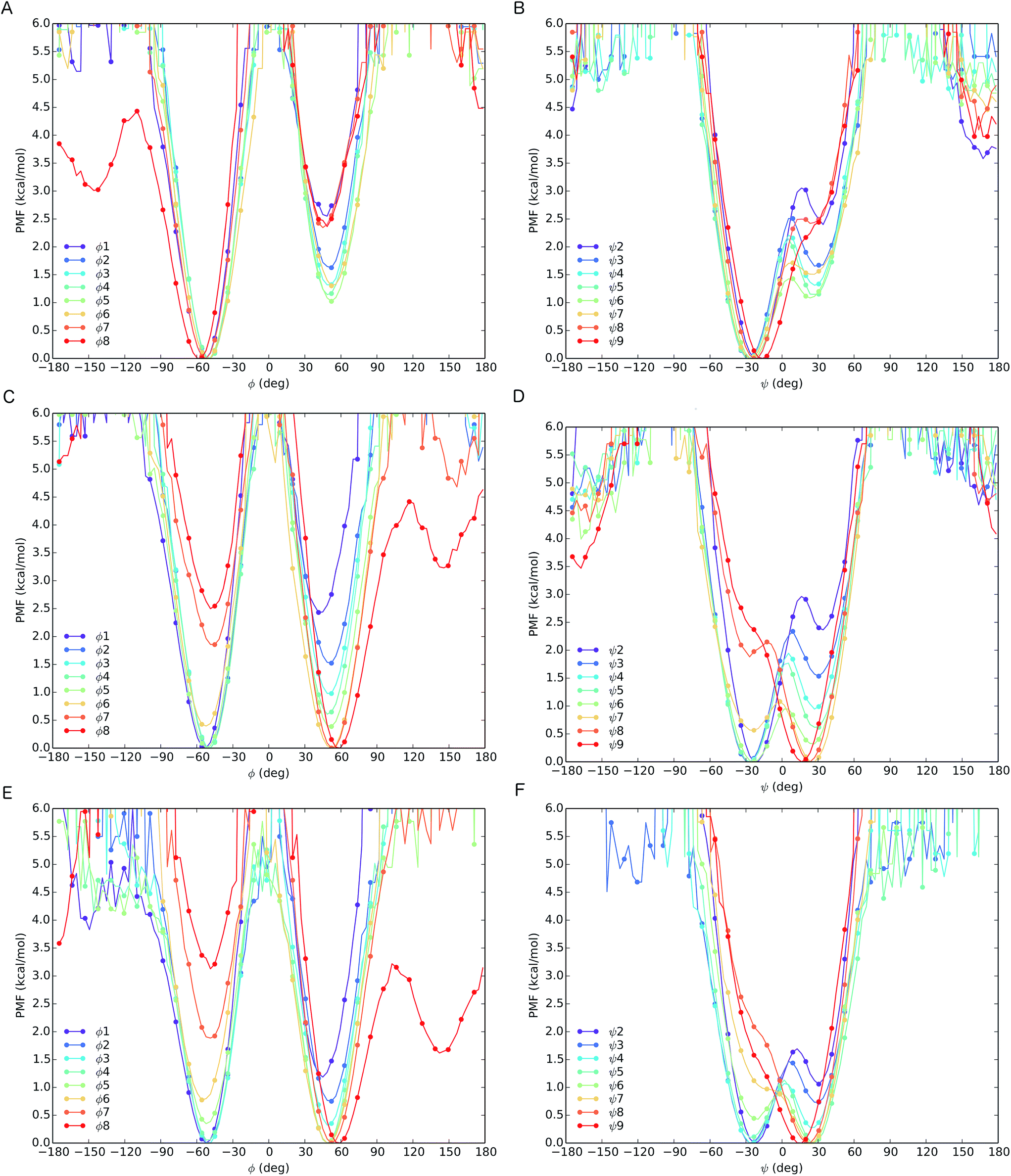 | ||
| Fig. 7 PMF as a function of φ (A) and ψ (B) dihedrals for oligomer 1a and of φ (C) and ψ (D) dihedrals for oligomer 1b from REMD simulations in implicit solvent. PMF as a function of φ (E) and ψ (F) dihedrals for oligomer 1b from REMD simulation in explicit MeOH. | ||
In order to model more closely the conformation in methanol solution, PMF profiles were also calculated for 1b in explicit methanol (Fig. 7). Both the energetic differences between the two screw-sense conformers and the barriers to screw-sense inversion were lower than in implicit solvent. Importantly, in explicit methanol the right-handed screw sense is favoured from the N-terminus only as far as Aib4. Aib5 has almost equal stability in either screw sense, and from Aib6 to the C-terminus the left-handed screw sense is favoured. Thus, overall, REMD simulations and PMF analysis suggest that in explicit methanol the screw-sense reversal is more mobile and more likely to be found closer to the centre of the oligo-Aib domain, close to Aib5, than in implicit solvent, where the reversal is found to be most likely between Aib6 and Aib7.
The populations at 300 K of left- and right-handed screw-sense conformers at each residue were calculated from the Boltzmann distributions that arise from these PMF energy differences (see ESI, Tables S3–S5†). For comparison, the values derived from NMR experiments are shown in Fig. 5 for 1a and 1b in implicit solvent. Values obtained from the implicit solvent simulation match better with the NMR data than those computed from the explicit methanol REMD simulation. However, the differences observed are within the internal error of the method and result from the statistical nature of PMF analysis.
Locating the screw sense reversal spectroscopically
In agreement with the NMR data (Fig. 5), the computational predictions suggested that in the solution phase the screw-sense reversal in 1b is mobile, but is most likely to be found in the vicinity of residues Aib5, Aib6 or Aib7 (in other words, between the middle of the oligo-Aib domain and its C-terminal end), depending on the method of simulation. Guided by this information we made a series of compounds with the aim of locating the reversal spectroscopically by methods that allow the differentiation of left- and right-handed screw-sense preference.Circular dichroism generally provides an overview of the global conformation of a peptide oligomer through bands located around 200–220 nm. Mazaleyrat et al. have used the conformationally responsive aromatic amino acid Bip 3 to provide a more detailed local probe of screw-sense preference in the form of the sign of its Cotton effect at 247 nm.68–70 We reasoned that a series of oligomers 4 in which a Bip probe was stationed at successive positions along the chain would allow us to extract local information about screw sense and thus gather evidence for the location of a trapped reversal. A series of three structures 4a–c were made in which a Bip residue forms the second, fourth or sixth of a series of seven achiral residues, the remainder being Aib (Fig. 8). Owing to the synthetic challenges associated with the repeated hindered couplings required to couple an αMeVal dimer to each end of the oligomer, structures 4a–c were made with a single L-αMeVal residue at the N-terminus and a single D-αMeVal at the C-terminus. Cbz-L-αMeVal is reported35 to induce a P screw-sense preference of ∼50% h.e. at a site four residues from the N-terminus of an Aib oligomer and D-αMeValNHR an M screw-sense preference of ∼70% h.e. at a site four residues from the C-terminus.36 The mismatch between these approximately equal and opposite screw-sense preferences should, as in 1b and 2b, trap a screw-sense reversal within the achiral domains of 4a–c.
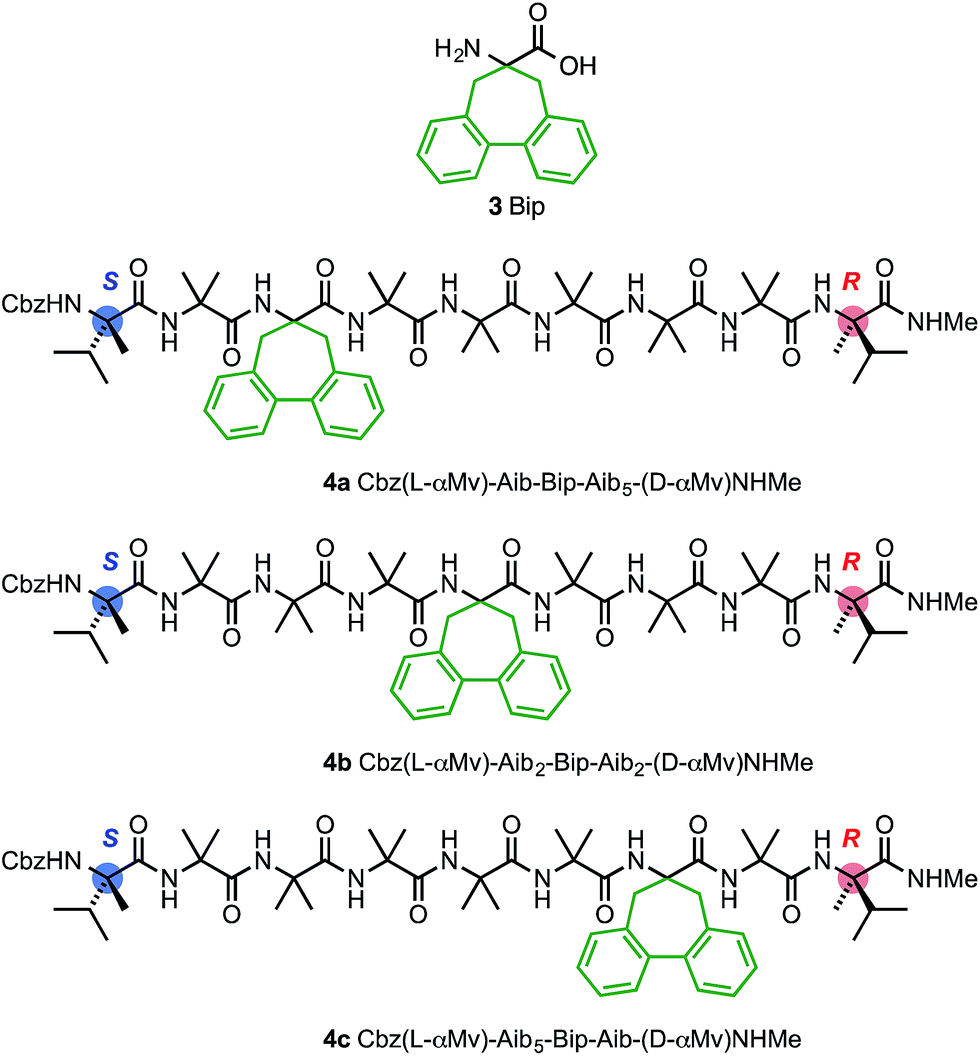 | ||
| Fig. 8 Bip 3 and Bip-containing oligomers 4a–c, with opposing terminal screw-sense preferences and the CD-responsive Bip residue at positions 3, 5 and 7. | ||
The CD spectra of 4a–c are shown in Fig. 9a. In each case a Cotton effect is evident, due to the Bip residue, with a positive or negative maximum at 247 nm.68–70 For the two compounds 4a and 4c in which the Bip residue is located close to the terminal controllers, the sign of the Cotton effect is consistent with the screw sense induced by the nearby controller, as expected, and both Cotton effects have more or less equal and opposite molar ellipticity. The CD spectrum of the oligomer 4b indicates that the screw sense at the location of the Bip residue, which lies in the middle of the chain, is right-handed. This suggests, that in these compounds, the screw-sense reversal lies between residue 5 and 7 of the oligomer, a result consistent with evidence from X-ray crystallography, NMR and computation that the screw-sense reversal of 1b lies closer to the C-terminus than to the N-terminus of the achiral central domain.
