
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica-supported isolated gallium sites as highly active, selective and stable propane dehydrogenation catalysts†
Keith
Searles
a,
Georges
Siddiqi
a,
Olga V.
Safonova
b and
Christophe
Copéret
 *a
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, CH-8093 Zürich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch
bPaul Scherrer Institute, CH-5232 Villigen, Switzerland
First published on 3rd January 2017
Abstract
Single-site gallium centers on the surface of silica are prepared via grafting of [Ga(OSi(OtBu)3)3(THF)] on SiO2–700 followed by a thermolysis step. The resulting surface species corresponds to well-defined tetra-coordinate gallium single-sites, [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3Ga(XOSi)] (X = –H or Si) according to IR, X-ray absorption near-edge structure and extended X-ray absorption fine structure analysis. These gallium sites show high activity, selectivity and stability for propane dehydrogenation with an initial turnover frequency of 20 per h per gallium center, propylene selectivity of ≥93% and remarkable stability over 20 h. The stability of the catalyst probably results from site-isolation of the active site on a non-reducible support such as silica, diminishing facile reduction typical of Ga2O3-based catalysts.
SiO)3Ga(XOSi)] (X = –H or Si) according to IR, X-ray absorption near-edge structure and extended X-ray absorption fine structure analysis. These gallium sites show high activity, selectivity and stability for propane dehydrogenation with an initial turnover frequency of 20 per h per gallium center, propylene selectivity of ≥93% and remarkable stability over 20 h. The stability of the catalyst probably results from site-isolation of the active site on a non-reducible support such as silica, diminishing facile reduction typical of Ga2O3-based catalysts.
Introduction
The increasing propylene demand combined with the use of alternative feedstocks such as shale gas has amplified the pressure towards propene production and the development of light alkane dehydrogenation processes.1 In particular, the recent conversion of cracking plants from naphtha to ethane, the second largest component of shale gas, has resulted in a decline in annual propylene production while demand continues to increase. As a result, on-site propane dehydrogenation (PDH) is of particular interest for current and future propylene production.1a,c,2 While the two principal industrial processes for PDH utilize Cr–Al2O3 (CATOFIN) and PtSn–Al2O3 (Oleflex) supported catalysts,1c,2 other materials such as gallium-based catalysts are receiving considerable attention as alternative catalysts.Gallium-based zeolites have already been implemented for the conversion of lightweight alkanes to aromatics and H2, a process proposed to involve a tandem dehydrogenation–aromatization process.3 Ga2O3 and related materials have also been investigated as catalysts for alkane conversion, specifically PDH due to the high selectivity for propylene.4 In this regard it is proposed that tetra-coordinate surface sites of Ga2O3 are the active sites for propane dehydrogenation. However, due to the reducibility of Ga2O3, these active sites typically suffer from facile reduction, ultimately resulting in catalyst deactivation.1c,5 This is supported by advantageous effects of co-feeding CO2 or H2O during PDH, which have been attributed to the re-oxidation of inactive Ga(I) sites and removal of coke from the surface.5a,6 Several computational and experimental studies have aimed at identifying these active sites,4f,h,i,7 which so far remains a topic of debate. Silica-supported gallium species have been recently prepared by electrostatic adsorption methods.7a While showing promising results, the structure of the active sites is unknown.
Surface organometallic chemistry (SOMC) allows for the generation of well-defined surface species on a variety of supports via grafting of tailored molecular precursors on supports with isolated –OH sites.8 Thus, we reasoned that the development of an appropriate molecular precursor would allow for generating well-defined isolated active sites of gallium outside of bulk Ga2O3. This would result in a material with high catalytic activity with respect to total metal loading, as inactive bulk Ga2O3 would not be present. Additionally, generating isolated gallium sites on a non-reducible support such as silica could diminish reduction processes and allow for the development of a catalyst with enhanced stability under PDH conditions. A two-step approach, involving both SOMC and the thermolytic molecular precursor approach has previously been implemented to generate isolated surface sites with controlled oxidation state and nuclearity.9 Herein we describe the synthesis, structural characterization of the molecular complex [Ga(OSi(OtBu)3)3(THF)] (1)10 and its utilization as a precursor for generating gallium single-sites on silica. These isolated gallium sites were achieved through grafting of 1 on silica followed by a thermolysis step at 500 °C under high vacuum. The presented gallium species display unprecedented catalytic performances, combining high activity, selectivity and stability in the dehydrogenation of propane.
Results and discussion
First, we developed the synthesis of [Ga(OSi(OtBu)3)3(THF)] (1) by reacting GaCl3 with Na(OSi(OtBu)3) in THF.10 A high-quality X-ray diffraction experiment provides reliable bond distances and angles for this precursor (Fig. 1). Opaque single crystals were grown from a saturated pentane solution cooled to −40 °C. This analysis reveals a distorted trigonal pyramidal geometry around the Ga(III) center (τ4 = 0.86).11 The THF ligand occupies the axial position (Ga1–O1THF, 1.963(2) Å) rendering the three siloxide ligands in the equatorial plane (Ga1–O2siloxide, 1.780(2) Å). This unusual geometry about the gallium metal center is attributed to the steric repulsion of the bulky siloxide ligands.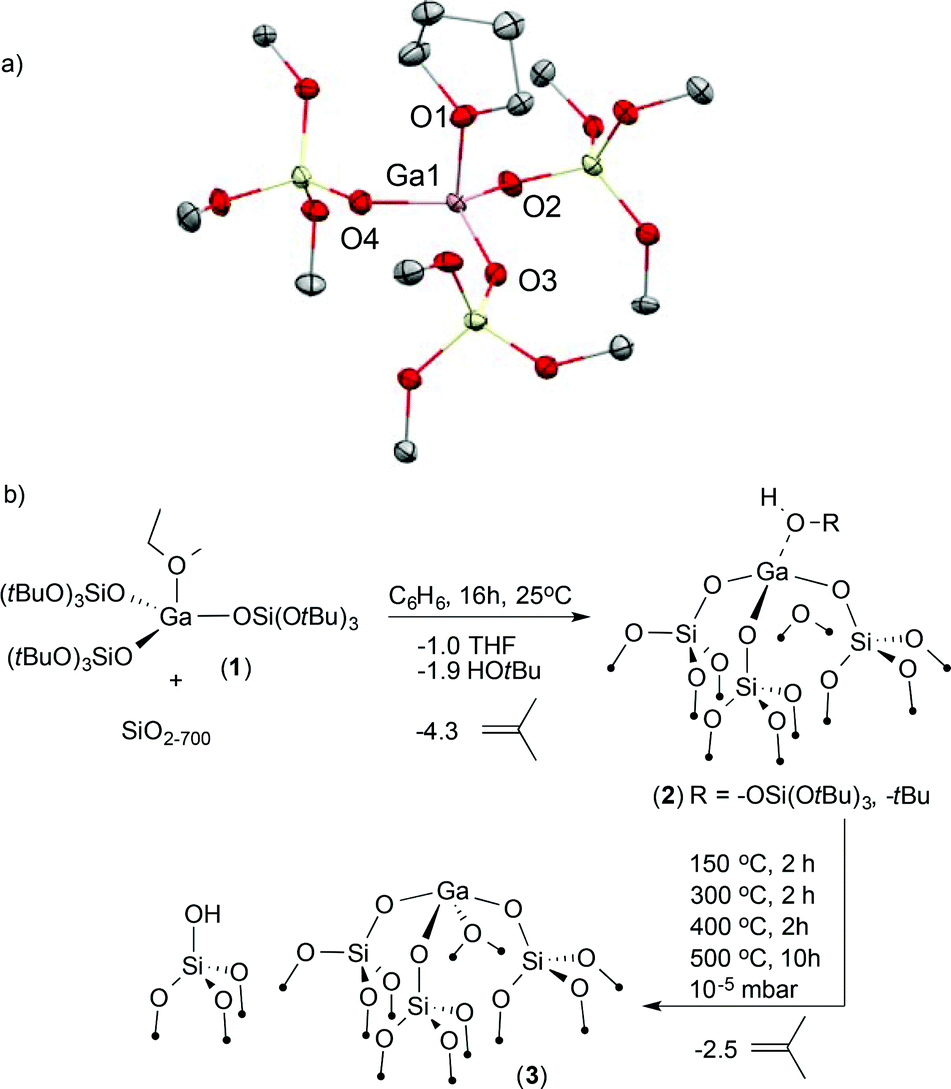 | ||
| Fig. 1 (a) Molecular structure of 1 obtained from X-ray diffraction studies. Ellipsoids are at the 50% probability and all H atoms and all –CH3 groups of the –OSi(OtBu)3 ligand have been omitted for clarity. (b) Synthesis of [(SiO)3Ga(HOR)], (2, R = –Si(OtBu)3 or –tBu) and thermal transformation under high vacuum yielding [(SiO)3Ga(XOSi)] (3, X = H or Si). | ||
The next step was the reaction of a benzene solution of 1 with SiO2–700 (0.31 mmol –OH per g)12 (Fig. 1). After 12 hours and subsequent washings of the material with benzene, 1H NMR spectroscopy revealed complete consumption of 1 and formation of isobutene, tert-butanol and THF in ratios of 4.3, 1.9, and 1.0 equivalents per consumed gallium complex. No detectable amount of HOSi(OtBu)3 was released after the grafting process, which is in sharp contrast to what was observed for similar Cr(III) or Fe(III) analogues.9b,h The aforementioned ratios indicate release of THF and thermal transformation of two –OSi(OtBu)3 ligands of 1 when contacted with the silica surface over the duration of 12 hours. The material was dried under high vacuum (10−5 mbar) for 12 hours and subsequent analysis by IR spectroscopy revealed two νOH bands in the range of 3800–3100 cm−1 that are attributed to regenerated surface –OH sites and HOR (R = –Si(OtBu)3 or –tBu) remaining on the surface (see ESI†). Additional νCH bands of an alkoxide ligand are observed in the range of 3050–2860 and 1550–1360 cm−1. 13C MAS SSNMR also revealed chemical shifts consistent with the presence of a remaining alcohol ligand on the surface of the material (see ESI†). Elemental analysis of the new material indicated a gallium loading of 1.53 weight% and 9.12 C atoms per Ga on the surface. These findings are consistent with a material possessing a remaining alcohol ligand per gallium immobilized on the support, specifically [(SiO)3Ga(HOR)], (2, R = –Si(OtBu)3 or –tBu).
The material 2 was then subjected to thermal treatment up to 500 °C for 10 hours under high vacuum (10−5 mbar). Analysis of the volatile components from thermolysis revealed 2.5 equivalents of isobutene per gallium on the surface gauged by 1H NMR spectroscopy. Quantification of ca. 3 C4 fragments per gallium, the absence of νCH and the regeneration of SiOH in the IR of the new material indicates that thermal elimination of aliphatics from 2 was achieved yielding [(SiO)3Ga(XOSi)] (3, X = H or Si)] along with the regeneration of SiOH groups (Fig. 1).
To understand the coordination environment of gallium on the silica surface, we performed X-ray absorption spectroscopy (XAS) measurements at Ga K-edge on materials 1–3. The edge position in the X-ray absorption near-edge structure (XANES) spectra of 2 and 3 was similar (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 374.8 and 10374.2 eV, respectively) as well as the position of a shoulder at higher energies (10380.8 and 10381.2 eV, respectively), which is consistent with tetra-coordinate Ga(III) sites on the surface (Fig. 2).7a,13
374.8 and 10374.2 eV, respectively) as well as the position of a shoulder at higher energies (10380.8 and 10381.2 eV, respectively), which is consistent with tetra-coordinate Ga(III) sites on the surface (Fig. 2).7a,13
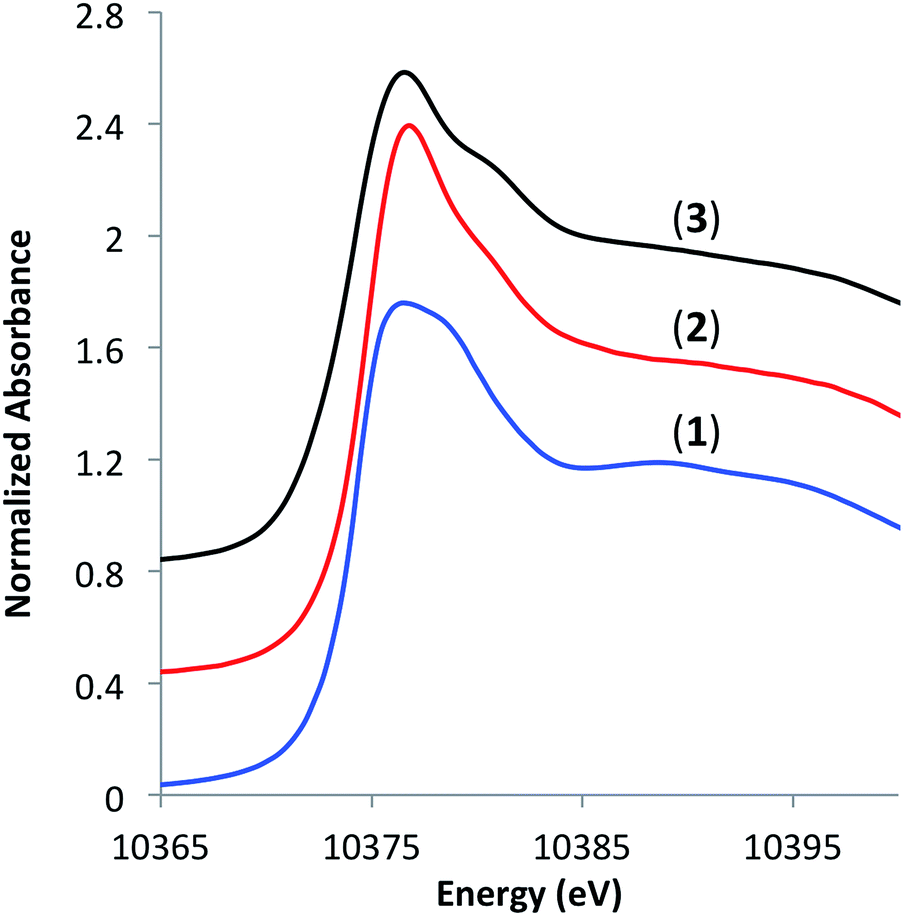 | ||
| Fig. 2 XANES spectra of [Ga(OSi(OtBu)3)3(THF)] (1), [(SiO)3Ga(HOR)], (2, R = –Si(OtBu)3 or –tBu) and [(SiO)3Ga(XOSi)] (3, X = H or Si). | ||
Fits of the extended X-ray absorption fine structure (EXAFS) data for complexes 1–3 were fitted in R-space (1.0–3.5 Å) after a Fourier transform (1 and 3, k = 3.0–12.0 Å−1; 2, k = 3.0–11.0 Å−1). The data are summarized in Table 1. For the molecular complex 1, two Ga–O scattering paths at distances of 1.79 Å (N = 3) and 2.01 Å (N = 1) were used for modeling of the 1st shell; a longer scattering path for Ga–Si of 3.15 Å (N = 3) was also employed for modeling of the 2nd shell. An additional multi-scattering path for Ga–O–Si of 3.25 Å (N = 6) and a long scattering path for Ga–O of 3.50 Å (N = 3) were required to produce a good fit. These distances match well with the X-ray single-crystal diffraction data obtained for 1. For the grafted complex 2 and the thermally treated complex 3, only Ga–O and Ga–Si scattering paths were used for fitting the experimental data. The fit for 2 indicates that Ga has 4.1 oxygen neighbors at 1.81 Å and 2.0 silicon neighbors at 3.17 Å. Similarly, the best fit for 3 is consistent with 3.6 oxygen neighbors at 1.80 Å and 1.4 silicon atoms at 3.08 Å.
SiO)3Ga(HOR)], (2, R = –Si(OtBu)3 or –tBu) and [(SiO)3Ga(XOSi)] (3, X = H or Si)a
| Sample | Neighbor | N | r [Å] | σ 2 [Å2] |
|---|---|---|---|---|
| a Samples were measured at 295 K in transmission mode. b Number of neighbors. c Distance between Ga and neighbor. d Debye–Waller factor. Set parameters are indicated by (*). | ||||
| 1 | O | 3* | 1.788(3) | 0.0035(2) |
| O | 1* | 2.01(1) | 0.0035(2) | |
| Si | 3* | 3.15(2) | 0.007(2) | |
| O–Si | 6* | 3.25(3) | 0.007(2) | |
| O | 3* | 3.50(2) | 0.007(2) | |
| 2 | O | 4.1(4) | 1.81(2) | 0.008(1) |
| Si | 2.0(1.2) | 3.17(1) | 0.011(6) | |
| 3 | O | 3.6(5) | 1.80(1) | 0.008(1) |
| Si | 1.4(6) | 3.08(3) | 0.011* | |
Given the possibility of generating gallium dimers upon grafting on the surface of silica,14 we have also probed the contribution of a Ga–Ga scattering path by performing a wavelet transform (WT) analysis15 on the EXAFS data for thermally treated 3 (Fig. 3). This analysis provides a correlation of R and k-space and ultimately aids in the distinction between two different atoms positioned at similar distances from the gallium metal center. The WT analysis of 3 shows a predominant feature with the maximum intensity in the range of R = 1.2–1.6 Å and k = 4.5–6.2 per Å, which is ascribed to the Ga–O scattering path. Scattering paths for Ga–Si and Ga–O–Si produce features in the range of R = 2.6–2.8 Å and k = 6.0–8.0 per Å for complex 3; no Ga–Ga path could be identified. Fig. 3 also shows the WT analysis of EXAFS data of a Ga2O3 reference sample. This was done to evaluate the presence of a Ga–Ga scattering path in 3. The WT analysis performed on the EXAFS data of Ga2O3 shows a similar feature for a Ga–O scattering path described for 3 and an additional intense feature for a Ga–Ga scattering path in the range of R = 2.5–3.0 Å and k = 9.0–10.0 per Å. This later feature is not observed for complex 3, implying that a Ga–Ga scattering path is not significant for fitting of the EXAFS data indicating that Ga2O3 domains are not present at the surface. Analysis of the XAS measurements for 1–3 strongly supports the formation of well-defined and dispersed tetra-coordinate gallium single-sites on the silica surface.
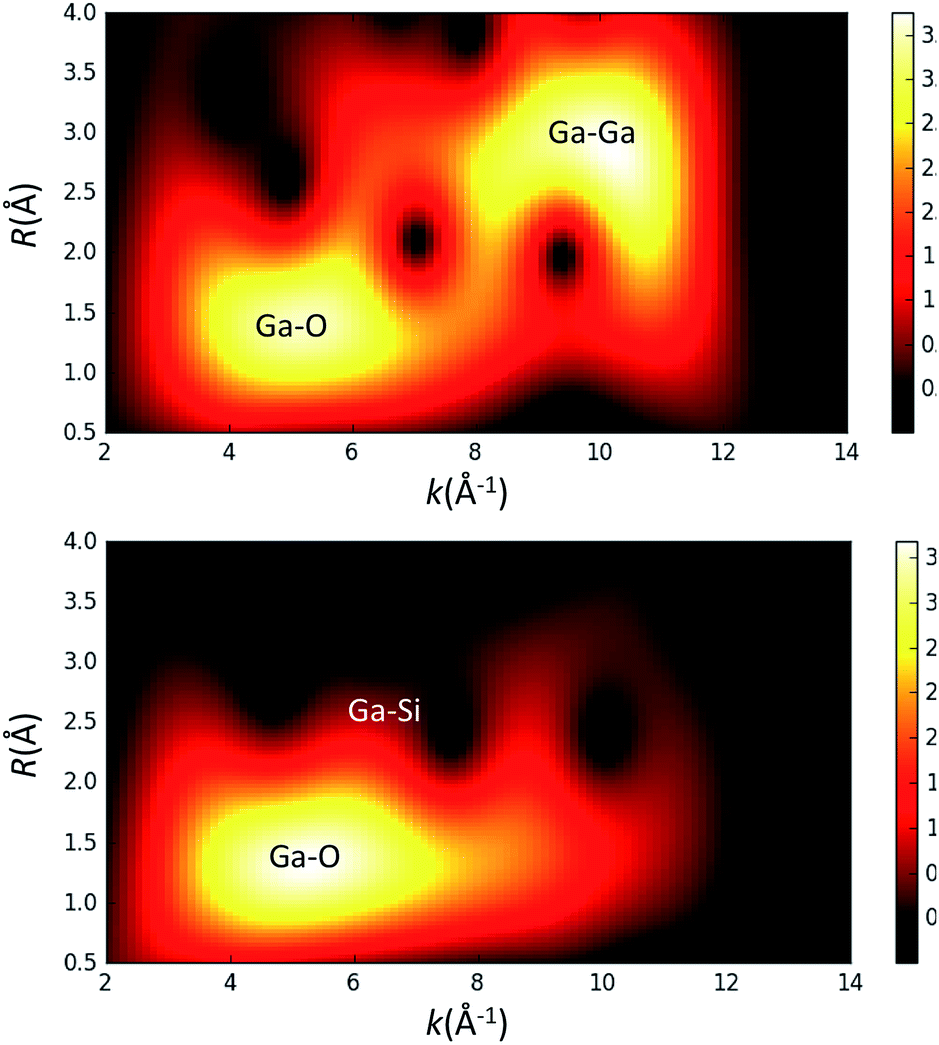 | ||
| Fig. 3 Wavelet transform (WT) analysis of EXAFS data for Ga2O3 (top) and [(SiO)3Ga(XOSi)] (3, X = H or Si) (bottom). | ||
To chemically probe the presence and accessibility of the gallium sites on the surface, pyridine adsorption studies were performed on 3 revealing three vibrational bands at 1621, 1493, 1458 cm−1 (see ESI†). These vibrational bands were retained upon heating to 500 °C under high vacuum indicating the presence of strong Lewis acid sites on the surface.7a,16 Additionally, no vibrational bands characteristic of the pyridinium ion were observed, indicating the absence of Brønsted acid surface sites.
We then investigated the catalytic performance of 3 towards PDH at 550 °C. Using a flow (10 mL per min; WHSV, 2.1 per h) consisting of 20% C3H8 in Ar, selectivity for propylene after 30 min was 94.3% with a TOF of 20.4 mol C3H6 per mol Ga per h and conversion of 9.3% (Fig. 4). After 20 hours of catalytic performance selectivity remained nearly constant (≥93%) with a TOF of 14.2 per h and conversion of 6.5%. The only other hydrocarbons detected during the duration of the experiment were methane and ethylene, determined to be secondary products of the reaction (see ESI†). After PDH only minimal darkening of the catalyst, as well as elemental analysis (0.18% C), suggests that coke formation is a negligible contribution to propane conversion. The initial TOF for 3 is ca. 11 times higher than recently reviewed Ga2O3 based materials (highest reported TOF of 1.8 per h; WHSV, 0.97 per h)1c and five times greater than isolated Ga sites prepared by electrostatic adsorption (TOF of 3.8 per h), which were tested at significantly lower space velocities.7a The higher activity of 3, with respect to the total metal loading of the catalysts, is attributed to negligible amounts of inactive bulk gallium-material being present in 3. Over the 20 hour experiment, 3 experiences a slow deactivation process (kd of 0.02 per h) which is an order of magnitude slower than that observed for Ga2O3 (kd of 0.67 per h) and β-Ga2O3 (kd of 0.21 per h) which have a catalyst life of 4 and 6 hours, respectively.1c,17 An industrial-like CrOX–Na/Al2O3 (20 wt% Cr, 1 wt% Na) catalyst displays an initial TOF of 0.027 per h and slightly lower selectivity (80%) for propylene at 550 °C with a WHSV of 0.12 per h.1c The considerably lower initial TOF in comparison to 3 highlights the effect of having significant amounts of inactive bulk metal in the catalyst. These catalytic comparisons are summarized in the ESI.†
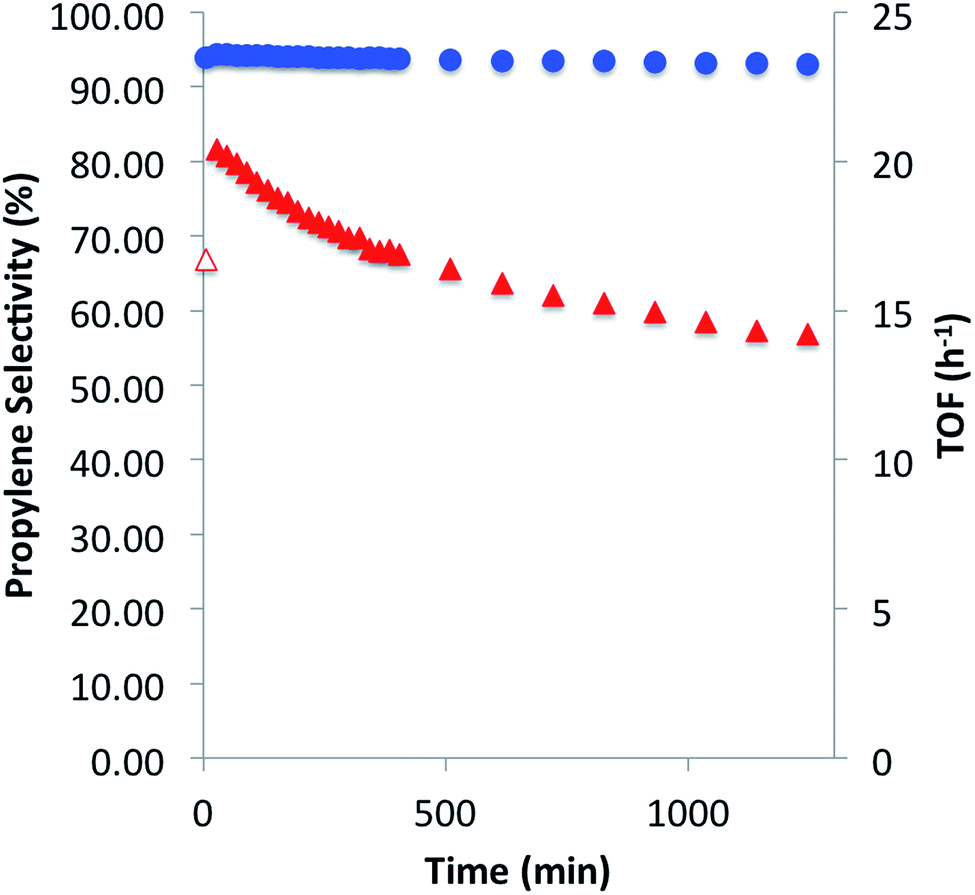 | ||
| Fig. 4 Catalytic performance of [(SiO)3Ga(XOSi)] (3, X = H or Si) for propane dehydrogenation at 550 °C with conversions in the range of 9.3–6.5%. Propylene selectivity (circles) and turnover frequency per hour (triangles), over a period of 20 hours. | ||
In an effort to understand the stability of 3 under PDH conditions, XAS measurements at Ga K-edge of the spent catalyst were performed. This analysis revealed a partial reduction of the catalyst as evident by a new feature with an edge position at 10370.6 eV in the XANES spectrum (Fig. 5). This shift to lower energies is attributed to partial formation of Ga(I) surface sites as no vibrational bands characteristic of surface Ga–H species were observed in the IR spectrum of the spent catalyst (see ESI†). Analysis of the EXAFS data after catalysis displayed no intense feature at higher R-values (2.2–3.5 Å) indicating that site-isolation of the gallium sites is retained after PDH. The partial reduction of the catalyst could explain the decrease in activity (ca. 30%) after 20 hours. The high TOF and selectivity (≥93%) maintained over 20 hours exemplifies the ability of SOMC methods to generate a stable material possessing highly active sites on the surface. It also indicates that site isolation and immobilization on a non-reducible support can diminish reduction processes that contribute to deactivation of other Ga2O3-based materials.
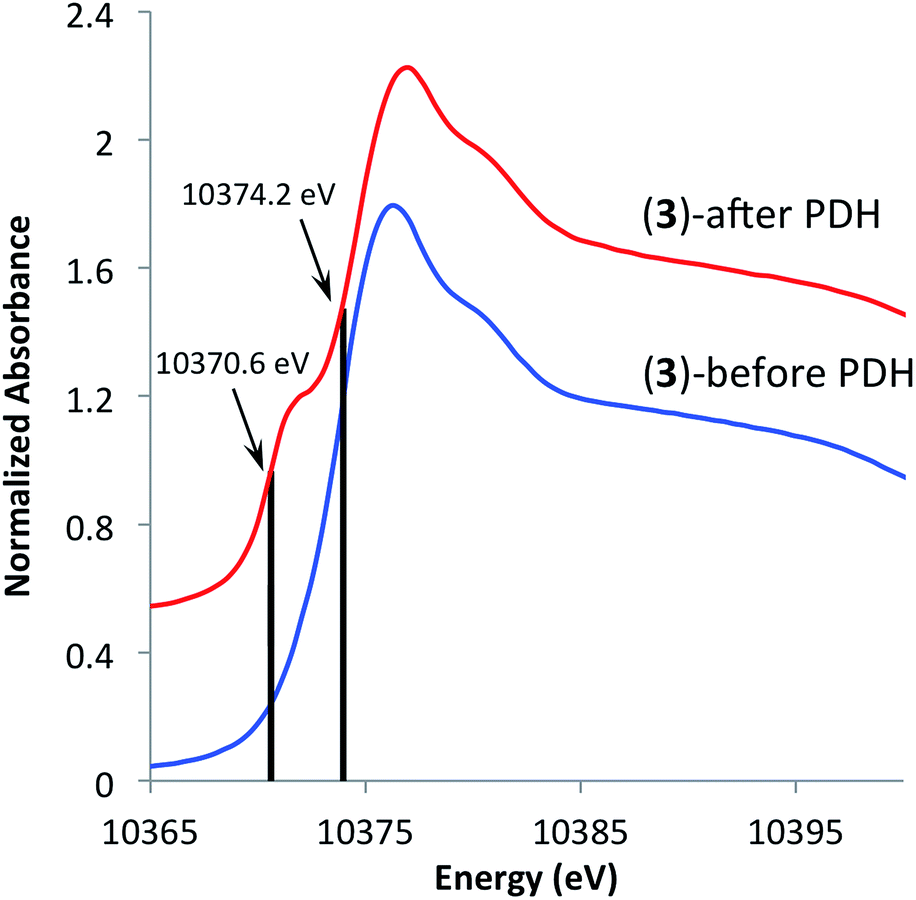 | ||
| Fig. 5 XANES spectra of [(SiO)3Ga(XOSi)] (3, X = H or Si) before (bottom) and after (top) propane dehydrogenation at 550 °C for 20 hours. | ||
Conclusions
In summary, we have synthesized silica-supported isolated tetra-coordinated gallium sites from a well-defined gallium-siloxide molecular precursor employing to a two-step process involving the grafting of a bulky siloxide molecular precursor 1 on highly dehydroxylated silica followed by thermolysis. XANES and EXAFS data, including a wavelet transform analysis, allowed for assignment of well-defined isolated gallium sites on the surface. Such material combines high activity towards propane dehydrogenation with high selectivity for propylene and stability over a 20 hour period. We attribute the prolonged stability of the catalyst to the generation of isolated Ga sites on a non-reducible support, thus diminishing facile reduction typical of Ga2O3. This study has thus opened new avenues to generate more efficient and rationally designed dehydrogenation catalysts, and we are currently exploring this area.Acknowledgements
The authors are grateful to the Swiss National Foundation (SNF) for financial support of this work (grant no. 200020_149704). We thank Professor Christoph R. Müller and Paula M. Abdala (ETH Zürich) for assistance in collecting XAS data. We also thank Florian Allouche (ETH Zürich) for assistance with X-ray crystallography analysis. We thank Alexander L. Trigub (Kurchatov Institute, Moscow, Russia) for providing software for WT analysis.References
- (a) Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CrossRef CAS; (b) D. Malakoff, Science, 2014, 344, 1464–1467 CrossRef CAS PubMed; (c) J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed; (d) P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed; (e) J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, Wiley-VCH, New York, 2nd edn, 2015 Search PubMed; (f) G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed; (g) K. Weisermel and H. J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 4th edn, 2003 Search PubMed.
- D. Sanfilippo and I. Miracca, Catal. Today, 2006, 111, 133–139 CrossRef CAS.
- (a) R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2406 CrossRef CAS PubMed; (b) G. L. Price and V. Kanazirev, J. Catal., 1990, 126, 267–278 CrossRef CAS; (c) P. Meriaudeau and C. Naccache, J. Mol. Catal., 1989, 50, L7–L10 CrossRef CAS; (d) N. S. Gnep, J. Y. Doyemet, A. M. Seco, F. R. Ribeiro and M. Guisnet, Appl. Catal., 1988, 43, 155–166 CrossRef CAS; (e) H. Kitagawa, Y. Sendoda and Y. Ono, J. Catal., 1986, 101, 12–18 CrossRef CAS; (f) J. M. Thomas and X. S. Liu, J. Phys. Chem., 1986, 90, 4843–4847 CrossRef CAS; (g) G. D. Meitzner, E. Iglesia, J. E. Baumgartner and E. S. Huang, J. Catal., 1993, 140, 209–225 CrossRef CAS.
- (a) S. Tan, S.-J. Kim, J. S. Moore, Y. Liu, R. S. Dixit, J. G. Pendergast, D. S. Sholl, S. Nair and C. W. Jones, ChemCatChem, 2016, 8, 214–221 CrossRef CAS; (b) S. Tan, L. B. Gil, N. Subramanian, D. S. Sholl, S. Nair, C. W. Jones, J. S. Moore, Y. Liu, R. S. Dixit and J. G. Pendergast, Appl. Catal., A, 2015, 498, 167–175 CrossRef CAS; (c) J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS PubMed; (d) J.-L. Wu, M. Chen, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Catal. Commun., 2013, 30, 61–65 CrossRef; (e) P. Michorczyk, P. Kuśtrowski, A. Kolak and M. Zimowska, Catal. Commun., 2013, 35, 95–100 CrossRef CAS; (f) M. Chen, J. Xu, F.-Z. Su, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS; (g) H. Li, Y. Yue, C. Miao, Z. Xie, W. Hua and Z. Gao, Catal. Commun., 2007, 8, 1317–1322 CrossRef CAS; (h) B. Xu, B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2006, 239, 470–477 CrossRef CAS; (i) B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2005, 232, 143–151 CrossRef CAS; (j) N. S. Nesterenko, O. A. Ponomoreva, V. V. Yuschenko, I. I. Ivanova, F. Testa, F. Di Renzo and F. Fajula, Appl. Catal., A, 2003, 254, 261–272 CrossRef CAS; (k) K. Nakagawa, C. Kajita, K. Okumura, N.-O. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Catal., 2001, 203, 87–93 CrossRef CAS; (l) K. Nakagawa, C. Kajita, Y. Ide, M. Okamura, S. Kato, H. Kasuya, N. O. Ikenaga, T. Kobayashi and T. Suzuki, Catal. Lett., 2000, 64, 215–221 CrossRef CAS; (m) K. Nakagawa, M. Okamura, N. Ikenaga, T. Suzuki, K. Nakagawa, M. Okamura, T. Suzuki, T. Kobayashi and T. Kobayashi, Chem. Commun., 1998, 1025–1026 RSC; (n) H. Xiao, J. Zhang, P. Wang, X. Wang, F. Pang, Z. Zhang and Y. Tan, Catal. Sci. Technol., 2016, 6, 5183–5195 RSC; (o) M. Saito, S. Watanabe, I. Takahara, M. Inaba and K. Murata, Catal. Lett., 2003, 89, 213–217 CrossRef CAS.
- (a) C. Copéret, Chem. Rev., 2010, 110, 656–680 CrossRef PubMed; (b) C. Copéret, D. P. Estes, K. Larmier and K. Searles, Chem. Rev., 2016, 116, 8463–8505 CrossRef PubMed.
- (a) E. A. Pidko and R. A. van Santen, J. Phys. Chem. C, 2009, 113, 4246–4249 CrossRef CAS; (b) E. J. M. Hensen, E. A. Pidko, N. Rane and R. A. van Santen, Angew. Chem., Int. Ed., 2007, 46, 7273–7276 CrossRef CAS PubMed; (c) E. A. Pidko, R. A. van Santen and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2009, 11, 2893–2902 RSC.
- (a) A. B. Getsoian, U. Das, J. Camacho-Bunquin, G. Zhang, J. R. Gallagher, B. Hu, S. Cheah, J. A. Schaidle, D. A. Ruddy, J. E. Hensley, T. R. Krause, L. A. Curtiss, J. T. Miller and A. S. Hock, Catal. Sci. Technol., 2016, 6, 6339–6353 RSC; (b) Y. Liu, Z. H. Li, J. Lu and K.-N. Fan, J. Phys. Chem. C, 2008, 112, 20382–20392 CrossRef CAS; (c) I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2004, 96, 29–32 CrossRef CAS; (d) P. Michorczyk and J. Ogonowski, Appl. Catal., A, 2003, 251, 425–433 CrossRef CAS; (e) A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev and A. G. Stepanov, Chem. Phys. Lett., 2010, 496 Search PubMed; (f) V. B. Kazansky, I. R. Subbotina, A. A. Pronin, R. Schlögl and F. C. Jentoft, J. Phys. Chem. B, 2006, 110, 7975–7978 CrossRef CAS PubMed; (g) U. Das, G. Zhang, B. Hu, A. S. Hock, P. C. Redfern, J. T. Miller and L. A. Curtiss, ACS Catal., 2015, 5, 7177–7185 CrossRef CAS.
- (a) C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed; (b) C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed; (c) S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS PubMed; (d) J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- (a) T. D. Tilley, J. Mol. Catal. A: Chem., 2002, 182–183, 17–24 CrossRef CAS; (b) C. Nozaki, C. G. Lugmair, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 13194–13203 CrossRef CAS PubMed; (c) J. Jarupatrakorn and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 8380–8388 CrossRef CAS PubMed; (d) K. L. Fujdala and T. D. Tilley, J. Catal., 2003, 216, 265–275 CrossRef CAS; (e) K. L. Fujdala, R. L. Brutchey and T. D. Tilley, Surface and Interfacial Organometallic Chemistry and Catalysis, Springer, Berlin, Heidelberg, 2005 Search PubMed; (f) A. W. Holland, G. Li, A. M. Shahin, G. J. Long, A. T. Bell and T. D. Tilley, J. Catal., 2005, 235, 150–163 CrossRef CAS; (g) M. P. Conley, M. F. Delley, G. Siddiqi, G. Lapadula, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Angew. Chem., Int. Ed., 2014, 53, 1872–1876 CrossRef PubMed; (h) M. F. Delley, F. Núñez-Zarur, M. P. Conley, A. Comas-Vives, G. Siddiqi, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11624–11629 CrossRef CAS PubMed; (i) V. Mougel, K.-W. Chan, G. Siddiqi, K. Kawakita, H. Nagae, H. Tsurugi, K. Mashima, O. Safonova and C. Copéret, ACS Cent. Sci., 2016, 138, 14987–14997 Search PubMed; (j) D. P. Estes, G. Siddiqi, F. Allouche, K. V. Kovtunov, O. V. Safonova, A. L. Trigub, I. V. Koptyug and C. Copéret, J. Am. Chem. Soc., 2016, 2, 569–576 Search PubMed; (k) R. L. Brutchey, C. G. Lugmair, L. O. Schebaum and T. D. Tilley, J. Catal., 2005, 229, 72–81 CrossRef CAS.
- [Ga(OSi(OtBu)3)3(THF)] was disclosed during the course of this study; J. P. Dombrowski, G. R. Johnson, A. T. Bell and T. D. Tilley, Dalton Trans., 2016, 11025–11034 RSC.
- M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 CrossRef CAS PubMed.
- Silica (Degauss Aerosol, 204 m2 g−1) thermally treated at 700 °C with a temperature ramp of 5 °C per min under high vacuum (10−5 mbar) yielded 0.31 mmol OH per g of SiO2 corresponding to 0.92 accessible OH groups per nm2. The slightly higher –OH density in comparison to that reported in literature for SiO2-700 is attributed to the slightly higher temperature ramp used during dehydroxylation (see ref. 8a).
- (a) K. Nishi, K.-i. Shimizu, M. Takamatsu, H. Yoshida, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, J. Phys. Chem. B, 1998, 102, 10190–10195 CrossRef CAS; (b) P. Behrens, H. Kosslick, T. Vu Anh, M. Fröba and F. Neissendorfer, Microporous Mater., 1995, 3, 433–441 CrossRef CAS; (c) K. A. Al-majnouni, N. D. Hould, W. W. Lonergan, D. G. Vlachos and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 19395–19405 CrossRef CAS; (d) R. I. Walton and D. O'Hare, J. Phys. Chem. Solids, 2001, 62, 1469–1479 CrossRef CAS.
- (a) Z. A. Taha, E. W. Deguns, S. Chattopadhyay and S. L. Scott, Organometallics, 2006, 25, 1891–1899 CrossRef CAS; (b) S. D. Fleischman and S. L. Scott, J. Am. Chem. Soc., 2011, 133, 4847–4855 CrossRef CAS PubMed.
- H. Funke, M. Chukalina and A. C. Scheinost, J. Synchrotron Radiat., 2007, 14, 426–432 CrossRef CAS PubMed.
- (a) A. Vimont, J. C. Lavalley, A. Sahibed-Dine, C. Otero Areán, M. Rodríguez Delgado and M. Daturi, J. Phys. Chem. B, 2005, 109, 9656–9664 CrossRef CAS PubMed; (b) E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
- k d is defined as ln(1 − convend/convend) − ln(1 − convstart/convstart)/t, where convstart is the conversion at start of experiment, convend is the conversion at end of experiment, and t is the duration of experiment in hours.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, material characterization data, catalytic measurement details and crystallographic details. CCDC 1499756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05178b |
| This journal is © The Royal Society of Chemistry 2017 |