
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium(I)-catalyzed stereoselective [4+2] cycloaddition of oxetanols with alkynes through C(sp3)–C(sp3) bond cleavage†
Rui
Guo‡
ab,
Xinxin
Zheng‡
c,
Dayong
Zhang
*c and
Guozhu
Zhang
 *ab
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: guozhuzhang@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cInstitute of Pharmaceutical Science, China Pharmaceutical University, Nanjing, P. R. China. E-mail: cpuzdy@163.com
First published on 23rd January 2017
Abstract
An efficient and convenient synthesis of highly functionalized dihydropyrans has been achieved through rhodium(I)-catalysed tandem C(sp3)–C(sp3) bond cleavage and annulation of oxetanols with alkynes. An enantioselective version was enabled using a Binaphine ligand. Excellent site-selectivity and remarkable enantioretention are obtained for 2-substituted oxetanols.
Introduction
Dihydropyran and its derivatives are ubiquitous molecular skeletons, which are widely observed in natural products1 (Fig. 1), and are advanced intermediates that can lead to substances of biological or medicinal importance.2 Thus, new methods which enable convenient access to this type of motif in a step-economic, flexible and stereoselective fashion are highly demanded. | ||
| Fig. 1 Representative natural products. | ||
The selective cleavage and functionalization of carbon–carbon single bonds by transition-metal catalysts has undergone significant progress in recent years, allowing ready access to a variety of synthetically useful molecular scaffolds.3 Among those, cyclobutanols are frequently employed as privileged building blocks for the construction of complex cyclic molecules.4 In 2012, Murakami and co-workers reported seminal work on rhodium-catalyzed tandem C–C single bond cleavage/formal cycloaddition of benzocyclobutanols with alkynes [eqn (1)].4b Since then, vinyl ketones,5 carbene precursors,6 and allenes7 have been proven to be suitable cycloaddition counterparts, demonstrating the broad applicability of this protocol [eqn (2)–(4)]. However, to the best of our knowledge, under rhodium catalyzed cycloaddition with unsaturated 2-π systems, ring opening of cyclobutanols exclusively occurred on the C(sp2)–C(sp3) bond adjacent to the hydroxyl group, with subsequent addition of the ipso carbon [i.e. (sp2)C] to the 2-π units.
Previous work: Rh catalysed C–C bond cleavage of 4-membered tert-alcohols
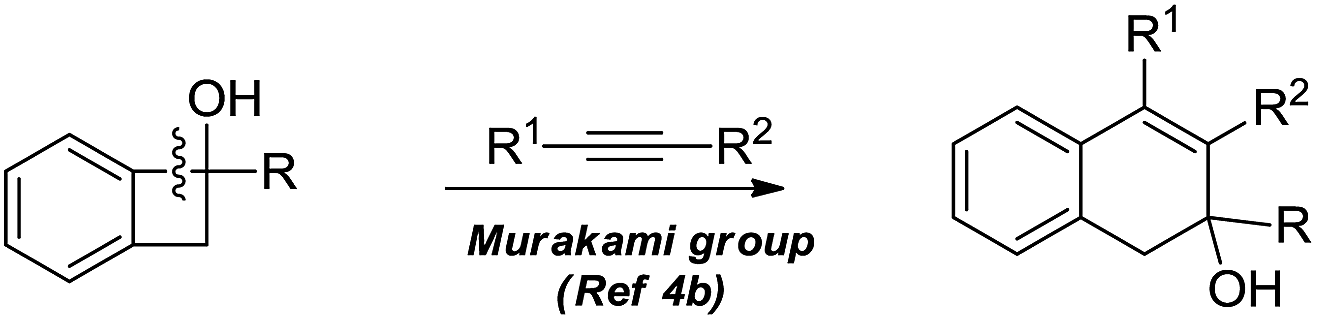 | (1) |
 | (2) |
 | (3) |
 | (4) |
This work: C(sp3)–C(sp3) cleavage and cycloaddition
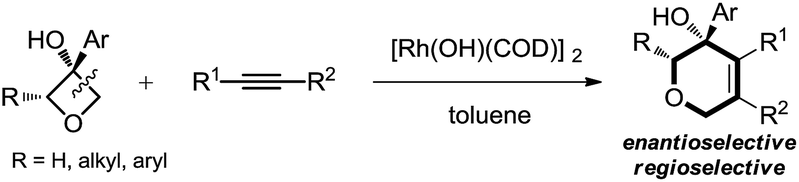 | (5) |
Therefore, the means to alter the hybridized nature of the additional carbon would offer a unique opportunity for chemists and would add to the fast expanding synthetic tool box, allowing the facile assembly of important molecular skeletons which are not easy to construct by conventional methods. Encouraged by the considerable medicinal utility and unique molecular skeleton of oxetane and its derivatives,8 we envisioned that they set a good stage for exploring new reactions. Herein, we report the highly efficient rhodium(I)-catalyzed cycloaddition of oxetanols with alkynes, via a tandem C(sp3)–C(sp3) bond cleavage and subsequent cross addition over a 2-π unit for the first time, leading to complex dihydropyran ring systems. Binaphine proves to be a viable chiral phosphine ligand to promote the enantioselective version of this transformation. Furthermore, for 2-substituted oxetanols, it was found that the ring opening selectively took place on the nonsubstituted side, with the subsequent ring closing proceeding in a highly diastereoselective manner [eqn (5)].
