
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a facile antibody–drug conjugate platform for increased stability and homogeneity†
Nimish
Gupta‡
 abd,
Johny
Kancharla‡§
a,
Shelly
Kaushik‡§
a,
Aasif
Ansari‡
a,
Samad
Hossain‡
abd,
Ravinder
Goyal‡
a,
Manoj
Pandey‡
a,
Jwala
Sivaccumar‡
a,
Sazid
Hussain‡
a,
Arindam
Sarkar‡
abd,
Aniruddha
Sengupta‡
abd,
Swadhin K.
Mandal‡
c,
Monideepa
Roy‡
*abd and
Shiladitya
Sengupta‡
*abef
abd,
Johny
Kancharla‡§
a,
Shelly
Kaushik‡§
a,
Aasif
Ansari‡
a,
Samad
Hossain‡
abd,
Ravinder
Goyal‡
a,
Manoj
Pandey‡
a,
Jwala
Sivaccumar‡
a,
Sazid
Hussain‡
a,
Arindam
Sarkar‡
abd,
Aniruddha
Sengupta‡
abd,
Swadhin K.
Mandal‡
c,
Monideepa
Roy‡
*abd and
Shiladitya
Sengupta‡
*abef
aInvictus Oncology Pvt. Ltd., Delhi-110092, India. E-mail: shiladit@mit.edu; mroy@invictusoncology.com
bIndia Innovation Research Center, Delhi-110092, India
cDepartment of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata, Mohanpur-741252, India
dHealth and Biomedical Sciences, Symbiosis International University, Pune-412115, India
eBrigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA
fHarvard – MIT Division of Health Sciences and Technology, Cambridge, MA 02139, USA
First published on 9th December 2016
Abstract
Despite the advances in the design of antibody–drug conjugates (ADCs), the search is still ongoing for novel approaches that lead to increased stability and homogeneity of the ADCs. We report, for the first time, an ADC platform technology using a platinum(II)-based linker that can re-bridge the inter-chain cysteines in the antibody, post-reduction. The strong platinum–sulfur interaction improves the stability of the ADC when compared with a standard maleimide-linked ADC thereby reducing the linker–drug exchange with albumin significantly. Moreover, due to the precise conserved locations of cysteines, both homogeneity and site-specificity are simultaneously achieved. Additionally, we demonstrate that our ADCs exhibit increased anticancer efficacy in vitro and in vivo. The Pt-based ADCs can emerge as a simple and exciting proposition to address the limitations of the current ADC linker technologies.
Introduction
Antibody–drug conjugates (ADCs) have emerged as a new genre of cancer therapeutics that involves the antibody-mediated delivery of highly potent cytotoxic agents specifically to the cancer cells expressing the target antigen. With more than 50 ADCs currently in the clinical pipeline,1 the prospects for the ADC field have escalated remarkably since the approvals of brentuximab vedotin (Adcetris™)2 for Hodgkin’s lymphoma in 2011 and ado-trastuzumab emtansine (Kadcyla™)3 for HER2+ breast cancer in 2013.The success of an ADC is dependent on various factors such as the target antigen, the antibody and the drug. However, the mainstay of an effective ADC lies in its linker—the glue that couples the drug and the antibody together until the ADC reaches the cancer cell. Conventionally, linker–drugs have been coupled to either lysines on the antibody, as in ado-trastuzumab emtansine,3 or to cysteines via maleimide chemistry, as in brentuximab vedotin (Scheme 1a and b).2 Both technologies have major limitations, such as the former producing heterogeneous ADCs4,5 and the latter being unstable during circulation.6 While heterogeneous ADCs have unpredictable safety and PK profiles and show batch-to-batch variability, the instability of maleimide chemistry results in off-target toxicity.4 These shortcomings with current linker technologies have hindered the concept of an ideal “magic bullet”, as envisioned by Paul Ehrlich,7 to be realized.
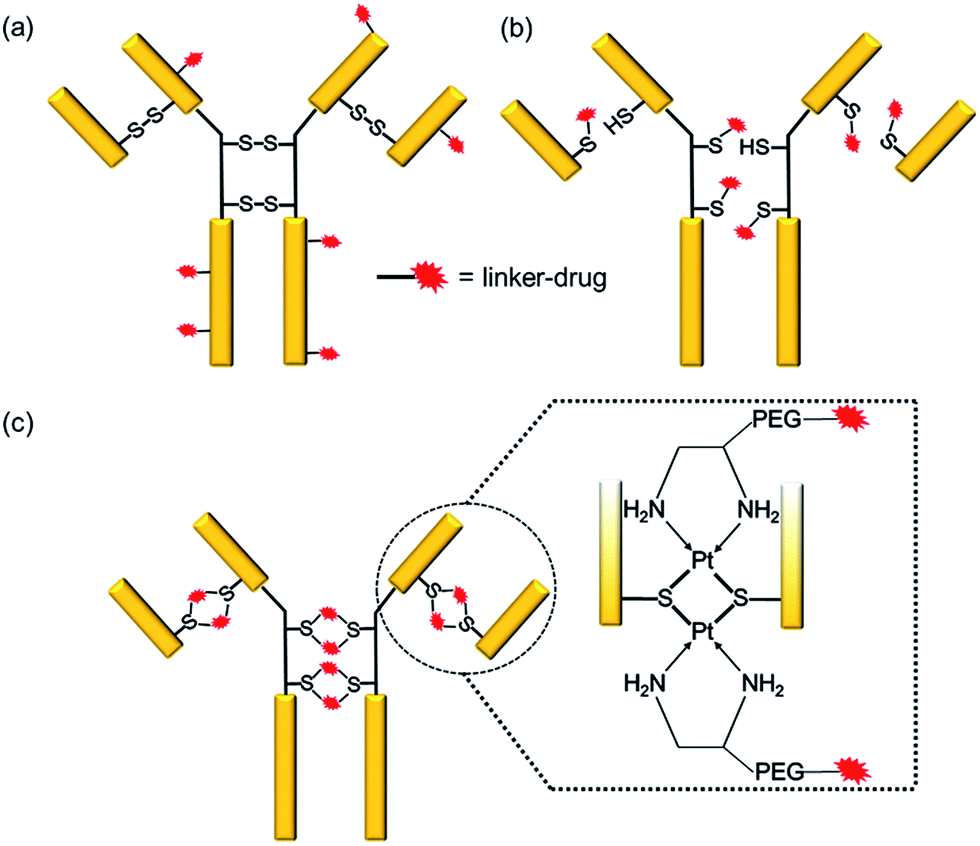 | ||
| Scheme 1 Comparison of conventional linker technologies with a Pt(II)-linker. (a) Conjugation to lysines results in a heterogeneous mixture. (b) Conjugation to cysteines via maleimide leads to reduced higher order structural stability and instability in the presence of albumin. (c) Pt(II)-linker re-bridges the antibody chains with strong Pt–S interaction imparting stability, homogeneity and site-specificity. | ||
In the last few years, several ideas have emerged to address these issues.8 The use of unnatural amino acids,9 additional cysteines10 and transglutaminase11 for site-specific conjugation have shown promise in reducing the heterogeneity of ADCs but require genetic engineering and involve complex processes. Furthermore, these methods pose challenges during the manufacturing and scale-up processes. Attempts to stabilize the maleimides by accelerating the thiosuccinimide ring hydrolysis have been shown to result in more efficacious and relatively safer ADCs.12 Alternative strategies to maleimide-based conjugations have also surfaced where the disulfides are re-bridged after cysteine reduction.4 Despite the potential advantages of these technologies, they all employ the C–S bond for conjugation, which is known to undergo a retro-Michael reaction resulting in the exchange of the linker–drug with thiol-containing molecules such as albumin.4 This not only leads to premature loss of ADC molecules from circulation but also causes toxic side effects.12 There is an imperative need to improve upon the conventional linker technologies with a goal to enhance their stability, site-specificity and ability to produce homogeneous ADCs in a facile manner.