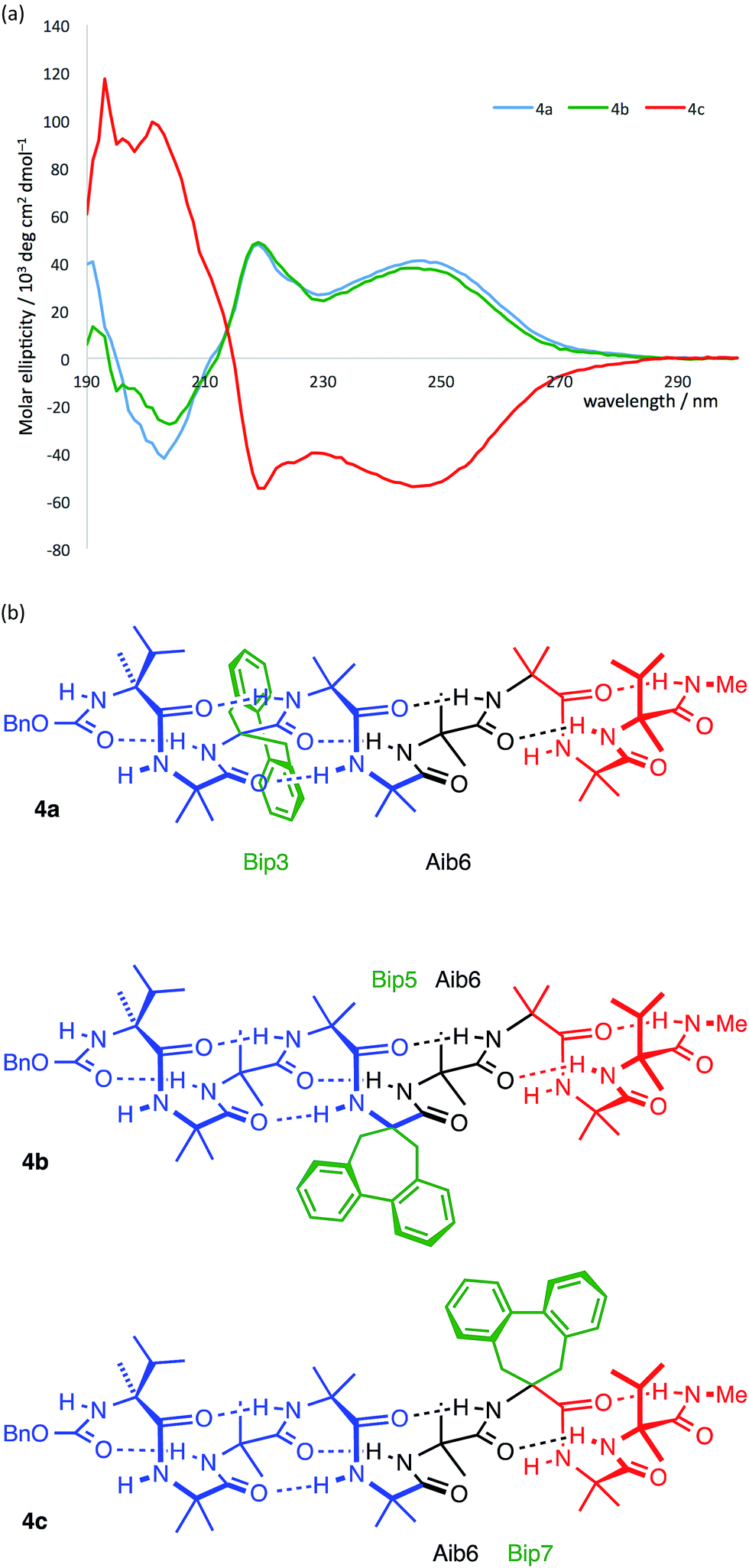 | ||
| Fig. 9 (a) CD spectra of compounds 4a–c indicating that the Bip residue is in a P environment in 4a and 4b and in an M environment in 4c; (b) presumed principal conformation of the three isomers, with the screw-sense reversal located at Aib6. Blue indicates right-handed screw sense; red indicates left-handed screw sense. | ||
Curiously, the degree of screw-sense induction does not decrease when the Bip residue is moved from position 3 to position 5, which suggests that in 4b the screw-sense reversal never lies to the N-terminal side of residue 5. This seemed surprising, but the bulky Bip residue is manifestly not a ‘silent’ local reporter of screw-sense preference, and could itself have a role to play in amplifying the screw-sense preference. We therefore turned to less intrusive NMR methods and to the analysis of chemical shift differences between 13C signals of enantioselectively isotopically enriched Aib residues in order to quantify changes in screw-sense preference along the chain.
The oligomer 5a, terminated at both ends by matched L-(α-MeVal) residues and carrying a Gly residue at the mid-point of the chain,71 showed a lower degree of anisochronicity in the 1H NMR signals of this Gly residue (Δδ = 200 ppb) than its ‘mismatched’ diastereoisomer 5b (Δδ = 290 ppb). The conformational flexibility associated with a Gly residue,53,56 along with its tendency to loosen a 310 helix towards an α-helix structure,60 makes it difficult to draw quantitative conclusions from these figures. The CD spectra of 5a and 5b (Fig. 10) suggest that 5a does indeed display more right-handed α-helical character than the all-Aib chain (the band at 220 nm is negative rather than positive as in 1a) and that 5b contains essentially equal amounts of left- and right-handed helical structures (its CD spectrum is much weaker than 5a).
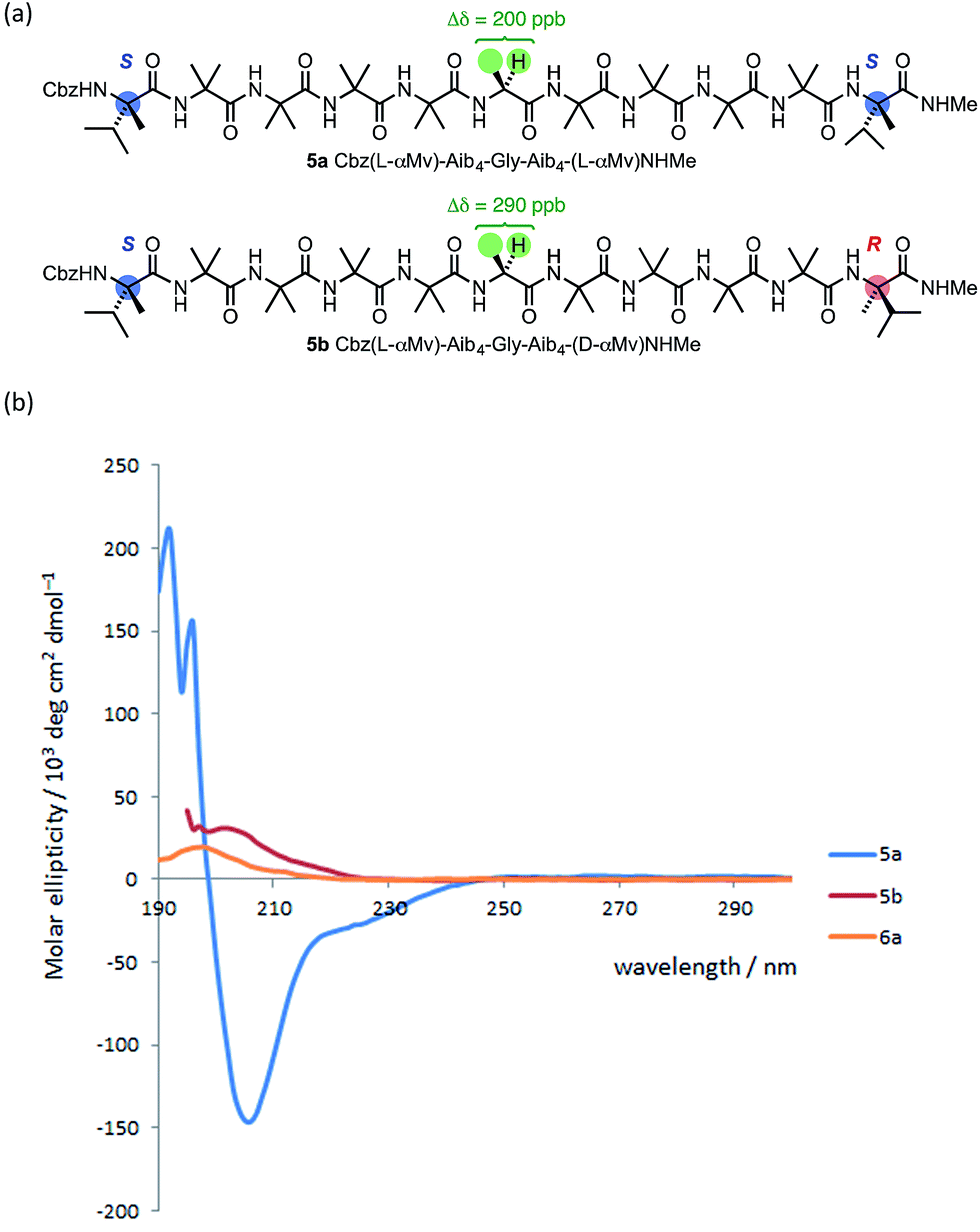 | ||
| Fig. 10 (a) Gly-containing compounds 5; (b) CD spectra of 5a, 5b and 6a recorded in MeOH at 20 °C. | ||
Raman spectroscopy of 5a and 5b showed in both cases a strong band centred at 1662 cm−1 (ESI, Fig. S2†). Band deconvolution on the amide 1 region showed two chief contributors to this band, at 1661 cm−1 and 1682 cm−1, in a 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for 5a and a 9:1 ratio for 5b. As found for 1a and 1b, these foldamers principally adopt 310 helical conformations, either in single or multiple domains. However the small contribution at 1682 cm−1 would be consistent with small regions adopting non-helical conformations. (The dimer N3Aib2OtBu, for example, which is too short to fold into a helix, shows only a single amide 1 band at 1681 cm−1 after deconvolution [ESI, Fig. S3†].) This contributor at 1682 cm−1 is more pronounced for the screw-sense mismatched foldamer 5b, consistent with a non-helical region possibly localized around the Gly residue.
:1 ratio for 5a and a 9:1 ratio for 5b. As found for 1a and 1b, these foldamers principally adopt 310 helical conformations, either in single or multiple domains. However the small contribution at 1682 cm−1 would be consistent with small regions adopting non-helical conformations. (The dimer N3Aib2OtBu, for example, which is too short to fold into a helix, shows only a single amide 1 band at 1681 cm−1 after deconvolution [ESI, Fig. S3†].) This contributor at 1682 cm−1 is more pronounced for the screw-sense mismatched foldamer 5b, consistent with a non-helical region possibly localized around the Gly residue.
To allow confidence in assigning absolute screw-sense preference at each point along the chain, compounds 6a–c were made, in which individual Aib residues were labelled with enantioselective 13C enrichment in one of their two Me groups (Fig. 11). To minimize the number of compounds needed, previous indications that the reversal was likely to reside between the Aib4 and Aib7 residues of the Aib heptamer fragment were used to narrow the search window to this area of the molecule. Oligomers 6a, 6b and 6c were made with (R)-mono-13CH3-Aib (Aib*, ca. 75:25 e.r.63) residues incorporated at positions 4, 5 and 6 respectively, with 6b additionally containing a second 13C-labelled Aib residue at position 7, and diluted to 50% total 13C abundance to allow the two labelled residues to be distinguished.22 The Aib* residues were introduced by means of labelled 2-azidoisobutyric acid derivatives, made by treatment of monolabelled Aib* with trifluoromethylsulfonyl azide.72,73 The CD spectrum of 6a (Fig. 10), like that of 5b, showed no strong preference for either screw sense, as expected for a structure adopting an M screw-sense at the N-terminus and a P screw-sense at the C-terminus.
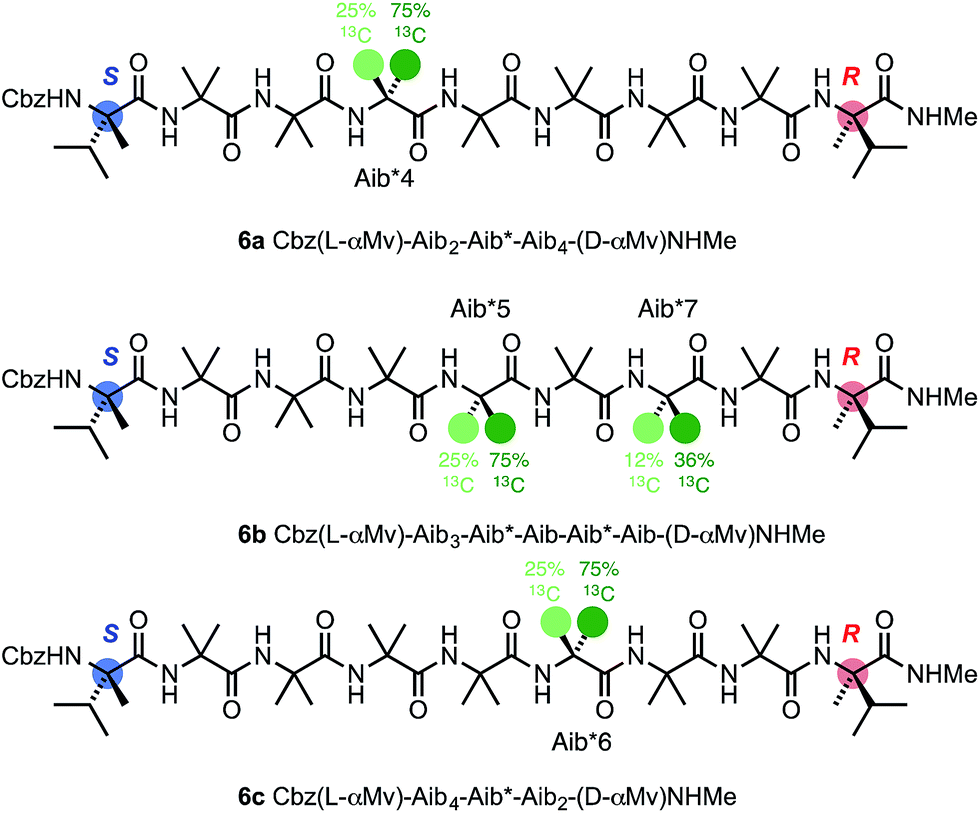 | ||
| Fig. 11 Oligomers containing 13C-labelled Aib residues enantioselectively isotopically enriched in their pro-R:pro-S methyl groups in a 3:1 ratio. The isotopic enrichment of residue 7 of 6b is half that of residue 5, to aid identification. | ||
NMR spectra of 6a–6c were run in MeOH and in MeCN and the results (shown in Fig. 12) are broadly comparable. The absolute screw sense is low at all four positions, with values of Δδ all less than 1500 ppb, corresponding to ca. 30% h.e. M screw sense (indicated by a downfield major signal, and represented here as a negative value for both Δδ and h.e.) dominating from the C-terminus to residue 5, with the decrease in chemical shift separation from Aib7 to Aib5 indicating a reduction in average screw-sense preference further from the C-terminus. By Aib4, the screw sense is essentially zero, with the spectrum in MeOH suggesting a P screw-sense is just beginning to dominate, while those in MeCN suggest that an M screw-sense persists even as far as Aib4.
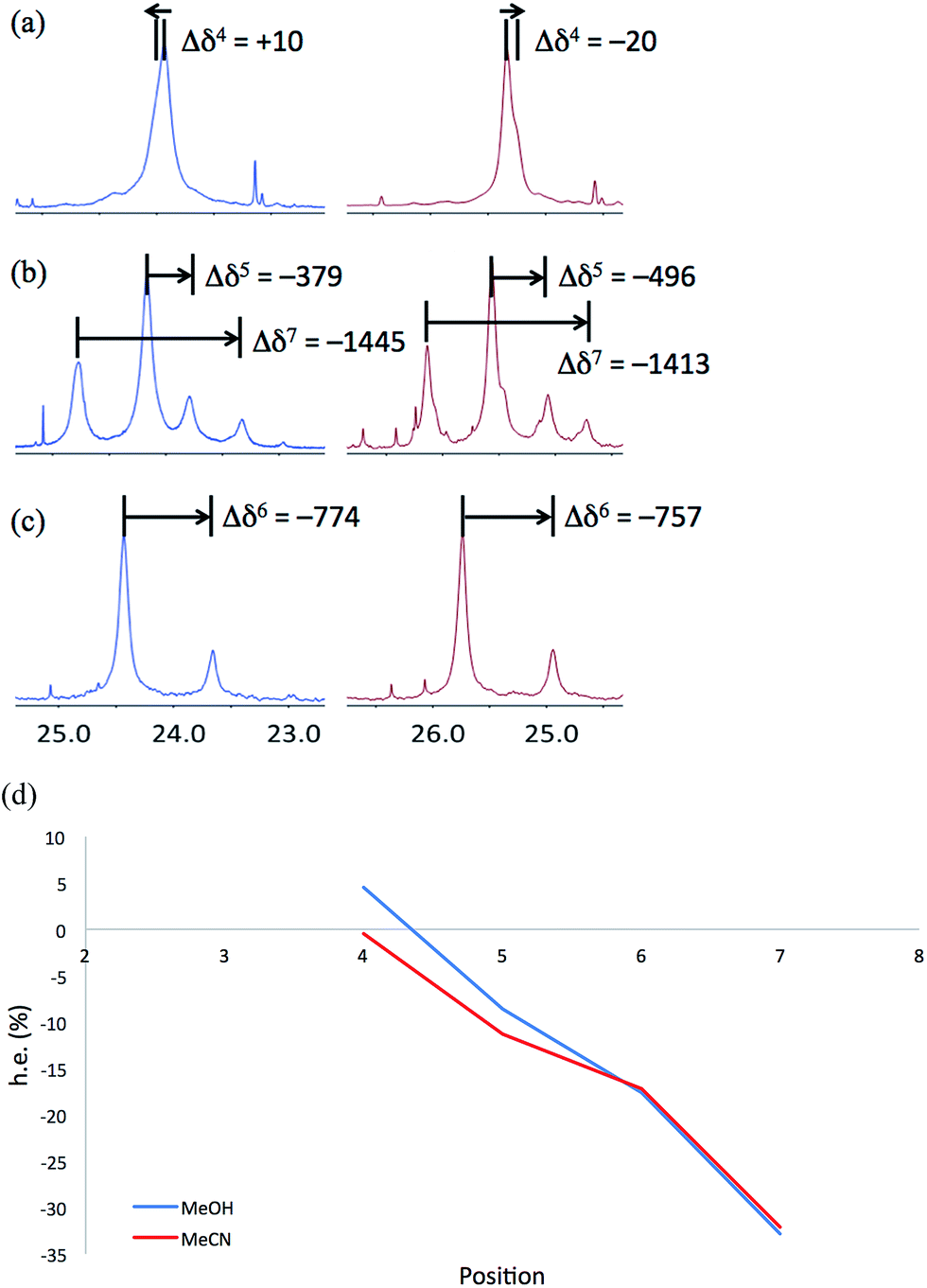 | ||
| Fig. 12 Portions of the 13C NMR spectra recorded at 23 °C in MeOH (blue, left) and MeCN (red, right), with Δδ values in ppb, of (a) 6a, (b) 6b and (c) 6c. (d) Plot of h.e., calculated from chemical shift difference Δδ in the 13C NMR spectrum recorded in methanol (blue) and acetonitrile (red), against the position in the chain, showing a change from an M to a P screw-sense at around Aib4. (Negative h.e. indicates an M screw-sense.) | ||
These data are consistent with our general interpretation of the experimental and computational data shown in Fig. 5: these show a gradual, rather than sudden, change in screw sense along the achiral domain of 1 and 6, with a reversal in the average screw-sense preference – and thus the average location of the screw-sense reversal – occurring somewhere close to the centre of the achiral domain, around Aib4 (in 6) or Aib6 (in 1).
Summary and conclusions
We studied a group of substrates of the general structure Cbz-Paa1-Xaa2-Aib3-Aib4-Aib6-Aib7-Yaa8-Maa9-NHMe, in which Paa and Maa are residues that induce preferred P and M screw-sense preferences respectively, and Xaa and Yaa may be either Aib or additional residues that reinforce those preferences. Using a variety of techniques, we located and characterized, in the solid state, in solution, and computationally, the reversal of screw sense that must occur in these terminally mismatched helical oligomers. The crystal structure of 1b suggests that the screw-sense reversal may take the form of an antisymmetric tendril perversion, accompanied by the loss of a single hydrogen bond. Computational studies in implicit solvent are consistent with this view of the screw-sense inversion in the absence of solvent. The minimum energy structures calculated for 1b are close to that observed crystallographically, with a reversal of screw sense located at Aib7.Studies using NMR spectroscopy allowed us not only to locate the average position of the screw-sense reversal, but also to assess its mobility and preferred habits. In compound 1b, the reversal happens on average between Aib6 and Aib7, but the less than quantitative preference for a P screw-sense at residues 3–6 suggests that the screw-sense inversion spends some of its time close to the N-terminus. The unchanging value of h.e. between residues 3 and 6 indicates that the reversal is not commonly found in this part of the structure.
Computational studies of 1b in explicit methanol support this interpretation. The most populated clusters of structures show screw-sense reversals at Aib6 or Aib7, consistent with the NMR data. A less populated, but still accessible, cluster shows a left-handed helical preference stretching as far as the N-terminus, also consistent with the NMR data. Calculation of the relative energies of the M and P screw-senses at each point in the chain show a steady decrease in energetic preference for a P screw-sense from Aib3 to Aib4, switching to a preference for an M screw-sense at Aib5. These screw-sense preferences show a good match to the solution state values. Computed structures in explicit methanol also suggest that screw-sense reversals are associated with γ-turns, and a γ-turn offers a relatively stable solution-state alternative to the non-hydrogen bonded structure adopted by the screw-sense reversal in the crystalline state.
The less powerfully induced terminal screw-sense preference of 6 results in an even more gradual variation of screw-sense preference between Aib3 and Aib7. NMR spectroscopy shows that the screw-sense reversal in 6 lies on average at Aib4, but that there is a smooth and gradual increase in preference for an M screw-sense between this point and the C-terminus, indicating that the screw-sense reversal is significantly more mobile in 6 than in 1, spending some of its time at most positions in the chain. The mobility of the reversal must be rapid on the NMR time scale, with migration of a screw-sense reversal from one end of a helical oligomer to the other providing a plausible mechanism by which left- and right-handed screw senses interconvert with one another on this rapid time scale.
Our strategy for exploring the helical reversal using mismatched induction of screw sense has potential for applicability across a wide range of helical polymers. Foldamer chemistry has classically sought to build conformationally well defined structures, but the enforced induction of this conformationally mobile feature makes possible the planned incorporation of well-defined, localised conformational flexibility into artificial structures. Evaluation of the relative population of rapidly interconverting conformers is made possible by reducing structural complexity to the point where quantified NMR analysis is facilitated. The simplification of these structures in order to reveal the fine details of conformation and to open up possibilities for using fine-grained control over the dynamics of biomimetic molecules is a concept ripe for further application.
Acknowledgements
This work was funded by the ERC (Advanced Grant ROCOCO), EPSRC and BBSRC. We are grateful to Prof J. H. Maddocks (École Polytechnique Fédérale de Lausanne) for helpful discussions.Notes and references
- C. McManus, Right Hand, Left Hand, Harvard University Press, 2003 Search PubMed.
- N. Chouaieb, A. Goriely and J. H. Maddocks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9398–9403 CrossRef CAS PubMed.
- C. Darwin, The movements and habits of climbing plants, 1882 Search PubMed.
- A. Goriely and M. Tabor, Phys. Rev. Lett., 1998, 80, 1564–1567 CrossRef CAS.
- T. McMillen and A. Goriely, J. Nonlinear Sci., 2002, 12, 241–281 CrossRef.
- J. Liu, J. Huang, T. Su, K. Bertoldi and D. R. Clarke, PLoS One, 2014, 9, e93183 Search PubMed.
- P. E. S. Silva, J. L. Trigueiros, A. C. Trindade, R. Simoes, R. G. Dias, M. H. Godinho and F. V. de Abreu, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed.
- E. Yashima, K. Maeda and T. Nishimura, Chem.–Eur. J., 2004, 10, 42–51 CrossRef CAS PubMed.
- D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349–1372 RSC.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- O. Pieroni, A. Fissi, C. Pratesi, P. A. Temussi and F. Ciardelli, J. Am. Chem. Soc., 1991, 113, 6338–6340 CrossRef CAS.
- S. Lifson, C. E. Felder and M. M. Green, Macromolecules, 1992, 25, 4142–4148 CrossRef CAS.
- A. Tuzi, M. Rosaria Ciajolo, D. Picone, O. Crescenzi, P. A. Temussi, A. Fissi and O. Pieroni, J. Pept. Sci., 1996, 2, 47–58 CAS.
- A. Pietropaolo and T. Nakano, J. Am. Chem. Soc., 2013, 135, 5509–5512 CrossRef CAS PubMed.
- M. Kudo, V. Maurizot, H. Masu, A. Tanatani and I. Huc, Chem. Commun., 2014, 50, 10090–10093 RSC.
- Y. Nagata, T. Yamada, T. Adachi, Y. Akai, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2013, 135, 10104–10113 CrossRef CAS PubMed.
- V. Jain, K. S. Cheon, K. Tang, S. Jha and M. M. Green, Isr. J. Chem., 2011, 51, 1067–1074 CrossRef CAS.
- B. A. F. Le Bailly, L. Byrne, V. Diemer, M. Foroozandeh, G. A. Morris and J. Clayden, Chem. Sci., 2015, 6, 2313–2322 RSC.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed.
- J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131–3152 CrossRef CAS PubMed.
- I. L. Karle and P. Balaram, Biochemistry, 1990, 29, 6747–6756 CrossRef CAS PubMed.
- B. V. Prasad and P. Balaram, CRC Crit. Rev. Biochem., 1984, 16, 307–348 CrossRef CAS PubMed.
- C. Toniolo and E. Benedetti, Trends Biochem. Sci., 1991, 16, 350–353 CrossRef CAS PubMed.
- C. Toniolo, M. Crisma, G. M. Bonora, E. Benedetti, B. Dl Blasio, V. Pavone, C. Pedone and A. Santini, Biopolymers, 1991, 31, 129–138 CrossRef CAS.
- C. Toniolo, G. M. Bonora, A. Bavoso, E. Benedetti, B. Di Blasio, V. Pavone and C. Pedone, Macromolecules, 1986, 19, 472–479 CrossRef CAS.
- C. Toniolo, G. M. Bonora, V. Barone, A. Bavoso, E. Benedetti, B. Di Blasio, P. Grimaldi, F. Lelj, V. Pavone and C. Pedone, Macromolecules, 1985, 18, 895–902 CrossRef CAS.
- R. P. Hummel, C. Toniolo and G. Jung, Angew. Chem., Int. Ed. Engl., 1987, 26, 1150–1152 CrossRef.
- J. Solà, G. A. Morris and J. Clayden, J. Am. Chem. Soc., 2011, 133, 3712–3715 CrossRef PubMed.
- B. Pengo, F. Formaggio, M. Crisma, C. Toniolo, G. M. Bonora, Q. B. Broxterman, J. Kamphuis, M. Saviano, R. Iacovino, F. Rossi and E. Benedetti, J. Chem. Soc., Perkin Trans. 2, 1998, 1651–1657 RSC.
- M. De Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef CAS PubMed.
- M. De Poli, L. Byrne, R. A. Brown, J. Solà, A. Castellanos, T. Boddaert, R. Wechsel, J. D. Beadle and J. Clayden, J. Org. Chem., 2014, 79, 4659–4675 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2014, 50, 7949–7952 RSC.
- R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS PubMed.
- R. A. Brown, T. Marcelli, M. De Poli, J. Solà and J. Clayden, Angew. Chem., Int. Ed., 2012, 51, 1395–1399 CrossRef CAS PubMed.
- Y. Inai, Y. Kurokawa and T. Hirabayashi, Biopolymers, 1999, 49, 551–564 CrossRef CAS.
- Y. Inai, Y. Kurokawa, A. Ida and T. Hirabayashi, Bull. Chem. Soc. Jpn., 1999, 72, 55–61 CrossRef CAS.
- Y. Inai, Y. Kurokawa and N. Kojima, J. Chem. Soc., Perkin Trans. 2, 2002, 1850–1857 RSC.
- Y. Inai, S. Ashitaka and T. Hirabayashi, Polym. J., 1999, 31, 246–253 CrossRef CAS.
- N. Ousaka, Y. Takeyama, H. Iida and E. Yashima, Nat. Chem., 2011, 3, 856–861 CrossRef CAS PubMed.
- J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680–6691 CrossRef CAS PubMed.
- Y. Inai, K. Tagawa, A. Takasu, T. Hirabayashi, T. Oshikawa and M. Yamashita, J. Am. Chem. Soc., 2000, 122, 11731–11732 CrossRef CAS.
- N. Ousaka and Y. Inai, J. Org. Chem., 2009, 74, 1429–1439 CrossRef CAS PubMed.
- Y. Inai and H. Komori, Biomacromolecules, 2004, 5, 1231–1240 CrossRef CAS PubMed.
- Y. Inai, N. Ousaka and T. Okabe, J. Am. Chem. Soc., 2003, 125, 8151–8162 CrossRef CAS PubMed.
- L. Byrne, J. Solà, T. Boddaert, T. Marcelli, R. W. Adams, G. A. Morris and J. Clayden, Angew. Chem., Int. Ed., 2014, 53, 151–155 CrossRef CAS PubMed.
- I. Maffucci, M. Tomsett, A. Contini and J. Clayden, manuscript in preparation.
- R. Armen, D. O. V. Alonso and V. Daggett, Protein Sci., 2003, 12, 1145–1157 CrossRef CAS PubMed.
- J. Clayden, A. Castellanos, J. Solà and G. A. Morris, Angew. Chem., Int. Ed., 2009, 48, 5962–5965 CrossRef CAS PubMed.
- J. Solà, S. P. Fletcher, A. Castellanos and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 6836–6839 CrossRef PubMed.
- G. Yoder, A. Polese, R. A. G. D. Silva, F. Formaggio, M. Crisma, Q. B. Broxterman, J. Kamphuis, C. Toniolo and T. A. Keiderling, J. Am. Chem. Soc., 1997, 119, 10278–10285 CrossRef CAS.
- C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, J. Am. Chem. Soc., 1996, 118, 2744–2745 CrossRef CAS.
- T. Boddaert, J. Solà, M. Helliwell and J. Clayden, Chem. Commun., 2012, 48, 3397–3399 RSC.
- C. Toniolo, F. Formaggio, B. Kaptein and Q. Broxterman, Synlett, 2006, 1295–1310 CrossRef CAS.
- A. I. Jiménez, G. Ballano and C. Cativiela, Angew. Chem., Int. Ed., 2005, 44, 396–399 CrossRef PubMed.
- R. Gessmann, H. Brückner and K. Petratos, J. Pept. Sci., 2003, 9, 753–762 CrossRef CAS PubMed.
- J. Solà, M. Helliwell and J. Clayden, Biopolymers, 2011, 95, 62–69 CrossRef PubMed.
- A. Lakhani, A. Roy, M. De Poli, M. Nakaema, F. Formaggio, C. Toniolo and T. A. Keiderling, J. Phys. Chem. B, 2011, 115, 6252–6264 CrossRef CAS PubMed.
- M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11–20 CrossRef CAS PubMed.
- S. P. Fletcher, J. Solà, D. Holt, R. A. Brown and J. Clayden, Beilstein J. Org. Chem., 2011, 7, 1304–1309 CrossRef CAS PubMed.
- Z. Dega Szafran, M. Szafran, J. Sitkowski and L. Stefaniak, J. Phys. Org. Chem., 1996, 9, 746–750 CrossRef CAS.
- J. A. Sogn, W. A. Gibbons and E. W. Randall, Biochemistry, 1973, 12, 2100–2105 CrossRef CAS PubMed.
- D. H. Live, D. G. Davis, W. C. Agosta and D. Cowburn, J. Am. Chem. Soc., 1984, 106, 1939–1941 CrossRef CAS.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS; C. Bergonzo, A. J. Campbell, R. C. Walker and C. Simmerling, Int. J. Quantum Chem., 2009, 109, 3781–3790 CrossRef PubMed; A. Baumketner and J. E. Shea, J. Mol. Biol., 2007, 366, 275–285 CrossRef PubMed; C. Bergonzo, A. J. Campbell, C. de los Santos, A. P. Grollman and C. Simmerling, J. Am. Chem. Soc., 2011, 133, 14504–14506 CrossRef PubMed; F. Ding, D. Tsao, H. Nie and N. V. Dokholyan, Structure, 2008, 16, 1010–1018 CrossRef PubMed; E. Lin and M. S. Shell, J. Chem. Theory Comput., 2009, 5, 2062–2073 CrossRef PubMed; I. Maffucci, S. Pellegrino, J. Clayden and A. Contini, J. Phys. Chem. B, 2015, 119, 1350–1361 CrossRef PubMed; H. Nymeyer and A. E. Garcı, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13934–13939 CrossRef PubMed; S. Pellegrino, A. Contini, F. Clerici, A. Gori, D. Nava and M. L. Gelmi, Chem.–Eur. J., 2012, 18, 8705–8715 CrossRef PubMed; X. Periole and A. E. Mark, J. Chem. Phys., 2007, 126, 014903 CrossRef PubMed; A. Rodriguez, P. Mokoema, F. Corcho, K. Bisetty and J. J. Perez, J. Phys. Chem. B, 2011, 115, 1440–1449 CrossRef PubMed; M. S. Shell, R. Ritterson and K. A. Dill, J. Phys. Chem. B, 2008, 112, 6878–6886 CrossRef PubMed; P. Tao, J. R. Parquette and C. M. Hadad, J. Chem. Theory Comput., 2012, 8, 5137–5149 CrossRef PubMed; K. P. Van Nostrand, S. D. Kennedy, D. H. Turner and D. H. Mathews, J. Chem. Theory Comput., 2011, 7, 3779–3792 CrossRef PubMed; K. Yoshida, T. Yamaguchi and Y. Okamoto, Chem. Phys. Lett., 2005, 412, 280–284 CrossRef; A. Ruffoni, A. Contini, R. Soave, L. Presti Lo, I. Esposto, I. Maffucci, D. Nava, S. Pellegrino, M. L. Gelmi and F. Clerici, RSC Adv., 2015, 5, 32643–32656 RSC; I. Maffucci and A. Contini, J. Chem. Theory Comput., 2016, 12, 714–727 CrossRef PubMed.
- L. Dutot, K. Wright, M. Wakselman, J.-P. Mazaleyrat, C. Peggion, M. De Zotti, F. Formaggio and C. Toniolo, Tetrahedron Lett., 2008, 49, 3475–3479 CrossRef CAS.
- J.-P. Mazaleyrat, K. Wright, A. Gaucher, N. Toulemonde, M. Wakselman, S. Oancea, C. Peggion, F. Formaggio, V. Setnička, T. A. Keiderling and C. Toniolo, J. Am. Chem. Soc., 2004, 126, 12874–12879 CrossRef CAS PubMed.
- F. Formaggio, M. Crisma, C. Toniolo, L. Tchertanov, J. Guilhem, J. P. Mazaleyrat, A. Gaucher and M. Wakselman, Tetrahedron, 2000, 56, 8721–8734 CrossRef CAS.
- J. Solà, M. Helliwell and J. Clayden, J. Am. Chem. Soc., 2010, 132, 4548–4549 CrossRef PubMed.
- H. Kim, J. K. Cho, S. Aimoto and Y.-S. Lee, Org. Lett., 2006, 8, 1149–1151 CrossRef CAS PubMed.
- P. B. Alper, S.-C. Hung and C.-H. Wong, Tetrahedron Lett., 1996, 37, 6029–6032 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC X-ray crystal data for 1a and 1b have been deposited with the CCDC, deposition numbers 1518806 and 1518807. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05474a |
| This journal is © The Royal Society of Chemistry 2017 |