Results and discussion
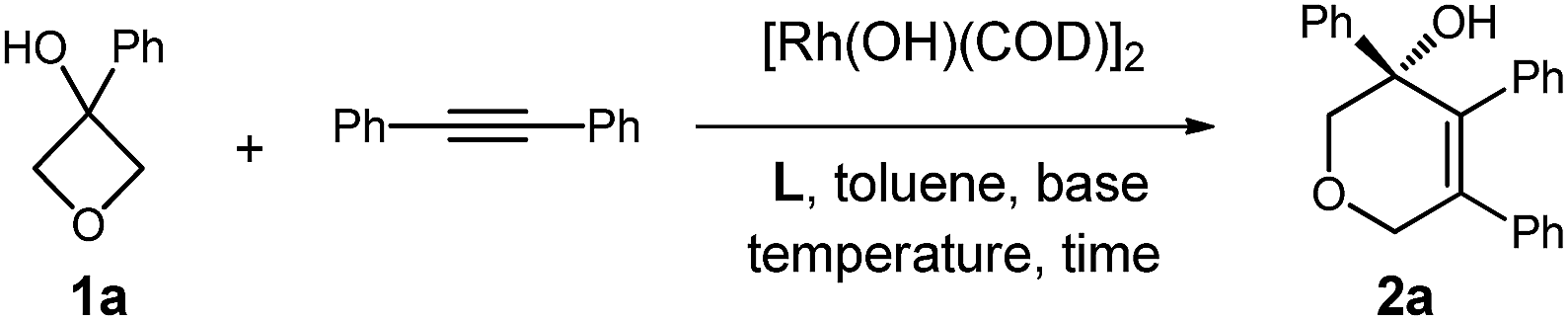
We initiated our studies by exploring the reaction of 3-phenyloxetan-3-ol, which was readily prepared from oxetan-3-one and 1,2-diphenylethyne (Table 1). After numerous trials, [Rh(OH)(COD)]2 proved to be the most effective catalyst, and the [4+2] cyclized product 2a was obtained in excellent yield (Table 1, entry 1). Emphasis was then put on the use of commercial chiral ligands for asymmetric carbon–carbon bond formation. BINAP and Segphos were not effective (Table 1, entry 2 and 3), with moderate yields and negligible enantiomer ratios being obtained. The reaction using Josiphos proceeded in good yield, albeit with only moderate enantioselectivity (Table 1, entry 4). To our delight, a much improved enantioselectivity of 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 er was obtained for 2a by using iPr-Duphos as the ligand (Table 1, entry 5). Further ligand examination identified Binaphine as the most effective ligand among those tested, which gave 2a in 91:9 er. Interestingly, analogous to Cramer's observations,4i the addition of 1.1 equiv. of K2CO3 significantly improved the reaction kinetics. Shortened reaction times and comparable product yields were achieved, suggesting base-facilitated ring opening and alkyne insertion. The er was further improved while lowering the temperature. Eventually, a 70% yield with 96.5:3.5 er of 2a was obtained when the reaction was conducted at 30 °C, however, a prolonged reaction time (72 hours) was required for full conversion of the starting material.
:18 er was obtained for 2a by using iPr-Duphos as the ligand (Table 1, entry 5). Further ligand examination identified Binaphine as the most effective ligand among those tested, which gave 2a in 91:9 er. Interestingly, analogous to Cramer's observations,4i the addition of 1.1 equiv. of K2CO3 significantly improved the reaction kinetics. Shortened reaction times and comparable product yields were achieved, suggesting base-facilitated ring opening and alkyne insertion. The er was further improved while lowering the temperature. Eventually, a 70% yield with 96.5:3.5 er of 2a was obtained when the reaction was conducted at 30 °C, however, a prolonged reaction time (72 hours) was required for full conversion of the starting material.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Base | Temperature | Time | Yielda [%] | erb |
| a Isolated yields. b The er was determined by chiral HPLC analysis. The absolute configuration of the products was assigned by single crystal X-ray analysis of 2i. c 1.1 equiv. of K2CO3 was used. | ||||||
| 1 | — | — | 110 °C | 6 h | 96 | — |
| 2 | L1 | — | 110 °C | 6 h | 75 | 55:45 |
| 3 | L2 | — | 110 °C | 6 h | 50 | 54:46 |
| 4 | L3 | — | 110 °C | 6 h | 45 | 37:63 |
| 5 | L4 | — | 110 °C | 6 h | 95 | 91:9 |
| 6c | L5 | K2CO3 | 110 °C | 6 h | 90 | 90.5:8.5 |
| 7c | L5 | K2CO3 | 70 °C | 12 h | 86 | 94:6 |
| 8c | L5 | K2CO3 | 50 °C | 24 h | 85 | 94:6 |
| 9c | L5 | K2CO3 | 30 °C | 72 h | 70 | 96.5:3.5 |
| 10 | L5 | — | 30 °C | 72 h | — | — |
|
||||||
With the optimized conditions in hand, we demonstrated the general applicability of our method with a range of oxetanols and alkynes (Table 2). We first evaluated the reactions of different oxetanols with diphenylethyne. Various arylated oxetanols bearing ortho-, para- and meta-substituted phenyl groups reacted smoothly to give the desired products in moderate to good yields with high enantioselectivities (er > 95:5). The substituted groups could be alkyl, methoxy, or fluoro groups. It needs to be pointed out that alkyl and alkenyl oxetanols did not undergo cycloaddition with alkynes under the current reaction conditions. The variations of diaryl alkynes were then briefly investigated and, to our delight, it was observed that alkynes with electron-donating methyl and methoxy groups at the para position can serve as suitable substrates and the corresponding dihydropyrans were isolated in moderate yields with high enantiomeric ratios.
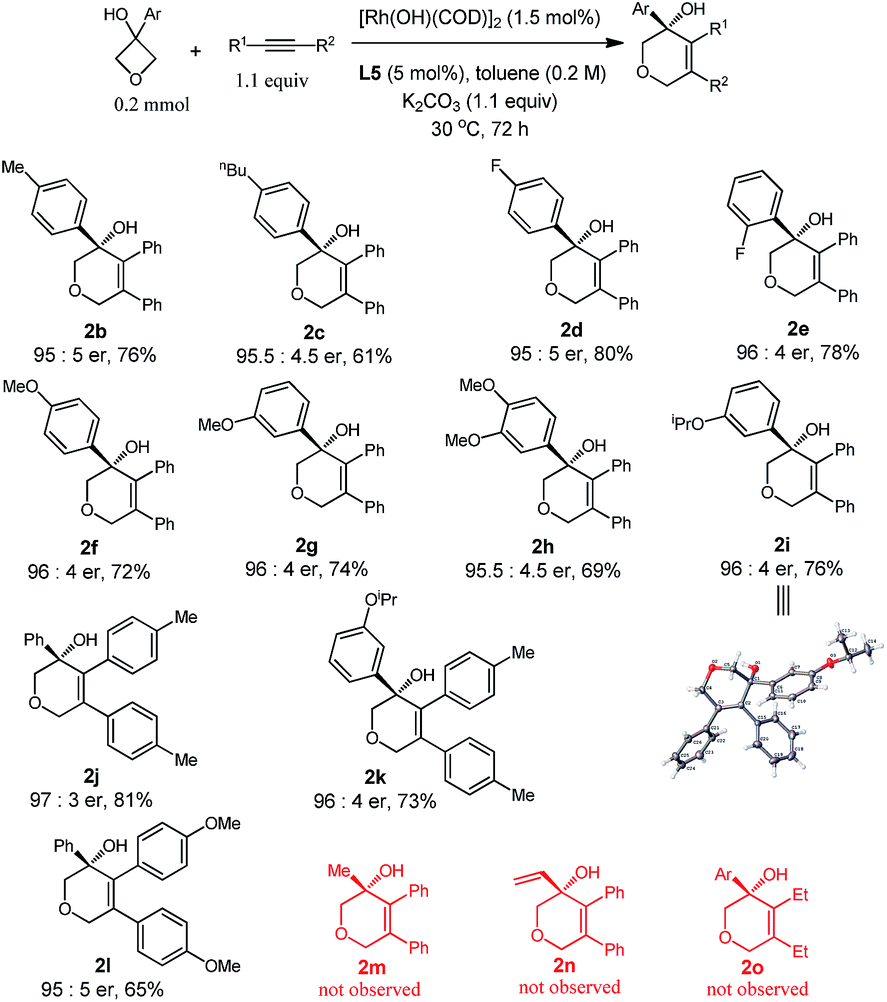
The attempts to further expand the substrate scope by changing the aryl substitution of cyclobutanols to alkyl or alkenyl groups were not successful (2m and 2n), suggesting that the aryl group might provide extra coordination to facilitate the β-carbon elimination.4b,p The reaction of 1a with 3-hexyne did proceed to give the racemic product 2o, however, no cyclization took place in the presence of the chiral ligand, probably due to the incompatibility of the ligand and the substrate.9
To address the question of regioselectivity in the ring opening of 2-substituted oxetanols, we synthesized a series of 2-substituted oxetanones from propargyl alcohols using Zhang's gold catalyzed procedure10 and treated them with the corresponding Grignard reagents or aryl lithium reagents. Gratifyingly, single diastereomers were obtained in high yields in all of the reactions of 2-substituted oxetanols with alkynes (Table 3). The above experimental result suggests that preferential cleavage of the C–C single bond between the hydroxyl carbon atom and the unsubstituted carbon atom of oxetanol takes place to afford the heterocyclic product. The scope of the site-selective insertion reaction is shown in Table 3. Various 2-alkylated phenyloxetan-3-ols bearing different linear alkyl, phenyl ethyl, and cyclic hexyl groups are cyclized readily with 1,2-diphenylethyne, giving the corresponding tetrahydropyrans in good yields as a single diastereomer (4a, 4b and 4f).
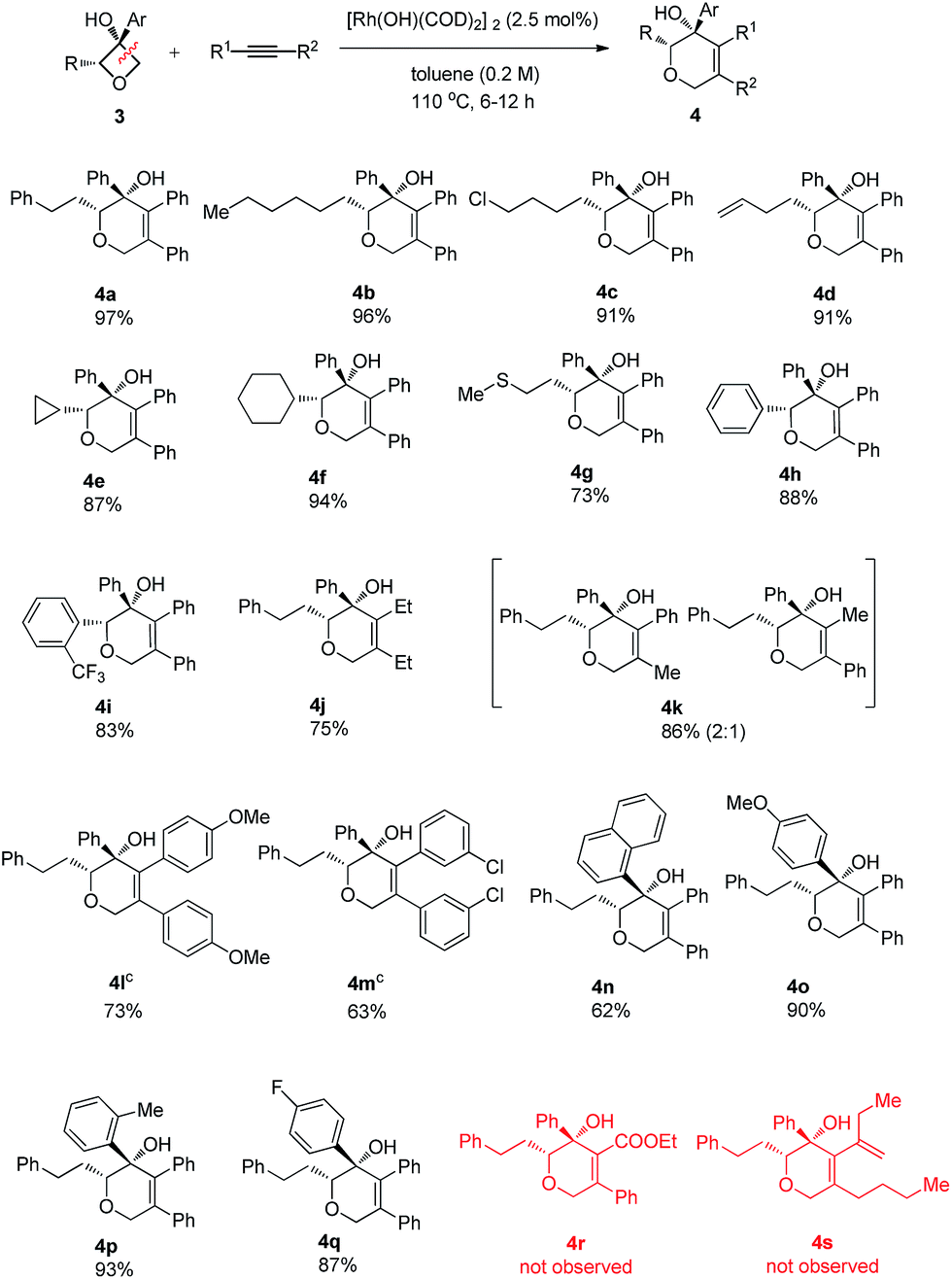
Notably, a cyclopropyl group which is sensitive to rhodium(I)-catalyzed reactions could also survive under the current conditions (4e). The relative configuration of the complex product was established by single crystal X-ray analysis of the product 4a-ent. Furthermore, terminal chloro, C–C double bond and methylthio groups remained intact under the optimized conditions (4c, 4d and 4g). Phenyl phenyloxetan-3-ol was also successfully engaged in the insertion reaction (4h). The site selective ring cyclization was observed even when a sterically demanding ortho-trifluoromethyl group was present (4i). Variations of the alkynes were also briefly investigated. The nature of the aryl substitution (electron-donating or -withdrawing) does not seem to affect the reaction (4l and 4m). Hex-3-yne was a good substrate for this reaction as well (4j). The reaction of an unsymmetric alkyne bearing methyl and phenyl substitution groups (3k) gave two inseparable regioisomers at a ratio of 2:1; this result indicates that the electronic and steric properties of the alkyne substitution groups have less effect on the site-selectivity compared to that in the Rh(I)-catalyzed cycloaddition of benzocyclobutanol with the same alkyne.4b,11 The aryl group at the 3 position of oxetanol could be functionalized, and naphthalene, para-methoxy phenyl, para-fluoro phenyl and ortho-methyl phenyl groups were all well tolerated (4n, 4o, 4p and 4q). A further substrate scope study showed that no desired cyclized product was observed from the reaction of ethyl 3-phenylpropiolate and 1,3-enyne (4r and 4s), and these results further suggest that the alkyl-rhodium species is more sensitive to electronic properties and steric hindrance than (sp2)C–rhodium species.9
 | (6) |
 | (7) |
 | (8) |
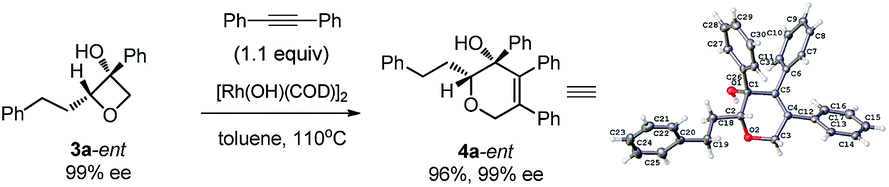
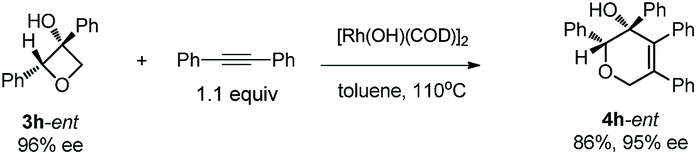
Excellent retention of enantiomeric purity was observed when enantiomerically pure 2-substituted oxetanols were used. Despite the potential epimerization challenges,7,12 the cycloaddition worked effectively to afford enantio-enriched cycloadducts in high yields [eqn (6)–(8)]. Both 2-alkyl substrates (3a-ent and 3r-ent) and a 2-aryl substrate (3h-ent) afforded regioselective cycloadducts in high yields with excellent enantioretention.
Based on previous studies and our own observations, a tentative mechanism is proposed (Scheme 1). First, simultaneous coordination of the Rh(I) center to both the hydroxy group and the arene moiety should be favored.4b,p Site-selective ring opening through β-carbon elimination should lead to II. Next, a cis-migratory insertion of C(sp3)–Rh(I) across the alkyne occurs to give III.4a,b The last ring closing takes place in a highly stereoselective manner. A four-center interaction of the carbonyl moiety with the carbon–rhodium bond might be involved in the transition state.13
 | ||
| Scheme 1 Proposed reaction mechanism. | ||
This reaction could be carried out at gram scale with the same high efficiency (Scheme 2). This series of highly functionalized dihydropyran products are synthetically versatile building blocks. For example, epoxidation of the alkene using m-CPBA produced the corresponding epoxide 5 in 62% yield with perfect diastereoselectivity (>20:1 dr) at gram scale. Treatment of the epoxide with a Lewis acid produced the ketone 6 in 78% yield through a semipinacol rearrangement. In contrast, treatment with strong base induced epoxide ring opening, giving rise to allylic alcohol 7. Our attempt to cleave the double bond by ozonolysis failed to provide the ring opening product, with lactone 8 being isolated instead, and so represented another type of important molecular scaffold which would be difficult to access using other methods.14
 | ||
| Scheme 2 Synthetic utilities of dihydropyrans. | ||
Conclusions
In summary, an efficient method has been developed to prepare highly functionalized dihydropyrans through ring opening of arylated oxetanols and cyclization with alkynes by Rh(I) catalysis. A high degree of enantioselective control was realized when using the chiral ligand Binaphine.15 Excellent site-selectivity and diastereoselectivity were observed in the cases of 2-substituted oxetanols. A high degree of retention of enantiomeric purity in the products was achieved in the reactions using optically pure oxetanols. The synthetic potential of the products was demonstrated in a gram scale operation and 4 facile derivatization reactions.Acknowledgements
We are grateful to NSFC-21421091, 30973607, 81172934, XDB20000000, the “Thousand Plan” Youth program, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. We would also like to thank Prof. Jiangchun Zhong of China Agricultural University for providing chiral propargyl alcohols as the starting materials, which was supported by the National Key Technology Research and Development Program (2015BAK45B01).Notes and references
- (a) Y. Yang, F. Yang, C. Miao, K. Liu, Q. Li, S. Qin, L. Zhao and Z. Ding, Phytochem. Lett., 2015, 11, 249 CrossRef CAS; (b) K. Krohn, J. Dai, U. Florke, H. J. Aust, S. Drager and B. Schulz, J. Nat. Prod., 2005, 68, 400 CrossRef CAS PubMed; (c) E. Quinoa, Y. Kakou and P. Crews, J. Org. Chem., 1988, 53, 3642 CrossRef CAS; (d) D. G. Corley, R. Herb, R. E. Moore, P. J. Scheuer and V. J. Paul, J. Org. Chem., 1988, 53, 3644 CrossRef CAS; (e) M. Ishibashi, R. E. Moore, G. M. L. Patterson, C. Xu and J. Clardy, J. Org. Chem., 1986, 51, 5300 CrossRef CAS.
- (a) J. Timonen, K. Vuolteenaho, T. Leppänen, R. Nieminen, E. Moilanen, P. Aulaskari and J. Jänis, J. Heterocycl. Chem., 2015, 52, 1286 CrossRef CAS; (b) I. Brito, T. Dias, A. R. Diaz-Marrero, J. Darias and M. Cueto, Tetrahedron, 2006, 62, 9655 CrossRef CAS; (c) F. Nagashima, Y. Murakami and Y. Asakawa, Chem. Pharm. Bull., 1999, 47, 139 CrossRef; (d) A. H. Lewin, S. L. Black, M. E. Bos, R. R. Goehring, X. Nair, G. Whiting, P. Bouquin, G. Tetrault and F. I. Carroll, Pharm. Res., 1995, 12, 983 CrossRef CAS PubMed; (e) J. Herscovici, C. Uriel, J. Welt and K. Antonakis, Bioorg. Med. Chem. Lett., 1994, 4, 421 CrossRef CAS; (f) J. Li, Z. Zhang, Z. Xia, C. Ni and Y. Wu, Acta Chim. Sin., 1987, 45, 558 CAS; (g) Z. Xia, Z. Zhang and J. Huang, Jiegou Huaxue, 1986, 5, 263 CAS.
- For selected reviews on C–C bond activation, see: (a) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed; (b) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CAS; (c) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (d) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC; (e) K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS; (f) C. Aïssa, Synthesis, 2011, 21, 3389 CrossRef; (g) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (h) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (i) H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 778 CrossRef CAS; (j) Y. J. Park, J.-W. Park and C.-H. Junn, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (k) M. Murakami, M. Makino, S. Ashida and T. Matsuda, Bull. Chem. Soc. Jpn., 2006, 79, 1315 CrossRef CAS; (l) J. A. Tunge and E. C. Burger, Eur. J. Org. Chem., 2005, 1715 CrossRef CAS; (m) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (n) C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Muller, T. Satoh and W. D. Jones, J. Mol. Catal. A: Chem., 2002, 189, 157 CrossRef CAS; (o) M. Murakami and Y. Ito, Top. Organomet. Chem., 1999, 3, 97 CrossRef CAS; (p) J. L. Beauchamps, Chem. Rev., 1990, 90, 629 CrossRef; (q) G. Dong, Topics in Current Chemistry 346, Springer-Verlag, Berlin Heidelberg, 2014 Search PubMed.
- (a) T. Matsuda and N. Miura, Org. Biomol. Chem., 2013, 11, 3424 RSC; (b) N. Ishida, S. Sawano, Y. Masuda and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17502 CrossRef CAS PubMed. For related works, see: (c) J. Yu, H. Yan and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 1143 CrossRef CAS PubMed; (d) R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef CAS PubMed; (e) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692 CrossRef CAS PubMed; (f) Q. Tian, B. Chen and G. Zhang, Green Chem., 2016, 18, 6236 RSC; (g) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (h) N. Ishida, D. Nečas, Y. Masuda and M. Murakami, Angew. Chem., Int. Ed., 2015, 54, 7418 CrossRef CAS PubMed; (i) L. Souillart and N. Cramer, Chem. Sci., 2014, 5, 837 RSC; (j) N. Ishida, Y. Nakanishi and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 11875 CrossRef CAS PubMed; (k) N. Ishida, Y. Shimamoto, T. Yano and M. Murakami, J. Am. Chem. Soc., 2013, 135, 19103 CrossRef CAS PubMed; (l) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed; (m) M. Shigeno, T. Yamamoto and M. Murakami, Chem.–Eur. J., 2009, 15, 12929 CrossRef CAS PubMed; (n) T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320 CrossRef CAS PubMed; (o) T. Seiser and N. Cramer, Angew. Chem., Int. Ed., 2008, 47, 5340 CrossRef PubMed; (p) T. Seiser and N. Cramer, Angew. Chem., Int. Ed., 2008, 47, 9294 CrossRef CAS PubMed; (q) T. Matsuda, M. Makino and M. Murakami, Org. Lett., 2004, 6, 1257 CrossRef CAS PubMed; (r) T. Nishimura, K. Ohe and S. Uemura, J. Am. Chem. Soc., 1999, 121, 2645 CrossRef CAS.
- N. Ishida, N. Ishikawa, S. Sawano, Y. Masuda and M. Murakami, Chem. Commun., 2015, 51, 1882 RSC.
- (a) Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2014, 136, 3013 CrossRef CAS PubMed; (b) A. Yada, S. Fujita and M. Murakami, J. Am. Chem. Soc., 2014, 136, 7217 CrossRef CAS PubMed.
- C. Zhao, L. C. Liu, J. Wang, C. Jiang, Q. W. Zhang and W. He, Org. Lett., 2016, 18, 328 CrossRef CAS PubMed.
- For selected reviews, see: (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed; (b) J. A. Burkhard, G. Wuitschik, M. RogersEvans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed. For selected recent examples, see: (c) W. Yang, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 6954 CrossRef CAS PubMed; (d) W. Yang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 1868 CrossRef PubMed; (e) J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adan and A. W. Kleij, Chem.–Eur. J., 2015, 21, 10754 CrossRef CAS PubMed; (f) Z. Wang, Z. Chen and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 6685 CrossRef CAS PubMed; (g) Z. Chen, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027 CrossRef CAS PubMed; (h) A. Thakur, M. E. Facer and J. Louie, Angew. Chem., Int. Ed., 2013, 52, 12161 CrossRef CAS PubMed; (i) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228 CrossRef CAS PubMed; (j) S. A. Ruider, S. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11908 CrossRef CAS PubMed; (k) K. Y. T. Ho and C. Aïssa, Chem.–Eur. J., 2012, 18, 3486 CrossRef CAS PubMed; (l) B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675 CrossRef CAS PubMed; (m) C. Gronnier, S. Kramer, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2012, 134, 828 CrossRef CAS PubMed; (n) W. Zhao, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 6209 CrossRef CAS PubMed; (o) J. A. Burkhard, B. H. Tchitchanov and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5379 CrossRef CAS PubMed; (p) R. Loy and E. Jacobsen, J. Am. Chem. Soc., 2009, 131, 2786 CrossRef CAS PubMed.
- See the ESI† for more detailed experimental data and tentative explanation for the unsuccessful substrates.
- (a) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550 CrossRef CAS PubMed; (b) A. S. K. Hashmi, A. Loos, A. Littmann, I. Braun, J. Knight, S. Doherty and F. Rominger, Adv. Synth. Catal., 2009, 351, 576 CrossRef CAS.
- For the reaction of diaryl alkyne with opposite electronic substitutions, low regioselectivity was observed.
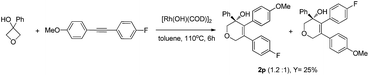 .
. - D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630 CrossRef CAS PubMed.
- (a) J. Saadi, C. Bentz, K. Redies, D. Lentz, R. Zimmer and H.-U. Reissig, Beilstein J. Org. Chem., 2016, 12, 1236 CrossRef CAS PubMed; (b) D. Solé, F. Mariani, I. Fernández and M. A. Sierra, J. Org. Chem., 2012, 77, 10272 CrossRef PubMed; (c) D. Solé, I. Fernández and M. A. Sierra, Chem.–Eur. J., 2012, 18, 6950 CrossRef PubMed; (d) D. Solé and I. Fernández, Acc. Chem. Res., 2014, 47, 168 CrossRef PubMed; (e) L. G. Quan, M. Lamrani and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 4827 CrossRef CAS.
- (a) Z.-Y. Yang, H.-Z. Liao, K. Sheng, Y.-F. Chen and Z.-J. Yao, Angew. Chem., Int. Ed., 2012, 51, 6484 CrossRef CAS PubMed; (b) H. Oguri, T. Hiruma, Y. Yamagishi, H. Oikawa, A. Ishiyama, K. Otoguro, H. Yamada and S. Omura, J. Am. Chem. Soc., 2011, 133, 7096 CrossRef CAS PubMed; (c) T. Kiyoi, M. Reid, S. Francis, K. Davies, S. Laats, D. McArthur, A.-M. Easson, Y. Kiyoi, G. Tarver, W. Caulfield, K. Gibson, G. Wishart, A. J. Morrison, J. M. Adam and P. Ray, Tetrahedron Lett., 2011, 52, 3413 CrossRef CAS; (d) C. Aouf, N. Galy, H. Doucet and M. Santelli, Appl. Organomet. Chem., 2010, 24, 794 CrossRef CAS; (e) K. C. Nicolaou, A. J. Roecker, M. Follmann and R. Baati, Angew. Chem., Int. Ed., 2002, 41, 2107 CrossRef CAS; (f) G. Appendino, E. Belloro, E. D. Grosso, A. Minassi and E. Bombardelli, Eur. J. Org. Chem., 2002, 277 CrossRef CAS; (g) M. M. Kabat, M. Lange, P. M. Wovkulich and M. R. Uskoković, Tetrahedron Lett., 1992, 33, 7701 CrossRef CAS; (h) M. M. Abelman, L. E. Overman and V. D. Tran, J. Am. Chem. Soc., 1990, 112, 6959 CrossRef CAS; (i) T. Tokoroyama, Y.Fukuyama and Y. Kotsuji, J. Chem. Soc., Perkin Trans. 1, 1988, 445 RSC; (j) J. Platzek and G. Snatzke, Tetrahedron, 1987, 43, 4947 CrossRef CAS; (k) M. Yamato, K. Hashigaki, M. Ikeda, H. Ohtake and K. Tasaka, Chem. Pharm. Bull., 1981, 29, 402 CrossRef CAS PubMed.
- (a) W. Tang, W. Wang, Y. Chi and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3509 CrossRef CAS PubMed; (b) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed.

Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1512439 and 1512440. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05246k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |