
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein modification via alkyne hydrosilylation using a substoichiometric amount of ruthenium(II) catalyst†‡
Terence T.-L.
Kwan
a,
Omar
Boutureira
 a,
Elizabeth C.
Frye
a,
Stephen J.
Walsh
a,
Moni K.
Gupta
a,
Stephen
Wallace
bc,
Yuteng
Wu
a,
Fengzhi
Zhang
a,
Hannah F.
Sore
a,
Warren R. J. D.
Galloway
a,
Jason W.
Chin
ab,
Martin
Welch
d,
Gonçalo J. L.
Bernardes
ae and
David R.
Spring
*a
a,
Elizabeth C.
Frye
a,
Stephen J.
Walsh
a,
Moni K.
Gupta
a,
Stephen
Wallace
bc,
Yuteng
Wu
a,
Fengzhi
Zhang
a,
Hannah F.
Sore
a,
Warren R. J. D.
Galloway
a,
Jason W.
Chin
ab,
Martin
Welch
d,
Gonçalo J. L.
Bernardes
ae and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Rd, Cambridge CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bMedical Research Council, Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, UK
cSchool of Biological Sciences, University of Edinburgh, The King's Buildings, Edinburgh, EH9 3FF, UK
dDepartment of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, UK
eInstituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Avenida Professor Egas Moniz, 1649-028, Lisboa, Portugal
First published on 14th March 2017
Abstract
Transition metal catalysis has emerged as a powerful strategy to expand synthetic flexibility of protein modification. Herein, we report a cationic Ru(II) system that enables the first example of alkyne hydrosilylation between dimethylarylsilanes and O-propargyl-functionalized proteins using a substoichiometric amount or low-loading of Ru(II) catalyst to achieve the first C–Si bond formation on full-length substrates. The reaction proceeds under physiological conditions at a rate comparable to other widely used bioorthogonal reactions. Moreover, the resultant gem-disubstituted vinylsilane linkage can be further elaborated through thiol–ene coupling or fluoride-induced protodesilylation, demonstrating its utility in further rounds of targeted modifications.
Introduction
The chemical modification of biomolecules has emerged as a powerful tool to study cellular systems.1–3 Alongside recombinant methods,4–7 advances in organic chemistry have fueled the development of an increasing number of chemical reactions capable of modifying proteins at both genetically and chemically predefined sites.8–13 These bioorthogonal reactions have transformed our ability to visualize cellular processes, and have enabled the development of new therapeutic strategies to treat diseases.14–16 Within this “toolbox” of bioorthogonal reactions, transition metal-mediated reactions are arguably the most underdeveloped.17–20 This is likely due to transition metals' propensity for unproductive chelation within the biological milieu, resulting in the need for high catalyst loadings to achieve acceptable reaction rates and labeling efficiency (Fig. 1a). Previous examples of transition metal-mediated reactions include Cu(I) azide–alkyne cycloaddition,21,22 Ru(II) cross-metathesis,23–26 Pd(II) cysteine bioconjugation,27 Suzuki–Miyaura28,29 and some of their intracellular variants.30,31 However, most published reaction conditions utilize high catalyst loading and the development of a truly catalytic transition metal-mediated bioconjugation strategy has received little attention. | ||
| Fig. 1 (a) General metal-mediated protein modification protocols using excess of metal catalysts and (b) our approach via Ru(II)-catalyzed alkyne hydrosilylation. | ||
Here we report a new Ru-catalyzed alkyne hydrosilylation reaction for protein modification. Using the water-soluble Ru catalyst [Cp*Ru(MeCN)3]PF6 (1) and dimethylaryl hydrosilane derivatives, this methodology enables the efficient labeling of multiple protein targets modified both stochastically and site-specifically with an alkyne-containing moiety (Fig. 1b). In addition, hydrosilylation has orthogonal chemical reactivity to ketone-hydrazine condensation reaction in vitro, and the resultant gem-disubstituted vinylsilane product can be further modified via thiol–ene coupling and fluoride-induced protodesilylation, demonstrating the potential of this methodology for use in both orthogonal dual labeling and single-site, multiple-probe imaging applications. To the best of our knowledge, this represents the first example of a C–Si bond formation on protein substrates using substoichiometric or low-loading of transition metal catalysts – a feature that we hope will reinstate this mode of catalysis as a viable avenue for future research in the field.
Results and discussion
Although hydrosilylation has gained widespread utility in organic synthesis and in the industrial production of organosilicon compounds,32–35 aqueous alkyne hydrosilylation is largely underdeveloped. Inspired by the development of a cationic ruthenium catalyst [Cp*Ru(MeCN)3]PF61 by Trost and Ball,36,37 we examined the catalyst's ability to catalyze hydrosilylation under biocompatible, aqueous conditions. We started our investigation by reacting 3,6,9,12-tetraoxapentadec-14-yne 2 as a model alkyne and a variety of water-soluble hydrosilanes with 1 (5 mol%). Despite previously reported reactivities of trialkoxy and trialkyl silanes, no vinylsilane products were observed under the reaction conditions tested (Table 1, entries 1–3).|
|
|||
|---|---|---|---|
| Entry | Silane (R3SiH) | Conversiona (%) | t (min) |
| a Determined by 1H-NMR using 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal standard. b Isolated yield. c 50% t-BuOH in PBS (pH 7.4). d Reaction conducted in the presence of 10% human plasma. e 10 mol% hippuric acid (BzNHCH2CO2H) as an additive. O/N: overnight; NR: no reaction (>95% starting alkyne recovered). | |||
| 1 | (EtO)3SiH | NR | O/N |
| 2 | (TMSO)3SiH | NR | O/N |
| 3 |

|
NR | O/N |
| 4 |

|
100 (92b) | <5 |
| 5 | 100 | 70 | |
| 6 | NR | O/N | |
| 7 | 24 | 80 | |
| 8c |

|
99b | 30 |
| 9c,d,e | 71b | 30 | |

Gratifyingly, hydrosilylation of dimethylaryl hydrosilane 4 with 2 proceeded smoothly (Table 1, entry 4), achieving full conversion with 92% isolated yield in less than 5 min. This apparent high reactivity may be attributed to the strong affinity for Cp*Ru complexes to coordinate with aromatic rings,37 allowing hydrosilylation to proceed rapidly in aqueous solution and open air, thus reinforcing the use of aryldialkyl hydrosilanes in further experiments. The reaction proceeded with a 2nd order rate constant k2 ∼ 1.0 M−1 s−1 (see ESI, Fig. S20‡), which is comparable to Ru(II) cross-metathesis and strain-promoted alkyne–azide cycloadditions. Furthermore, 4 was found to be stable in buffered conditions at neutral pH, with a half-life (t1/2) > 1 week (see ESI, Fig. S21‡).
One of the side reactions of aqueous hydrosilylation is the hydrolysis of hydrosilane to form silanol (Si–OH). In an effort to reduce silanol formation, we installed substituents adjacent to the Si–H bond with varying degree of steric hindrance (compounds 5–7) in the hope to increase selectivity for hydrosilylation over silanol formation. However, none of the tested analogues gave better selectivity or reaction rates (Table 1, entries 5–7). In particular, 6 and 7 showed incomplete conversion despite prolonged reaction times (Table 1, entries 6 and 7).
The hydrophobicity of chemical probes and modifications often require the use of organic co-solvents in the reaction mixture. Alcohol-based solvents were found to be tolerated as co-solvents in aqueous hydrosilylation and achieved similar reaction rates to those using pure water (see ESI, Fig. S22‡). Thus, the hydrosilylation of 2 with triethylene glycol hydrosilane derivative 8 in 50% t-BuOH in phosphate buffered saline (PBS) at pH 7.4 gave the corresponding vinylsilane in 99% isolated yield (Table 1, entry 8). In the presence of human plasma, the reaction initially proceeded extremely slowly and gave only trace of product. It was suspected that 1 is inactive in hydrosilylation due to nonproductive chelation to ruthenium in 10% human plasma. Remarkably, the addition of hippuric acid (BzNHCH2CO2H) as an additive/ligand helped to stabilize the Ru(II) complex from rapid exchange processes with, for example, histidine38,39 and aspartic acid40 residues in plasma protein and restored the activity of 1, with the corresponding vinylsilane product isolated in a good yield (Table 1, entry 9). This result demonstrates that our novel hydrosilylation methodology for protein modification can proceed under physiological conditions.
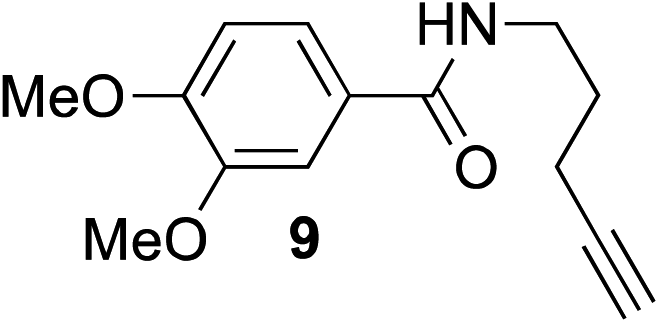
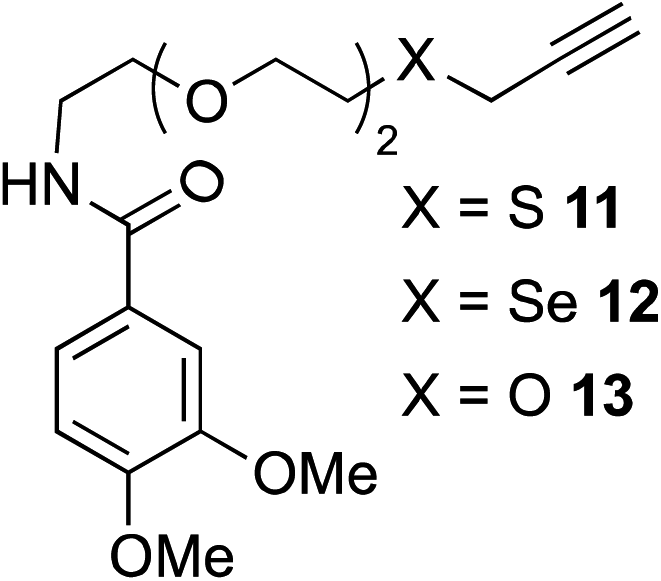
The scope of this reaction was further evaluated using a variety of small molecule alkynes representative of amino acids, carbohydrates, and hydrophobic drugs such as alkynes 9, 11–13 that may be considered substructural motifs of 3-O-methyl-DOPA (3-OMD), which is one of the most important metabolites of L-DOPA. We first investigated whether nearby chalcogens on the terminal alkyne group could increase the rate of hydrosilylation. With no nearby coordinating groups, the reaction with alkyne 9 proceeded slowly, requiring a long reaction time to reach 68% yield (Table 2, entry 1). Contrary to the reported chalcogen effect in protein cross-metathesis,23,26S-propargyl 11 and Se-propargyl 12 inhibited hydrosilylation and the respective vinylsilane products were not detected, despite extended reaction times (Table 2, entries 2 and 3). Surprisingly, O-propargyl 13 showed the most promise, affording vinylsilane 14 in 91% isolated yield (Table 2, entry 4). This is likely due to the intricate balance between ruthenium-coordination (X = O) and inhibition (X = S, Se).
|
|
|||
|---|---|---|---|
| Entry | Silane (ArMe2SiH) | Alkyne | Producta (yield%) |
| a Isolated yield. b 5 h reaction time. c 10 mol% hippuric acid added as an additive. NR: no reaction (>95% starting alkyne recovered). | |||
| 1b | 8 |
|
10 (68%) |
| 2 | 8 |
|
NR |
| 3 | 8 | NR | |
| 4 | 8 | 14 (91%) | |
| 5c | PhMe2SiH (15) |

|
17 (98%) |
| 6c | 15 | 19 (29%) | |
| 7c | 15 |

|
NR |
| 8c | 8 |

|
22 (85%) |
| 9c | 8 |
|
24 (81%) |
| 10 | 8 |

|
26 (82%) |

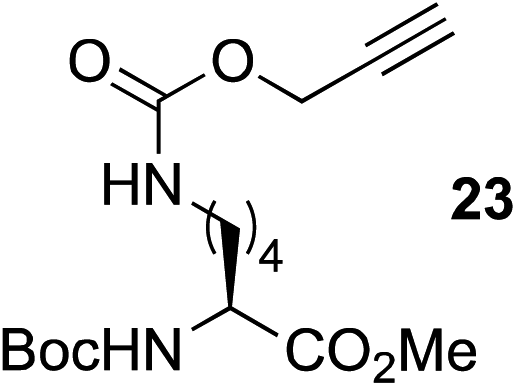
This observation was further confirmed by the decreasing isolated yields when reacting PhMe2SiH 15 with O-propargyl-serine 16, S-propargyl-cysteine 18, and Se-propargyl-selenocysteine 20 derivatives (Table 2, entries 5–7). Nonetheless, hydrosilylation proceeded smoothly on amino acids 21 and 23, as well as alkyne-sugar derivative 25, affording the corresponding products in excellent isolated yields (Table 2, entries 8–10). These examples are of particular importance, as strategies for the in vivo incorporation of such moieties into proteins and cell-surface glycans have been developed.41–43 Full conversion was also achieved on a model peptide 27 with biotinylated hydrosilane 29, demonstrating the potential for Ru(II) aqueous hydrosilylation protocol to modify more complex biomolecules (Scheme 1). Furthermore, the stability of the resulting gem-disubstituted vinylsilane moiety was assessed under physiological conditions and in the presence of biological thiols, with no observable degradation at 37 °C for up to 24 h (see ESI, Fig. S24‡).
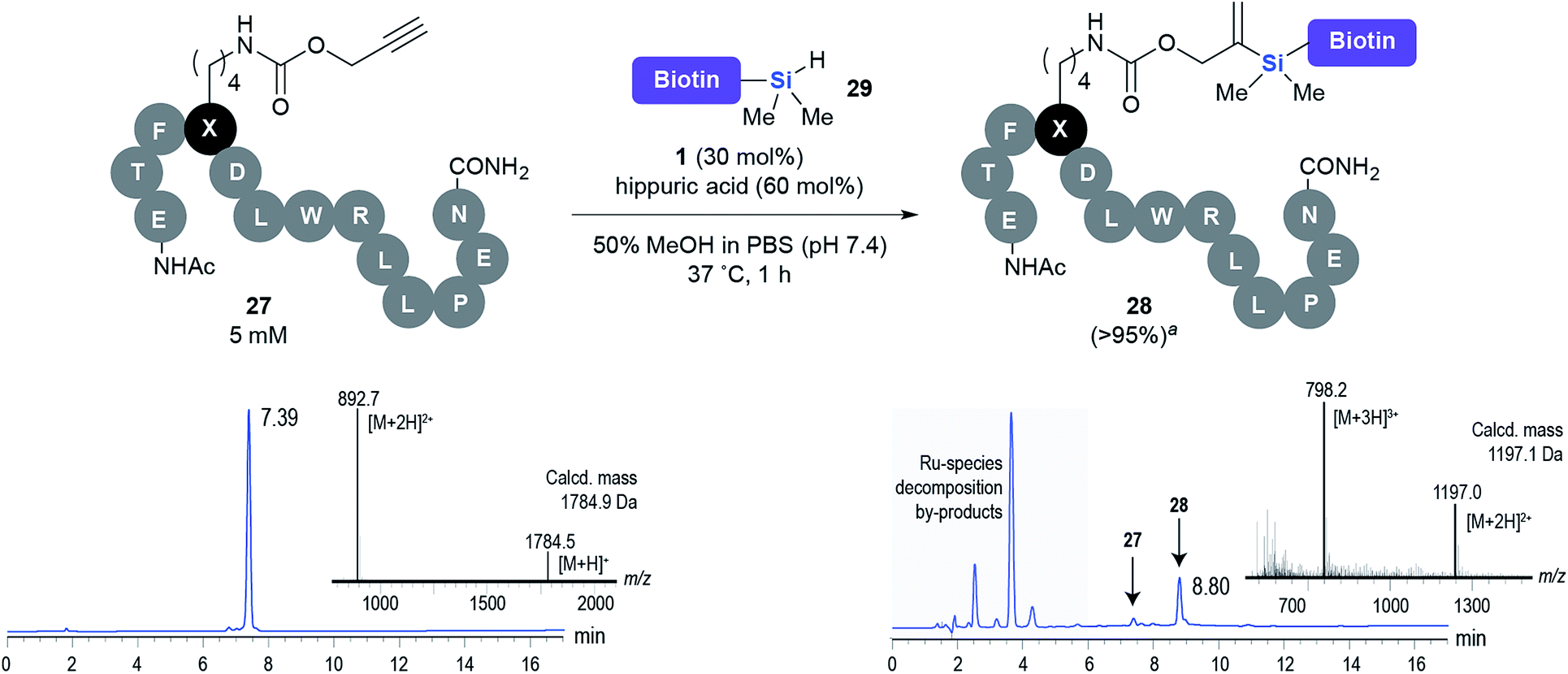 | ||
| Scheme 1 Hydrosilylation of peptide 27 with biotinylated hydrosilane 29. aConversion (%) determined by HPLC. | ||
Biocompatible chemical transformations are often most powerful when used in conjunction with each other, allowing for the site-specific incorporation of multiple chemical modifications into a single biomolecule.44 Encouragingly, hydrosilylation was found to be compatible with the widely used α-substituted amine/carbonyl condensation,45–48 where O-propargyl 13 reacted smoothly with hydrosilane 15 in the presence of ketone 30, achieving >95% conversion to desired vinylsilane 31 in 30 min. Subsequent addition of hydrazine 33 resulted in complete conversion of 30 to hydrazone 32. This demonstrates the utility of tandem hydrosilylation and condensation reactions via a step-wise, one-pot ligation strategy without any undesired interference to their reactivities (Scheme 2a).
 | ||
| Scheme 2 (a) Tandem hydrosilylation and hydrazine condensation reaction. (b) gem-Disubstituted vinylsilane reactivity under thiol–ene coupling conditions and fluoride-induced protodesilylation, giving thioether 34 and O-allyl 35 in 81% and 85% isolated yields, respectively. aConversion (%) determined by HPLC. | ||
Most site-selective dual-labeling efforts require the incorporation of two unique bioorthogonal functional groups49,50 or the use of bifunctional substrates,51,52 which can be a synthetic challenge and limits the wide adoption of such methods. Moreover, it would be advantageous to have the ability to selectively remove synthetically incorporated chemical modifications, allowing for potential “switch on/off” applications. To address these issues, we sought to further elaborate the gem-disubstituted vinylsilane linkage via radical thiol–ene53–55 and fluoride-induced protodesilylation reactions56,57 (Scheme 2b). To illustrate the dual-labeling methodology, we incubated model vinylsilane substrate 31 with benzyl mercaptan, 10 mol% 2,2-dimethoxy-2-phenylacetophenone (DMPA) and irradiated at 365 nm to give doubly-modified derivative 34 in excellent isolated yield (81%). Furthermore, the gem-disubstituted vinylsilane linkage can be cleaved by treatment with TBAF to give the corresponding O-allyl 35, demonstrating the potential for chemical Si-modifications installed via hydrosilylation to be selectively removed.
With these promising results in hand, we conducted protein-labeling experiments via hydrosilylation on different protein systems. First, lysine residues on lysozyme (Lyz) were non-selectively modified with 36 to give O-propargyl modified Lyz (OP-Lyz) (Fig. 2a and b). When treated with biotinylated hydrosilane 29 and only 10 mol% of 1, we were pleased to observe selective labeling of OP-Lyz over Lyz with negligible background labeling (Fig. 2c). Similarly, when the reaction time or concentration of 29 was held constant (1 h and 250 μM, respectively), dose- and time-dependent labeling was observed, even at very low catalyst loading (2 mol%) (see ESI, Fig. S1‡). Inductively coupled plasma-mass spectrometry (ICP-MS) determined that ruthenium content was <10 ppb after purification when using 10 mol% catalyst (see ESI‡ for details). To the best of our knowledge this result is the first demonstration of a protein modification protocol mediated by a substoichiometric amount of transition metal catalyst.
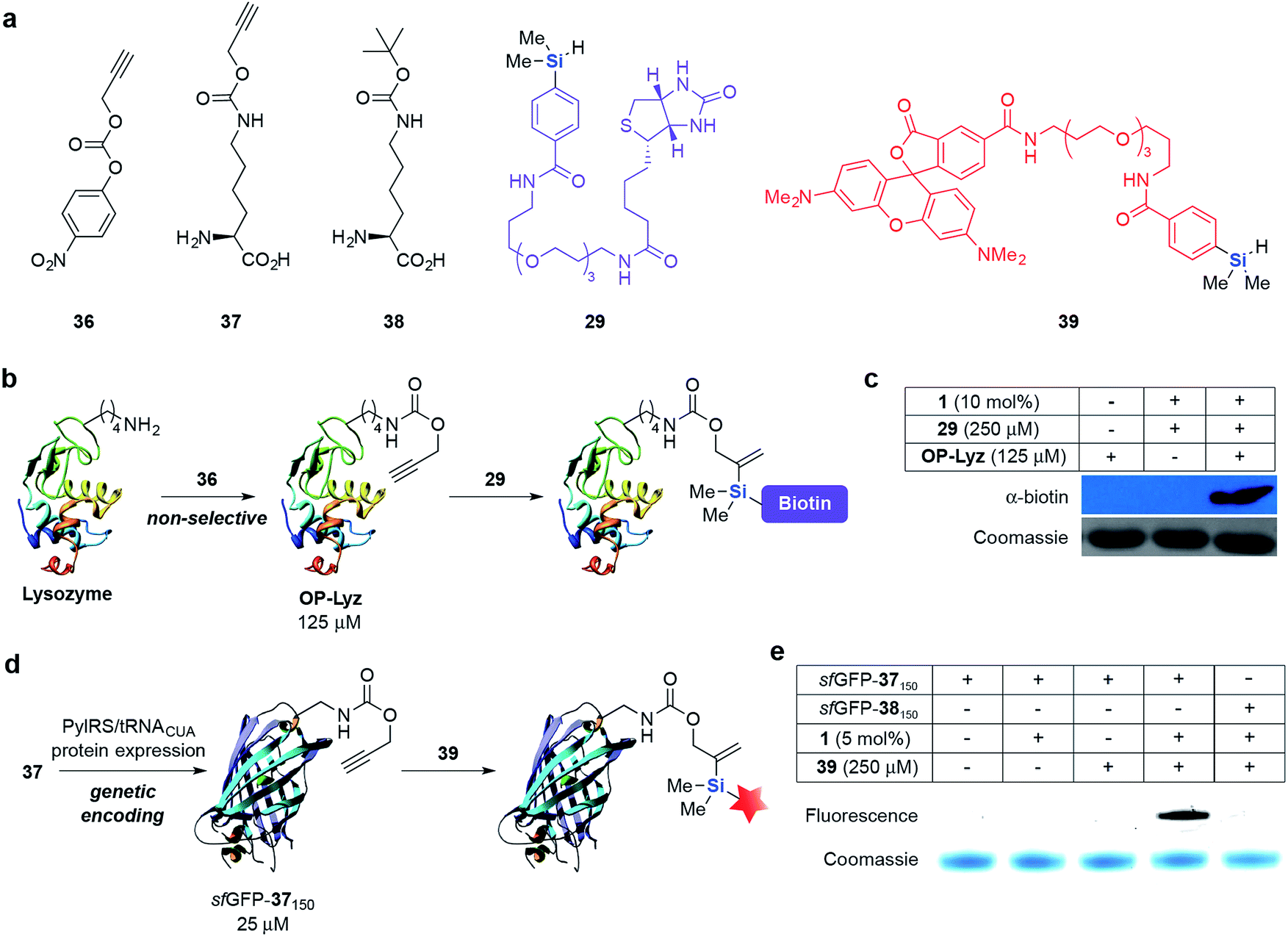 | ||
| Fig. 2 Selective labeling of O-propargyl (OP) modified protein substrates via hydrosilylation. (a) The structures of unnatural amino acids 37 and 38 and other reagents used in this study. (b) Modification of solvent-exposed lysine residues on lysozyme (Lyz) with 36 and subsequent labeling with 29. (c) Selective labeling of OP-Lyz via hydrosilylation with 29 and 1. Lyz (−) and OP-Lyz (+) (125 μM) was independently incubated with 29 (250 μM) and 1 (10 mol%) for 2 h at 37 °C and the presence of biotinylated protein was detected by Western blot using α-biotin-HRP conjugated antibody. (d) Genetic encoding and fluorescent labeling of 37via hydrosilylation. (e) In-gel fluorescence demonstrating specific labeling of sfGFP-37150 with 39. In (c) and (e), equal protein loading was verified by Coomassie staining. | ||
Next, we incorporated O-propargyl groups site-specifically into a super-folder GFP (sfGFP) protein (Fig. 2d).58 Briefly, PylRS/pylT pair, the wild-type orthogonal Methanosarcina barkeri pyrrolysyl-tRNA synthetase and tRNACUA pair and C-terminally hexahistidine-tagged sfGFP containing an amber codon (TAG) at position 150 (sfGFP150TAGHis6) were introduced into E. coli. Addition of 37 (5 mM) led to the amino acid dependent synthesis of full-length sfGFP-37150 in good yield (15 mg L−1 of culture). A similar approach was used to obtain sfGFP-38150 as a negative control for labeling experiments (Fig. S2 and S3‡). We subsequently incubated sfGFP-37150 with fluorescent hydrosilane 39 and 1 (5 mol%) in PBS (pH 7.4) at 37 °C. A fluorescent band was detected after 24 h of incubation with limited background fluorescence observed. This result is particularly noteworthy because no fluorescence was observed when sfGFP-38150 was reacted under the same conditions, highlighting the bioorthogonality and specificity of this reaction towards O-propargyl groups (Fig. 2e). The formation of the expected ligated protein was further confirmed by LC-MS (see ESI, Fig. S4‡).
As an alternative to recombinant techniques, we also site-specifically incorporated the alkyne handle through chemical modifications at cysteine. Using the methodology developed by Davis and co-workers,59 the single cysteine mutant of the C2A domain of Synaptotagmin I C2Am (eukaryotic marker of apoptosis) was converted to OP-C2Am via the dehydroalanine-tagged protein intermediate in >95% conversion (Scheme 3a). Gratifyingly, 1 successfully mediated hydrosilylation of OP-C2Am with 8 at 37 °C for 1 h to afford VS-C2Am as detected by LC-MS (Scheme 3b). Compared to the high loading of transition metal complexes used in typical metal-mediated protein modification protocols, Ru(II) complex 1 mediated aqueous hydrosilylation offers a milder alternative and these results highlight its potential for site-specific chemical protein modification using either substoichiometric or low-catalyst loading systems.
 | ||
Scheme 3 (a) Site-specific incorporation of 40 into C2Am via C2Am-Dha and (b) subsequent hydrosilylation with 8. Found masses corresponding to OP-C2Am (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 394 Da), Oxidized-OP-C2Am (16435 Da), and VS-C2Am (16704 Da). 394 Da), Oxidized-OP-C2Am (16435 Da), and VS-C2Am (16704 Da). | ||
Having succinctly demonstrated the ability to carry out alkyne hydrosilylation on numerous protein systems, efforts were then directed towards ascertaining whether it was possible to modify the protein-incorporated vinylsilane through our earlier described radical thiol–ene and protodesilylation reactions. Vinylsilane-modified lysozyme (VS-Lyz) was chosen for initial studies. We found that treatment of VS-Lyz with a protected cysteine amino acid and DMPA under hν irradiation yielded Cys-Lyz, as detected by LC-MS (see ESI, Fig. S18‡). Similarly, treatment of VS-Lyz with TBAF·3H2O afforded Ene-Lyz (see ESI, Fig. S19‡). These proof-of-principle experiments show that the vinylsilane can be further modified after its introduction on a protein through Ru(II)-catalyzed aqueous hydrosilylation.
Conclusions
In conclusion, we have demonstrated that O-propargyl groups and dimethylaryl hydrosilanes (HSiMe2Ar) are effective coupling partners for Ru(II) complex 1 catalyzed aqueous hydrosilylation, where alkyne-labeled small-molecules and peptides are site-specifically modified in good to excellent yields. Furthermore, hydrosilylation is shown to have orthogonal reactivity to the widely used bioorthogonal hydrazine condensation reaction, giving rise to potential biomolecule dual-labeling applications. Furthermore, O-propargyl tagged proteins (via chemical and genetic strategies) successfully undergo site-specific hydrosilylation in the presence of substoichiometric or low loading of 1 to achieve the first C–Si bond on protein substrates. Finally, the resultant gem-disubstituted vinylsilane linkage serves as a reactive chemical handle for thiol–ene coupling and protodesilylation, highlighting the potential for single-site dual-modification and the selective removal of vinylsilane modifications. Hence, we believe this work greatly expands the reaction conditions and substrate complexity of hydrosilylation and complements the growing interest in metal-mediated protein modification strategies.Acknowledgements
This work was supported by the EU, EPSRC, BBSRC, MRC, Wellcome Trust and ERC (FP7/2007-2013; 279337/DOS). We thank Dr André Neves and Prof. Kevin Brindle for providing the C2Am protein. T. T.-L. Kwan acknowledges a scholarship from the Cambridge Trusts and the Croucher Foundation of Hong Kong and O. B. thanks the European Commission (Marie Curie IEF) for financial support. S. J. W. acknowledges a scholarship from AstraZeneca and the Cambridge Trusts. S. W. is the recipient of a Career Development Fellowship from the Medical Research Council. G. J. L. B. is a Royal Society University Research Fellow and the recipient of an ERC Staring Grant (TagIt). Work in the Chin lab was supported by the Medical Research Council, UK (MC_U105181009 and MC_UP_A024_1008) to J. W. C. Data accessibility: all data supporting this study are provided as ESI‡ accompanying this paper.Notes and references
- E. Baslé, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef PubMed.
- D. P. Gamblin, S. I. van Kasteren, J. M. Chalker and B. G. Davis, FEBS J., 2008, 275, 1949–1959 CrossRef CAS PubMed.
- I. S. Carrico, Chem. Soc. Rev., 2008, 37, 1423–1431 RSC.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87–91 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589–1600 RSC.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- A. M. Elsohly and M. B. Francis, Acc. Chem. Res., 2015, 48, 1971–1978 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2015, 8, 103–113 Search PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253–262 CrossRef CAS PubMed.
- S. Kolodych, E. Rasolofonjatovo, M. Chaumontet, M.-C. Nevers, C. Créminon and F. Taran, Angew. Chem., Int. Ed., 2013, 1–6 Search PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- J. M. Chalker, Y. A. Lin, O. Boutureira and B. G. Davis, Chem. Commun., 2009, 3714–3716 RSC.
- Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805–16811 CrossRef CAS PubMed.
- Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156–12159 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed.
- B. Marciniec, in Hydrosilylation, ed. B. Marciniec, Springer Netherlands, Dordrecht, 2009, pp. 53–86 Search PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- E. C. Frye, C. J. O'Connor, D. G. Twigg, B. Elbert, L. Laraia, D. G. Hulcoop, A. R. Venkitaraman and D. R. Spring, Chem.–Eur. J., 2012, 18, 8774–8779 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726–12727 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS PubMed.
- M. Chaves-Ferreira, I. S. Albuquerque, D. Matak-Vinkovic, A. C. Coelho, S. M. Carvalho, L. M. Saraiva, C. C. Romão and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2015, 54, 1172–1175 CrossRef CAS PubMed.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- L. Messori and A. Merlino, Dalton Trans., 2014, 43, 6128–6131 RSC.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- T.-L. Hsu, S. R. Hanson, K. Kishikawa, S.-K. Wang, M. Sawa and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2614–2619 CrossRef CAS PubMed.
- D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141–149 CrossRef CAS PubMed.
- D. Rideout, Science, 1986, 233, 561–563 CAS.
- L. K. Mahal and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS PubMed.
- R. Sadamoto, K. Niikura, T. Ueda, K. Monde, N. Fukuhara and S.-I. Nishimura, J. Am. Chem. Soc., 2004, 126, 3755–3761 CrossRef CAS PubMed.
- L. Tang, Q. Yin, Y. Xu, Q. Zhou, K. Cai, J. Yen, L. W. Dobrucki and J. Cheng, Chem. Sci., 2015, 6, 2182–2186 RSC.
- A. Sachdeva, K. Wang, T. Elliott and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 7785–7788 CrossRef CAS PubMed.
- M. Mühlberg, M. G. Hoesl, C. Kuehne, J. Dernedde, N. Budisa and C. P. R. Hackenberger, Beilstein J. Org. Chem., 2015, 11, 784–791 CrossRef PubMed.
- M. Rashidian, S. C. Kumarapperuma, K. Gabrielse, A. Fegan, C. R. Wagner and M. D. Distefano, J. Am. Chem. Soc., 2013, 135, 16388–16396 CrossRef CAS PubMed.
- L. I. Willems, N. Li, B. I. Florea, M. Ruben, G. A. van der Marel and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 4431–4434 CrossRef CAS PubMed.
- K. Griesbaum, Angew. Chem., Int. Ed., 1970, 9, 273–287 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- Y. Li, M. Yang, Y. Huang, X. Song, L. Liu and P. R. Chen, Chem. Sci., 2012, 3, 2766 RSC.
- H. Oda, M. Sato, Y. Morizawa, K. Oshima and H. Nozaki, Tetrahedron Lett., 1983, 24, 2877–2880 CrossRef CAS.
- I. Fleming, J. Dunoguès and R. Smithers, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1989, pp. 57–575 Search PubMed.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
Footnotes |
| † Dedicated to Professor Stuart L. Schreiber on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05313k |
| This journal is © The Royal Society of Chemistry 2017 |