Overcoming the current challenges with ADC linker technologies requires meticulous thought into the design aspects of the linker–drug, particularly, the strength of the antibody–linker bond. It is known that platinum(II) ions can form strong edge-sharing binuclear Pt(d8) complexes with sulfur bridges while reacting with thiols at a physiological pH.13–16 We report a novel technology employing Pt(II) as a linker that is able to re-bridge the antibody chains after conjugation (Scheme 1c). We rationalized that the Pt(II) mediated binding with the antibody should render the required stability to the ADC during circulation thereby improving upon the conventional maleimide chemistry. Using cetuximab, an antibody that inhibits the epidermal growth factor receptor (EGFR), trastuzumab, which blocks the human epidermal growth factor receptor 2 (HER2), and rituximab, which binds to CD20, as the antibodies and camptothecin (CPT) as a ‘proof of concept’ payload, here we report the engineering of novel Pt(II)-based prototype ADCs. The Pt-based ADCs exhibit superior stability when compared to maleimide-based ADCs, as well as being potent in antigen +ve cells and efficacious in vivo in a lung adenocarcinoma xenograft model. The Pt(II) linker technology has the potential to surmount the bottlenecks of instability and heterogeneity with the existing linker technologies.
Results
Pt(II)-linker–drug design
We rationalized that the high affinity of platinum for sulfur could lead to a superior stability of the Pt-linker-based ADC. This strong affinity is due to soft–soft interaction between the Pt(II) and sulfide ions as per the well-known hard–soft acids and bases (HSAB) principle. The bond energy of a Pt–S bond (114 kcal mol−1)17 is significantly higher than that of a C–S bond (∼70–80 kcal mol−1)18 present in the case of maleimide linkers or existing re-bridging technologies.4 Keeping this strong Pt–S interaction in mind, we designed and synthesized a Pt-based linker–drug (Pt–PEG–CPT) (Scheme 2, see ESI† for details), that takes advantage of the two labile Pt–Cl bonds which can be easily cleaved to form Pt–S bonds on treatment with a reduced antibody in aqueous medium. | ||
| Scheme 2 Structure, design and synthesis of Pt–PEG–CPT. (Reagents and all intermediates are shown in the ESI;† PEG: polyethyleneglycol.) | ||
We used camptothecin (CPT), an easily available topoisomerase-I inhibitor with nanomolar range potency, as the ‘proof of concept’ payload for this study. Additionally, camptothecin has a characteristic UV spectrum and fluoresces when excited at 369 nm, which facilitates downstream analytical characterization. Since CPT is a hydrophobic drug with poor solubility in an aqueous environment, ADCs with CPT are expected to be hydrophobic and could result in immunogenic aggregates4 that can undergo rapid plasma clearance.19 To overcome this issue, a hydrophilic polyethyleneglycol (PEG) unit was incorporated by coupling to the hydroxyl group of CPT via an ester bond (Scheme 2). It should be noted that such a construct can impart stability to the CPT molecule during circulation,20 and the ester bond can be easily cleaved by esterases present in the tumour cellular environment, thereby releasing the active camptothecin moiety from the linker.21 The platinum(II) ion was incorporated by the reaction between PEG–CPT and the amine ligand, resulting in the formation of a Pt–PEG–CPT molecule with two labile Pt–Cl bonds that would enable Pt–S bridge formation in the presence of thiol groups. The molecule was HPLC purified and has been characterized by 1H, 13C and 195Pt NMR, IR spectroscopy and mass spectrometry (see ESI†). A single and sharp signal at −2977 ppm in the195Pt NMR spectrum established diamino dichloro platinum coordination.22 In order to compare our Pt-based technology with state-of-the-art maleimide chemistry, a similar linker–drug, mal–PEG–CPT, replacing the Pt(II) with 6-maleimidocaproic acid, was also synthesized and fully characterized (see ESI†).
Preparation and characterization of the ADCs
ADCs are designed using antibodies against well-known antigens that are over-expressed on cancer cells compared to normal cells. The antibody is selected for high specificity and affinity towards the antigen, which can minimize the off-target effects. Three such antibodies, cetuximab (Ctx), trastuzumab (Tra) and rituximab (Rtx), which target EGFR, HER2 and CD20 respectively, are ideal candidates fulfilling both criteria and were therefore used in this study to prove the concept. Conjugation was performed by reducing the antibodies in the presence of an excess amount of tris(2-carboxyethyl)phosphine (TCEP) to expose all the 8 inter-chain thiols and incubating them with excess Pt–PEG–CPT or mal–PEG–CPT. Purification by buffer-exchange ensured the removal of any unbound linker–drug. Furthermore, the conjugation to the antibody was confirmed by the inherent fluorescence of CPT as monitored inline via size-exclusion HPLC (SE-HPLC). The SE-HPLC result of Ctx–Pt–PEG–CPT suggests the conjugation of the linker–drug to cetuximab as observed by the characteristic fluorescence of CPT corresponding to the antibody peak (Fig. 1a and b). The conjugates were highly pure with less than 1% aggregates (measured by absorbance at 280 nm) and less than 1% free linker–drug in the ADC (measured by absorbance at 369 nm). Similarly, conjugates were also prepared with trastuzumab (Tra–Pt–PEG–CPT) and rituximab (Rtx–Pt–PEG–CPT) which demonstrated good purity by SE-HPLC (Fig. 1c and d; ESI Fig. S1†). | ||
| Fig. 1 SE-HPLC profiles of conjugates. Chromatogram of Ctx–Pt–PEG–CPT (a) at 280 nm showing low presence of aggregates (<1%) and (b) fluorescence of CPT corresponding to the antibody peak with negligible presence of free camptothecin (<1%). Chromatogram of Tra–Pt–PEG–CPT (c) at 280 nm showing low presence of aggregates (2%) and (d) fluorescence of CPT corresponding to the antibody peak with negligible presence of free camptothecin (<1%). | ||
The quantification of the bound CPT concentration was carried out by UV spectroscopy whereas the concentration of Pt was assessed by atomic absorption spectroscopy (AAS). Drug-to-antibody ratios (DARs) were calculated by measuring the absorbance of ADC at 254 nm and 280 nm and the extinction coefficients of the respective linker–drug and the antibody at both wavelengths.23 The concentration of Pt corroborates well with the CPT concentration which demonstrates the presence of the two species in the ADC in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. As an example, for one batch of Tra–Pt–PEG–CPT, the CPT concentration by UV spectrometry was measured as 164 μM while the Pt-concentration by AAS was determined to be 161 μM. The addition of an excess of Pt–PEG–CPT resulted in a maximum DAR of ∼8 with all three antibodies. Since there are 4 disulfide bonds per antibody, this implies a stoichiometry of 2 Pt–PEG–CPT molecules per disulfide bridge as anticipated. Similarly, with mal–PEG–CPT, an addition of excess linker–drug results in a maximum DAR of ∼8 with a stoichiometry of 1 mal–PEG–CPT per reduced cysteine. Previously it had been suggested that the optimal DAR is ∼4 for in vivo studies as higher DAR ADCs result in altered pharmacokinetics due to their hydrophobic nature.23 The conjugation reaction, thus, was stoichiometrically controlled to ensure DARs of 2.5–4.
:1 molar ratio. As an example, for one batch of Tra–Pt–PEG–CPT, the CPT concentration by UV spectrometry was measured as 164 μM while the Pt-concentration by AAS was determined to be 161 μM. The addition of an excess of Pt–PEG–CPT resulted in a maximum DAR of ∼8 with all three antibodies. Since there are 4 disulfide bonds per antibody, this implies a stoichiometry of 2 Pt–PEG–CPT molecules per disulfide bridge as anticipated. Similarly, with mal–PEG–CPT, an addition of excess linker–drug results in a maximum DAR of ∼8 with a stoichiometry of 1 mal–PEG–CPT per reduced cysteine. Previously it had been suggested that the optimal DAR is ∼4 for in vivo studies as higher DAR ADCs result in altered pharmacokinetics due to their hydrophobic nature.23 The conjugation reaction, thus, was stoichiometrically controlled to ensure DARs of 2.5–4.
The Pt(II)-linker was designed to bind to the exposed thiols of the reduced antibody. However, since it is known that Pt(II) can also bind to various amino acids such as histidine, methionine, cysteine etc.,24,25 we confirmed that our Pt–PEG–CPT molecule binds only to the reduced antibody upon exposure of cysteine thiols i.e., Pt–PEG–CPT showed negligible association (DAR: 0.26) with the non-reduced antibody, thus corroborating our design.
A concern with any ADC is the potential conformational change due to the conjugation of hydrophobic drugs which results in an altered antigen binding ability. To understand the impact of Pt-based conjugation on the protein conformation, an ELISA was performed by capturing antibodies and their respective ADCs using their target (Fig. 2). The affinity constant (Kd) was found to be similar for both the antibody and the ADC and comparable to the reported Kd values,26–28 suggesting that the conformational change, if any, is local and does not impact the antigen-binding domains of the antibody.
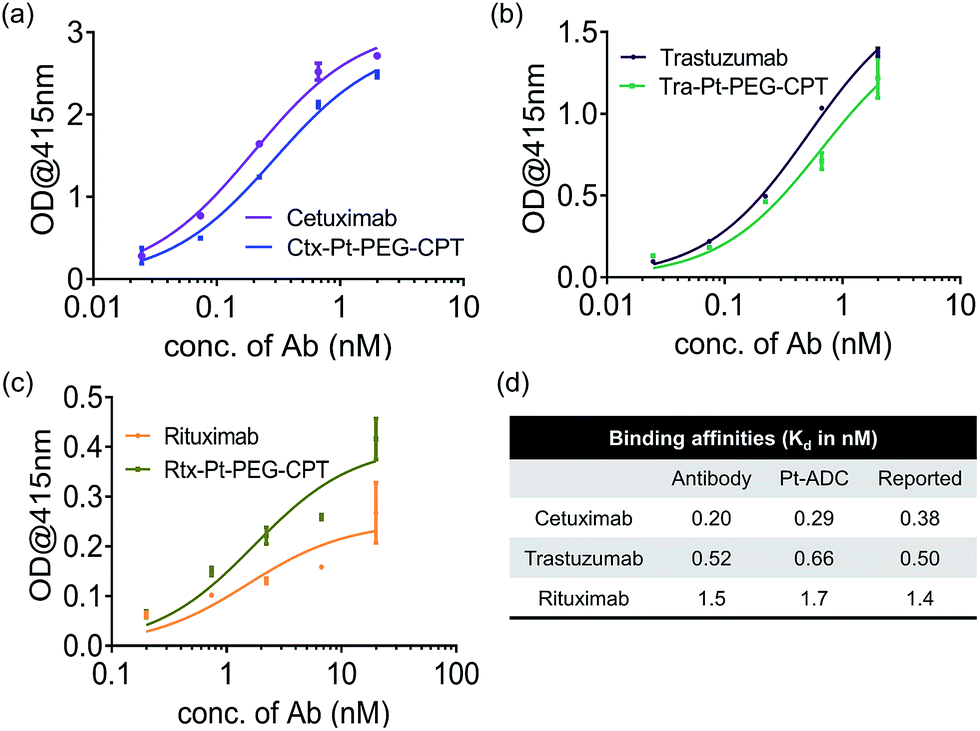 | ||
| Fig. 2 Antigen-binding ability. ELISA demonstrating that the affinity of the antibody is not compromised upon Pt(II)-based conjugation. (a) Ctx and Ctx–Pt–PEG–CPT binding to EGFR. (b) Tra and Tra–Pt–PEG–CPT binding to HER2. (c) Rtx and Rtx–Pt–PEG–CPT binding to CD20. (d) Calculated Kd values compared with reported binding affinities.26–28 Error bars represent standard error of the mean. | ||
Preparation of Fab–drug conjugate and heterogeneity
Heterogeneity is a vital parameter for any ADC technology as variations in the site of attachment lead to batch-to-batch disparities, resulting in inconsistent efficacy and safety profiles.6 ADCs prepared through conventional linker technologies result in a heterogeneous product with multiple species. To understand the heterogeneity of ADCs made with the Pt(II) linker, a trastuzumab-Fab fragment (TraFab) was prepared and conjugated to Pt–PEG–CPT and mal–PEG–CPT. As the Fab arm contains only one inter-chain disulfide, the analysis of the resulting Fab–drug conjugate (TraFab–Pt–PEG–CPT or TraFab–mal–PEG–CPT) was simplified. With excess linker–drug, the maximum achievable DAR, as measured by UV spectroscopy, was 1.9 for TraFab–Pt–PEG–CPT validating that two Pt–PEG–CPT molecules bind to each disulfide. With TraFab–mal–PEG–CPT, the maximum achievable DAR was 1.7.The Fab–drug conjugates were assayed by analytical hydrophobic interaction chromatography (HIC). TraFab–Pt–PEG–CPT (DAR: 1.9 by UV) showed two conjugated species (Fig. 3a). Both of the peaks were successfully separated by HIC (Fig. 3b) and were found to have a DAR of ∼2 by UV spectroscopy. The DAR by HIC was calculated to be 1.9 and the efficiency of the conjugation was 95%. Similarly, TraFab–mal–PEG–CPT (DAR: 0.65 by UV) showed two conjugated species (Fig. 3c) which were separated by HIC. The DAR by HIC was calculated to be 0.59. However, the peak at ∼24 min (Fig. 3c, peak 4) had a DAR of 1 whereas the peak at 30 min (Fig. 3c, peak 5) had a DAR of 2. These results imply that the Pt(II) linker yields more homogeneous ADCs with predominantly bi-conjugated ADC species compared to a heterogeneous mixture obtained with the maleimide linker. Therefore, the effective concentration of active molecules is enriched in the mixture, avoiding the need for further downstream purification procedures, thus ensuring an economical process.
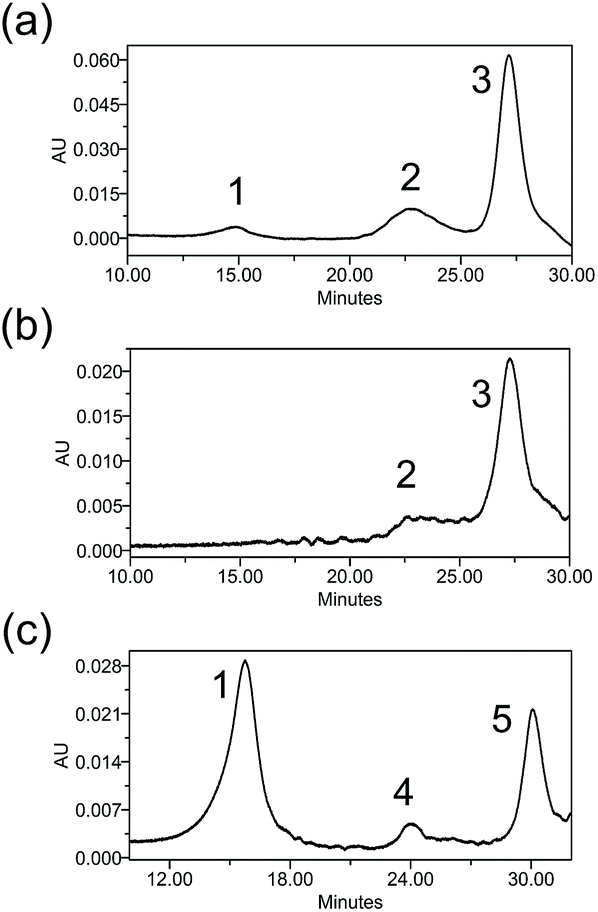 | ||
| Fig. 3 HIC profiles of TraFab–drug conjugates. (a) A representative HIC profile at 280 nm of TraFab–Pt–PEG–CPT (DAR: 1.9 by UV), demonstrating > 95% conjugation efficiency and homogeneity. The DAR was calculated to be 1.9 by HIC. (b) A representative HIC profile at 280 nm of the purified peak of TraFab–Pt–PEG–CPT (27.8 min). The DAR was verified by UV spectroscopy as 2. (c) A representative HIC profile at 280 nm of TraFab–mal–PEG–CPT (DAR: 0.65 by UV) demonstrating heterogeneity with the presence of both DAR1 and DAR2 species. The DAR was calculated to be 0.59 by HIC. (1) Unconjugated Fab; (2) TraFab–Pt–PEG–CPT (DAR: 2); (3) TraFab–Pt–PEG–CPT (DAR: 2); (4) TraFab–mal–PEG–CPT (DAR: 1); (5) TraFab–mal–PEG–CPT (DAR: 2). | ||
Stability of the ADCs
One of the critical features that contributes to the design of a successful ADC is a linker that is stable during circulation, yet able to specifically release the payload efficiently at the target site. It is known that maleimide-based ADCs tend to exchange the linker–drug with the free thiol present on albumin, resulting in off-target effects and a depletion of ADC levels in vivo.29 This instability is due to a retro-Michael reaction that regenerates the reactive maleimide (Scheme 3a). To investigate the stability of the ADCs in the presence of human serum albumin (HSA), both cetuximab ADCs, Ctx–Pt–PEG–CPT and Ctx–mal–PEG–CPT, and both trastuzumab ADCs, Tra–Pt–PEG–CPT and Tra–mal–PEG–CPT, were incubated in the presence of HSA. Albumin was purified and the sample was assayed by SE-HPLC with inline fluorescence monitoring. As camptothecin is known to non-covalently bind to albumin,30 isopropanol was used in the mobile phase for dissociating loosely bound CPT from albumin which eluted after 8 min on the SEC column. Albumin showed a retention time of 6.4 min on the SEC column with inherent fluorescence which was subtracted as background during calculations (ESI† Fig. S2). After 72 h of incubation, the fluorescence peak intensity of albumin was significantly higher in the case of the maleimide-based ADC as compared to the Pt-based ADC (Fig. 4). Our findings in the present study demonstrated that there was at least a 4-fold higher linker–drug exchange to albumin with the maleimide-based ADC than with the Pt-based ADC, hence validating the superior stability of the Pt(II) linker technology (Scheme 3b).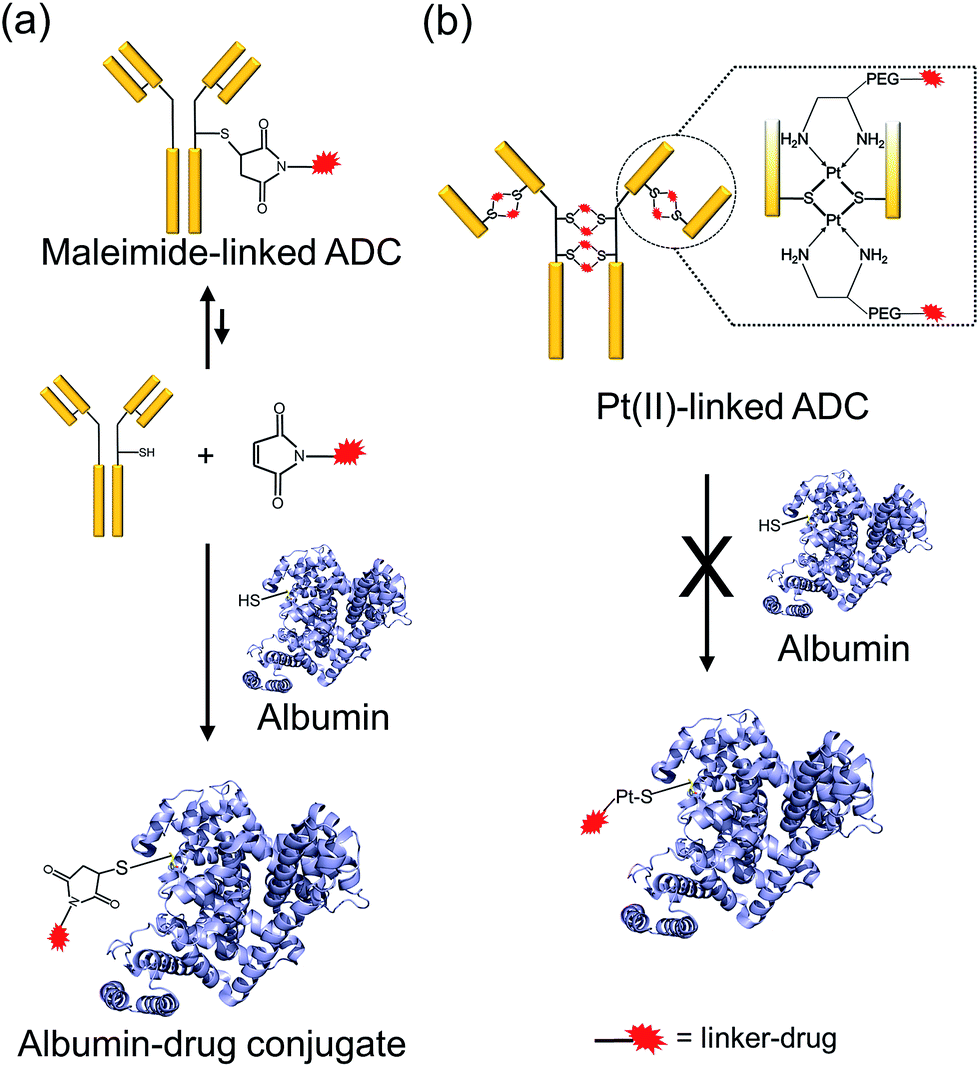 | ||
| Scheme 3 ADC stability in the presence of albumin. (a) Maleimide linkers undergo a retro-Michael reaction regenerating the maleimide which can react with the free thiol present on albumin. (b) Pt(II)-linker is significantly less reactive towards the albumin thiol. | ||
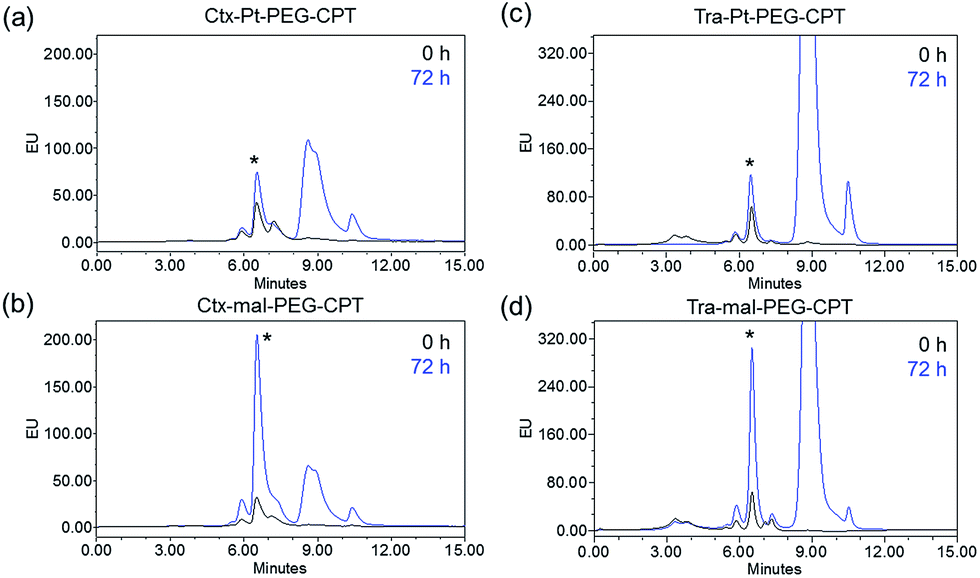 | ||
| Fig. 4 Stability in the presence of albumin. An overlay of the SE-HPLC fluorescence chromatogram of the albumin fraction post-incubation with (a) Ctx–Pt–PEG–CPT, (b) Ctx–mal–PEG–CPT, (c) Tra–Pt–PEG–CPT and (d) Tra–mal–PEG–CPT at 0 h and 72 h demonstrates that at least 4-fold more linker–drug attaches to albumin covalently in the case of mal–PEG–CPT when compared with Pt–PEG–CPT. (*): albumin peak; the peaks eluting post 8 min are unconjugated free low molecular weight species. | ||
The conventionally used maleimide linkers reduce the structural stability of the antibody due to the absence of covalent inter-chain bonds.29 This results in the separation of the heavy chain from the light chain of the antibody when analyzed on a denaturing SDS-PAGE gel. However, with the Pt(II) linker, we observe that the heavy chain and light chain of all the ADCs, whether with the whole antibody or with the Fab fragment, do not separate on the gel and the majority of the ADC appears as a single band corresponding to the intact molecular weight (Fig. 5; Lane 3 vs. Lane 4). The bands in the ADC lanes (Fig. 5; Lane 3 and Lane 4) show fluorescence when the gel is viewed under UV light, ensuring that the CPT remains conjugated to the antibody on the gel. This data suggests the presence of strong interactions between the antibody chains in Pt(II)–ADC unlike in maleimide–ADC thereby preserving the higher order structural stability.
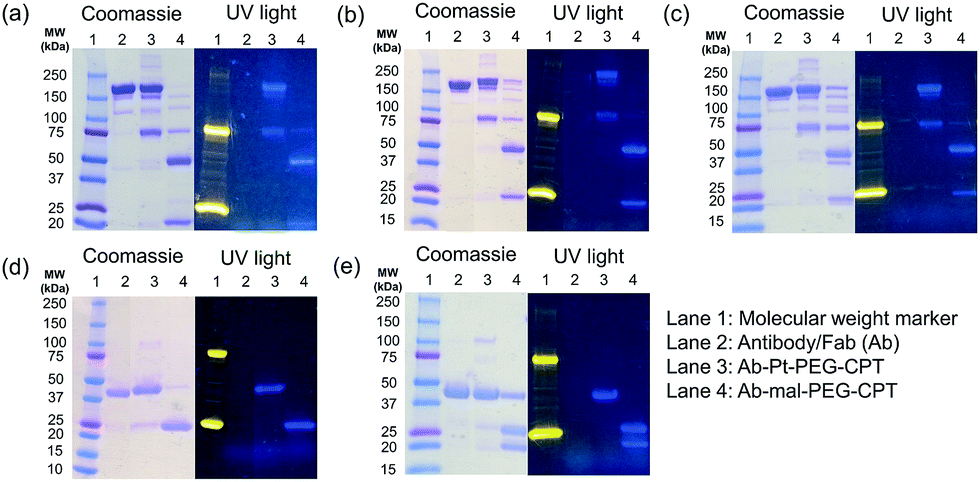 | ||
| Fig. 5 Higher-order structural stability. SDS-PAGE gel (Coomassie-stained) under non-reducing conditions, showing that the antibody light chain and heavy chain are held together by strong forces via the Pt(II) linker. Fluorescence corresponding to CPT can be visualized on the gel under UV light. (a) Cetuximab, (b) trastuzumab, (c) rituximab, (d) trastuzumab Fab and (e) cetuximab Fab. | ||
Encouraged by the stability of the Pt-based ADC in the presence of albumin and its conserved higher order structure, we next assessed its stability in the presence of human and mouse plasma. Instability of the linker technology during circulation in the blood stream would result in the premature release of the drug, leading to dose-limiting side effects. Ctx–Pt–PEG–CPT and Tra–Pt–PEG–CPT were incubated in human and mouse plasma at 37 °C and samples were withdrawn at various time-points. Extracted camptothecin species from the samples were analyzed by reverse-phase HPLC (RP-HPLC) using a fluorescence detector and the platinum content in the samples was measured by Pt–AAS. The stability of Ctx–Pt–PEG–CPT and Tra–Pt–PEG–CPT in human plasma was demonstrated by an extended half-life of 94 h and 133 h, respectively, in vitro (Fig. 6a and c) which is a significant improvement when compared with IMMU-132, a maleimide-based ADC with SN-38 (camptothecin derivative) in clinical trials, having a half-life of only 24 h in human serum.31 In mouse plasma, the half-lives were 12.4 h and 9.01 h for Ctx–Pt–PEG–CPT and Tra–Pt–PEG–CPT respectively (Fig. 6b and c). The lower half-life in mouse plasma is expected due to the presence of elevated levels of esterase.32 In one report, for an ADC, a 70-fold lower half-life of only 4 h was observed in mouse plasma than in human plasma.33 It is also interesting to note that only CPT and not Pt–PEG–CPT was detectable by HPLC in all samples and no platinum was detected in any of the samples by Pt-AAS, which suggests that Pt–PEG–CPT does not release from the ADC and the cleavage is at the ester bond only. This further establishes that the Pt-based ADC can resist even the harsh environment of the plasma.
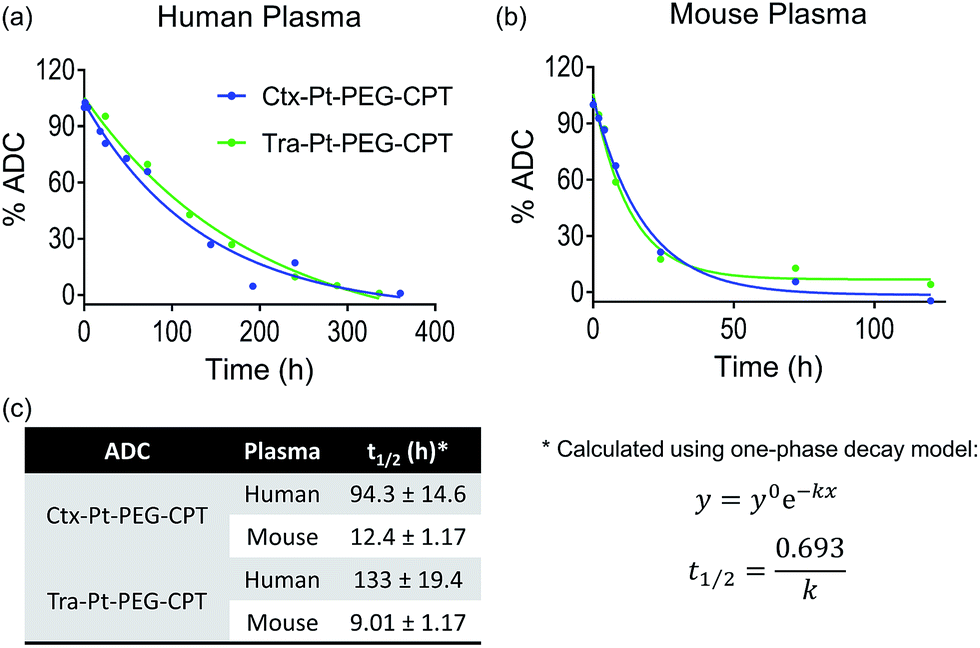 | ||
| Fig. 6 Plasma stability. The stability of Ctx–Pt–PEG–CPT and Tra–Pt–PEG–CPT in (a) human plasma and (b) mouse plasma demonstrates the superior stability of the Pt-based conjugation with extended half-lives (c). | ||
Biological characterization of the ADCs
The goal of any ADC is to combine the specificity of the antibody with the potency of the drug which was elucidated by in vitro cytotoxicity assays in EGFR or HER2-amplified cell lines with Ctx–Pt–PEG–CPT and Tra–Pt–PEG–CPT respectively. The non-targeted effects of conjugates were assessed on cell lines expressing low levels of antigen. Our findings revealed that the conjugates of Pt–PEG–CPT with both cetuximab and trastuzumab, have demonstrated excellent specificity and potency in vitro. Ctx–Pt–PEG–CPT treatment resulted in a 118-fold lower IC50 in MDA-MB-468 (EGFR high) as compared to in MCF-7 (EGFR low) (Fig. 7a) while Tra–Pt–PEG–CPT showed a 327-fold lower IC50 in SK-BR-3 (HER2 high) than in MDA-MB-453 (HER2 low) (Fig. 7b). We evaluated the potency in several other cell lines which showed similar trends (Fig. 7c). These results indicate the specificity of the present ADCs towards the target cells and that the payload is released from the linker upon internalization in the active form.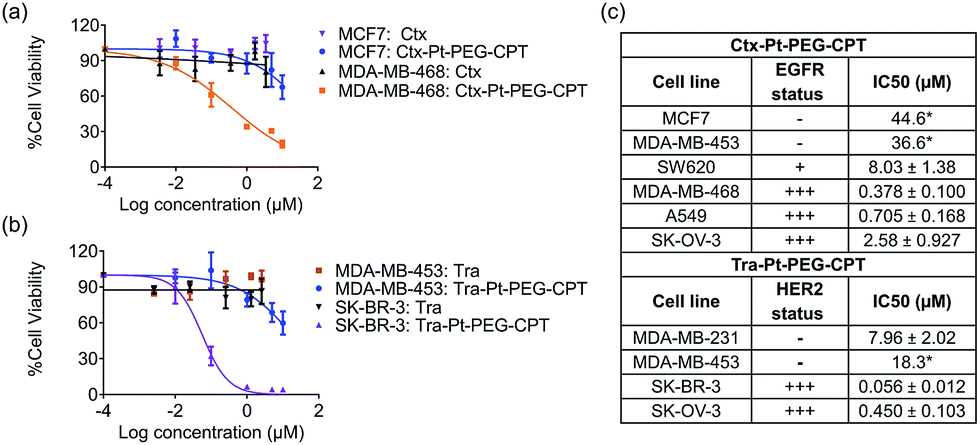 | ||
| Fig. 7 In vitro potency of ADCs. Cell viability was assessed using an MTT assay and IC50 values were calculated. (a) The Ctx–Pt–PEG–CPT conjugate showed a 118-fold increase in anti-proliferative activity on EGFR-high cells (MDA-MB-468) as compared to EGFR-low cells (MCF7). (b) The Tra–Pt–PEG–CPT conjugate showed a 327-fold increase in anti-proliferative activity on HER2-high cells (SK-BR-3) as compared to on HER2-low (MDA-MB-453) cells. Error bars represent standard error of the mean for at least 3 independent experiments. (c) IC50 values (drug-equivalent concentration of ADC) obtained for various cell lines demonstrate the specificity and potency of the ADCs. Standard errors reported from at least 3 independent experiments. *Extrapolated values. | ||
We next tested the specificity and potency of the Pt-based ADC in vivo in a A549 non-small cell lung cancer (NSCLC) model wherein a tumour growth inhibition (TGI) of 55% was achieved as compared to the control (Fig. 8a). Up to 10 doses of 20 mg kg−1 Ctx–Pt–PEG–CPT could be administered without any loss in body weight (Fig. 8b), suggesting a higher therapeutic window of the ADC. A TUNEL assay demonstrated that the tumour sections from ADC-treated mice had ∼80% apoptotic cells, while sections from those treated with cetuximab had only ∼20% apoptotic cells (Fig. 8c and d).
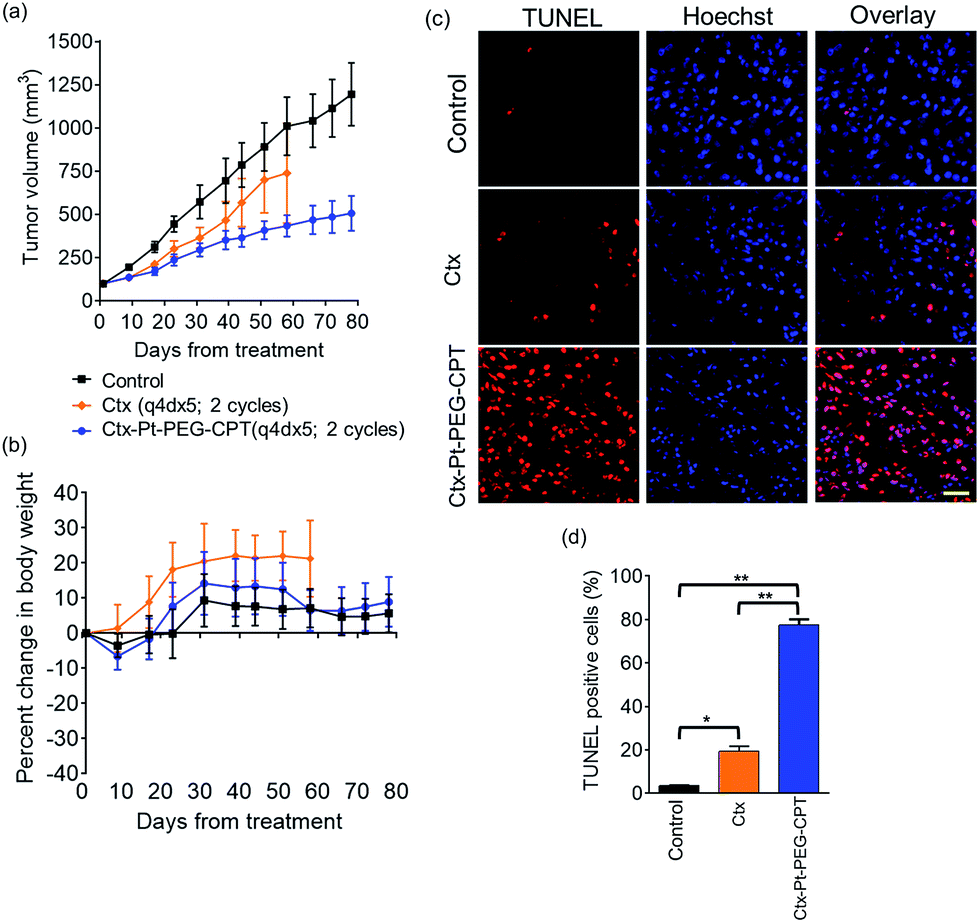 | ||
| Fig. 8 In vivo efficacy. Antitumour activity of Ctx–Pt–PEG–CPT was evaluated in an A549 xenograft model in SCID Beige mice. The mice were implanted with 5 × 106 A549 cells and grouped as untreated (control), cetuximab (Ctx) or Ctx–Pt–PEG–CPT (ADC) on a dosing schedule of q4dx5 for 2 cycles at 20 mg kg−1 of test article. (a) The Ctx–Pt–PEG–CPT treated animals showed significant differences in tumour volume compared with the control group (P < 0.016), with a tumour growth inhibition (TGI) of 55%. (b) No significant difference in body weight was observed across the groups. (c) Tumour sections examined using a TUNEL assay (TUNEL, TMR-Red; Hoechst, blue) show a higher number of apoptotic cells in ADC treated tumours. Scale bar, 20 μm. (d) The percentage of TUNEL-positive cells in ADC treated tumour sections is significantly higher than those observed for cetuximab treated samples. **p < 0.0001; *p < 0.005. | ||
Discussion
ADCs hold the promise of revolutionizing cancer treatment by increasing antitumour efficacy while overcoming the collateral damage associated with conventional chemotherapeutic drugs via targeted delivery. Although the approvals of ado-trastuzumab emtansine and brentuximab vedotin in this decade have set the tone for the future of ADCs,1 adverse effects are known to occur before the optimal therapeutic dose is reached with these drugs, hence limiting their clinical outcome. In order to widen the therapeutic window, strategies involving target selection, innovative linker technologies and payload potency need to evolve to design the ideal “magic bullet”. Here we report a novel Pt(II) based linker that re-bridges the antibody chains post reduction and provides a facile alternative to address the key issues of ADC linkers. The presence of strong interactions between the antibody chains in the Pt-based ADC preserves the higher order structural stability when compared to the maleimide-based ADC, in which non-covalent forces exist as elucidated with three different antibodies. The labile C–S bond in the case of maleimide, is overcome by the strength of the Pt–S interaction imparting the necessary stability to the ADC. Additionally, for any IgG1, the location of inter-chain cysteines is conserved and hence, the attachment of the linker–drug is site-specific and the high efficiency of the Pt–S reaction ensures that the resulting ADC is homogeneous.While the premature release of the linker–drug from the antibody would result in off-target effects, a very stable linker would be non-efficacious. Thus, careful selection and subsequent optimization of the payload and linker–payload combination is required to generate ADCs with maximal therapeutic efficacy. Another crucial factor for achieving success at the clinic is the requirement for the production of homogeneous ADCs, which may even become a prerequisite for future FDA approvals. However, ADCs prepared through conventional linker technologies, such as conjugation to lysines of the antibody, exhibit heterogeneity with multiple species, which is addressed by the Pt-based re-bridging of the disulfides on defined cysteines. On the other hand, conjugation to cysteines via maleimides can lead to the exchange of the linker with the free thiol present on albumin, thus resulting in off-target effects and a depletion of ADC levels in vivo. Since albumin is the most abundant protein in plasma and is ubiquitously taken up by all cells, this poses a significant challenge, especially when picomolar potency drugs are employed. Interestingly, our findings in the present study demonstrate that in comparison to the Pt-linker, at least four times more linker–drug exchange to albumin occurs in the case of the maleimide-linked ADC, indicating that the Pt(II) linker technology could lead to more of the intact ADC being available for delivery to cancer cells in vivo. The increased stability is consistent with the high affinity that platinum has for sulfur. It is well known that in the case of chemotherapeutic platinum drugs, such as cisplatin, a major fraction of the administered drug binds to sulfur-containing molecules such as glutathione, albumin, methionine etc., and is unavailable to reach and bind to its target (DNA).13 Indeed, sulfur-containing molecules, such as WR-2721, are used as chemoprotectants for Pt–drugs due to their ability to quench excess drug and alleviate toxicity.13 The Pt-linked conjugate described here already employs a strong Pt–S interaction thereby preventing competition from other sulfur-containing molecules and providing it extra stability during circulation. Additionally, the potency of the ADC in the in vitro studies, together with the in vivo efficacy, implies that the chemotherapeutic drug is optimally released within the cell. Taken together, our results indicate that the Pt-based ADC platform meets the requirement of stability in addition to homogeneity.
The stability of the linker coupled with the choice of payload is instrumental in determining the therapeutic window.34,35 The initial development with ADCs used nanomolar range cytotoxics such as doxorubicin, which did not succeed in translating to clinical development. It was speculated that the low potency of the drug was responsible for the lack of efficacy. It was later discovered that the main reason for its failure was the choice of the antigen rather than the cytotoxic itself.36,37 The current trend with ADCs is to employ drugs effective in a sub-nanomolar range such as calicheamicins, maytansinoids and auristatins.1 However, using such highly potent cytotoxics implies that even small amounts of the drug leaching out from the ADC can result in significant off-target toxicity, as was observed in Mylotarg™, an ADC that employs a low-stability pH-sensitive linker with calicheamicin as the payload, which was withdrawn from the market due to toxicity issues.34 With a better choice of antigens and antibodies and the optimization of the linker technology, it has been realized that drugs with nanomolar potency, such as camptothecin, could also prove efficacious. In fact, ADCs employing SN-38, a camptothecin derivative, are in phase 1/2 of clinical development1 and offer the advantage of a wider therapeutic window.37 Indeed, we observed that we could administer 10 doses of our ADC at 20 mg kg−1 without any visible adverse effects, potentially arising from the enhanced stability of the Pt–S interaction and the use of a nanomolar potency drug in camptothecin.
It has been reported that an ADC with a DAR of 8 is less effective in vivo than one with a DAR of 4,23 which has led to the norm of targeting a DAR of 4 for an ADC. However, recent findings suggest that by reducing the hydrophobicity of the ADC, the ADC with a DAR of 8 performs much better in vivo19 and is a feasible alternative. Thus, the higher DAR would not be a disadvantage if the linker was made hydrophilic by the use of PEG, for example, to alter the pharmacokinetic properties. For instance, the Fleximer technology conjugates up to 20 drug molecules per antibody without concerns due to the presence of a very hydrophilic polymer.38 With the Pt(II) linker, as it is possible to achieve a maximum DAR of 8, we included a PEG chain in our linker-design to compensate for the drug's hydrophobicity, which decreased aggregation of the resulting ADCs. If more hydrophobic drugs were to be employed or higher DARs were desired, higher chain length of PEG could be built-in to the design. Pt(II)-linked ADCs thus allow the flexibility of higher loading of drug per antibody resulting in an enhanced delivery of the payload. Overall, higher-DAR ADCs coupled with a stable linker like Pt(II) and a nanomolar potency drug such as camptothecin would provide a wider therapeutic window, and can potentially circumvent the use of extremely potent cytotoxics that are associated with dose-limiting toxicities in the clinic.
Conclusions
In summary, we have envisioned a novel platform technology using Pt(II) as a linker, which can be applied to generate stable ADCs that are effective in vitro as well as in vivo with a broad range of antibodies. Pt(II) provides a facile avenue to not only conjugate drugs to an antibody but also could be easily extended to the broad spectrum of bioconjugation processes in general. Most biomolecules possess amenable groups such as the thiols of the cysteines that would have strong interaction with Pt(II), thereby allowing for stable coupling products. The Pt–S interaction as employed here would add to the repertoire of robust linkers available for designing novel and efficient cancer therapeutics.Acknowledgements
We thank Dr Sudip Roy for critically reviewing the manuscript. This work was partially supported by grants from the DBT-SBIRI program, Government of India (BT/SBIRI/879/33-B17/2011). The authors gratefully acknowledge use of the services and analytical facilities of IISER Kolkata, funded by IISER-IOPL Technical grant.Notes and references
- B. E. de Goeij and J. M. Lambert, Curr. Opin. Immunol., 2016, 40, 14–23 CrossRef CAS PubMed.
- P. D. Senter and E. L. Sievers, Nat. Biotechnol., 2012, 30, 631–637 CrossRef CAS PubMed.
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS PubMed.
- M. T.-J. Kim, Y. Chen, J. Marhoul and F. Jacobson, Bioconjugate Chem., 2014, 25, 1223–1232 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- R. S. Schwartz, N. Engl. J. Med., 2004, 350, 1079–1080 CrossRef CAS PubMed.
- C. R. Behrens and B. Liu, mAbs, 2014, 6, 46–53 CrossRef PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 1–7 Search PubMed.
- X. Wang and Z. Guo, Anti-Cancer Agents Med. Chem., 2007, 7, 19–34 CrossRef.
- P. Papadia, N. Margiotta, A. Bergamo, G. Sava and G. Natile, J. Med. Chem., 2005, 48, 3364–3371 CrossRef CAS PubMed.
- G. Aullón, G. Ujaque, A. Lledós and S. Alvarez, Chem.–Eur. J., 1999, 5, 1391–1410 CrossRef.
- J. M. Gonzales, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 4908–4914 CrossRef CAS.
- T. Zimmermann, M. Zeizinger and J. V. Burda, J. Inorg. Biochem., 2005, 99, 2184–2196 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 95th edn, 2014 Search PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 8, 1–4 Search PubMed.
- H. Zhao, C. Lee, P. Sai, Y. H. Choe, M. Boro, A. Pendri, S. Guan and R. B. Greenwald, J. Org. Chem., 2000, 65, 4601–4606 CrossRef CAS PubMed.
- S. V. Govindan and D. M. Goldenberg, Expert Opin. Biol. Ther., 2012, 12, 873–890 CrossRef CAS PubMed.
- E. Gabano, E. Marengo, M. Bobba, E. Robotti, C. Cassino, M. Botta and D. Osella, Coord. Chem. Rev., 2006, 250, 2158–2174 CrossRef CAS.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS PubMed.
- D. C. J. Waalboer, J. a. Muns, N. J. Sijbrandi, R. B. M. Schasfoort, R. Haselberg, G. W. Somsen, H.-J. Houthoff and G. a. M. S. van Dongen, ChemMedChem, 2015, 10, 797–803 CrossRef CAS PubMed.
- D. Patel, A. Lahiji, S. Patel, M. Franklin, X. Jimenez, D. J. Hicklin and X. Kang, Anticancer Res., 2007, 27, 3355–3366 CAS.
- T. Kute, C. M. Lack, M. Willingham, B. Bishwokama, H. Williams, K. Barrett, T. Mitchell and J. P. Vaughn, Cytometry, Part B, 2004, 57A, 86–93 CrossRef CAS PubMed.
- M. Nishida, K. Teshigawara, O. Niwa, S. Usuda, T. Nakamura, P. Ralph, R. Newman and E. A. Padlan, Int. J. Oncol., 1992, 32, 1263–1274 CrossRef.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- T. G. Burke and Z. Mi, J. Med. Chem., 1994, 37, 40–46 CrossRef CAS PubMed.
- D. M. Goldenberg, T. M. Cardillo, S. V. Govindan, E. A. Rossi and R. M. Sharkey, Oncotarget, 2015, 6, 22496–22512 CrossRef PubMed.
- B. Li, M. Sedlacek, I. Manoharan, R. Boopathy, E. G. Duysen, P. Masson and O. Lockridge, Biochem. Pharmacol., 2005, 70, 1673–1684 CrossRef CAS PubMed.
- R. C. Elgersma, R. G. E. Coumans, T. Huijbregts, W. M. P. B. Menge, J. A. F. Joosten, H. J. Spijker, F. M. H. de Groot, M. M. C. van der Lee, R. Ubink, D. J. van den Dobbelsteen, D. F. Egging, W. H. A. Dokter, G. F. M. Verheijden, J. M. Lemmens, C. M. Timmers and P. H. Beusker, Mol. Pharmaceutics, 2015, 12, 1813–1835 CrossRef CAS PubMed.
- M. Gordon, M. Canakci, L. Li, J. Zhuang, B. A. Osborne and S. Thayumanavan, Bioconjugate Chem., 2015, 26, 2198–2215 CrossRef CAS PubMed.
- L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5–13 CrossRef CAS PubMed.
- A. W. Tolcher, S. Sugarman, K. A. Gelmon, R. Cohen, M. Saleh, C. Isaacs, L. Young, D. Healey, N. Onetto and W. Slichenmyer, J. Clin. Oncol., 1999, 17, 478–484 CAS.
- S. V. Govindan, T. M. Cardillo, E. A. Rossi, P. Trisal, W. J. McBride, R. M. Sharkey and D. M. Goldenberg, Mol. Pharmaceutics, 2015, 12, 1836–1847 CrossRef CAS PubMed.
- A. V. Yurkovetskiy, M. Yin, N. Bodyak, C. A. Stevenson, J. D. Thomas, C. E. Hammond, L. Qin, B. Zhu, D. R. Gumerov, E. Ter-Ovanesyan, A. Uttard and T. B. Lowinger, Cancer Res., 2015, 75, 3365–3372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic schemes and characterization data, experimental procedures, Fig. S1 and S2. See DOI: 10.1039/c6sc05149a |
| ‡ All authors are either employees/collaborators with Invictus Oncology Pvt. Ltd. and/or hold equity of IOPL. |
| § J. K. and S. K. contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |