
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective anomeric acetylation of unprotected sugars in water†
David
Lim
 a and
Antony J.
Fairbanks
*ab
a and
Antony J.
Fairbanks
*ab
aDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand. E-mail: antony.fairbanks@canterbury.ac.nz
bBiomolecular Interaction Centre, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand
First published on 11th November 2016
Abstract
High yielding selective acetylation of only the anomeric hydroxyl of unprotected sugars is possible in aqueous solution using 2-chloro-1,3-dimethylimidazolinium chloride (DMC), thioacetic acid, and a suitable base. The reaction, which may be performed on a multi-gram scale, is stereoselective for sugars that possess a hydroxyl group at position-2, exclusively yielding the 1,2-trans products. The use of an iterative reagent addition procedure allows the use of sodium carbonate as the base, avoiding the formation of triethylammonium salts, which may hamper product purification. The glycosyl acetate products may be used as donor substrates for glycosidase-catalysed synthesis. The crude aqueous acetylation reaction mixture may also be used for this purpose.
Introduction
Efficient differentiation between the multiple hydroxyl groups of an unprotected carbohydrate is difficult without the aid of enzymatic catalysis.1 Selective reaction of only the anomeric hydroxyl group is however possible, for example by way of its reactivity as a hemi-acetal as in Fischer glycosylation,2 a process that has remained as the only widely used method for selective protection of the anomeric centre of unprotected sugars since its inception in 1893.However, fundamentally different, yet selective, chemical processes may also be possible. For an unprotected sugar the significant difference between the pKa of the anomeric hydroxyl (pKa ∼ 12.1–12.5)3 and the other less acidic hydroxyl groups means that its selective reaction as a nucleophile can sometimes be effected.4 For example, anomeric O-alkylation, which has been applied as a means of glycoside and oligosaccharide synthesis using partially protected sugars,5 was investigated using completely unprotected sugars by Schmidt in the 1990's.6 However these reactions only provided modest yields of products (typically 50–60%), and required the use of organic solvents such as DMPU; the approach therefore did not find any significant applications.
Regioselective acylation of the anomeric hydroxyl group of a completely unprotected sugar was first reported by Pfander in the 1970's.7 The idea was further developed by Plusquellec8 and co-workers who reported a method for the mono-acylation of only the anomeric hydroxyl of a small selection of unprotected sugars (maltose, glucose, lactose), using a series of activated amides, thio- and arylesters as acylating agents, in the presence of 0.16 equiv. of a base (typically NaH), and pyridine as the reaction solvent. The modest yields for these processes, calculated based on the quantity of acylating agent used, could only be obtained by using an excess of the carbohydrate (typically 3–4 equivalents). Since that point in time, now some 30 years ago, no further reports on selective acylation of the anomeric hydroxyl group have appeared.
Recently the potential utility of performing selective reactions on completely unprotected sugars, particularly in aqueous solution, has once again become the subject of scientific interest. In 2009 Shoda and co-workers introduced the dehydrating reagent 2-chloro-1,3-dimethylimidazolinium chloride 1 (DMC),9 into the carbohydrate field, initially for the direct synthesis of glycosyl oxazolines from reducing sugars in water.10 Subsequently DMC and similar derivatives have been applied to the direct synthesis of glycosyl azides,11 including the one-pot conjugation of unprotected sugars to other species via Click chemistry of glycosyl azides made in situ.12 All of these processes rely on initial nucleophilic attack of the anomeric hydroxyl group on DMC; a process which activates it to subsequent displacement. Further applications of DMC-activation of unprotected sugars have allowed selective nucleophilic substitution processes at the anomeric centre in water, including the synthesis of pyridyl thioglycosides,13 and the linking of peptides to carbohydrates via the side chains of cysteine residues.14
The two key components to the success of these DMC-mediated processes in aqueous solution are: (a) the greater acidity of the anomeric hydroxyl group, which, under the basic reaction conditions may be selectively de-protonated and so can outcompete both solvent water and other sugar hydroxyl-groups as a nucleophile for reaction with DMC; (b) the addition of a good nucleophile following activation of the anomeric hydroxyl, which is itself capable of outcompeting interception of the activated intermediate by both solvent water and other sugar hydroxyl-groups.15
Herein we report a simple method for the synthesis of glycosyl acetates from the corresponding unprotected sugars, remarkably in water as the reaction solvent, which also relies on the use of DMC as an activating agent, but which operates by a different mechanism.
Results and discussion
As part of on-going studies into applications of DMC and derivatives for selective transformations of unprotected sugars in water,16 we found that reaction of DMC 1 with GlcNAc 2 in water, in the presence of an excess of thioacetic acid and triethylamine, unexpectedly led to the formation of the glycosyl acetate 3 in 86% yield, as a mixture of anomers (Scheme 1).‡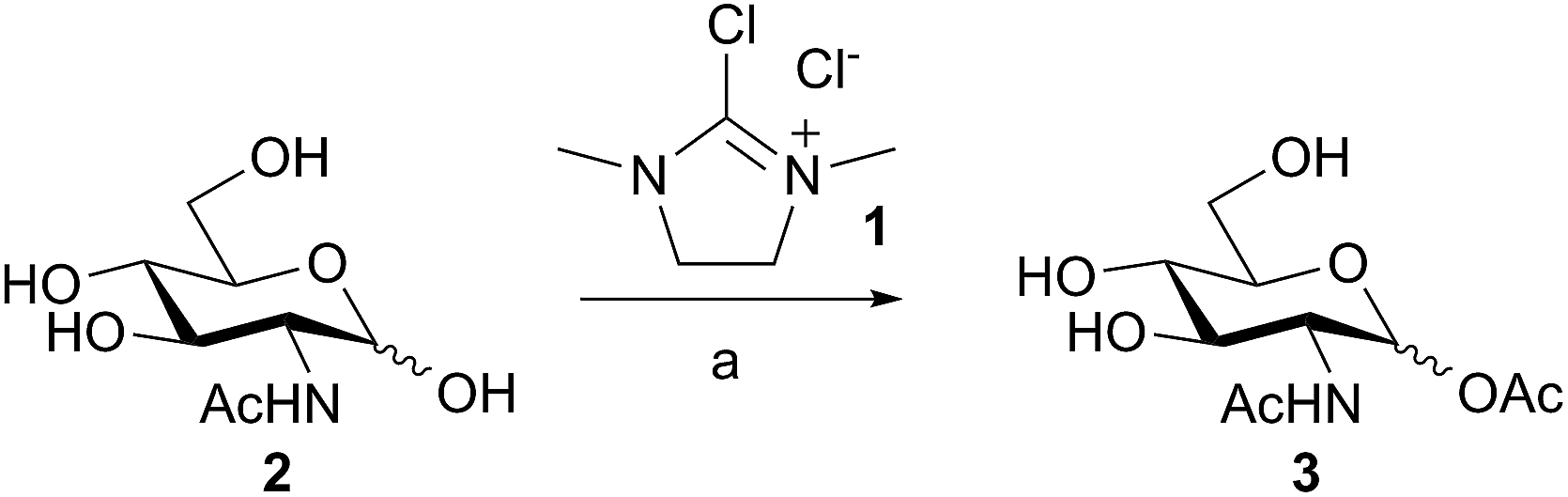 | ||
| Scheme 1 Selective acetylation of the anomeric hydroxyl group of GlcNAc in water. Reagents and conditions: (a) Et3N (10 equiv.), AcSH (5 equiv.), H2O, 0 °C, then 1 (3 equiv.), 30 min, 86%. | ||
Importantly the reaction outcome was dependent on the order of addition of the reagents; in the case in question the DMC was slowly added last to a stirred mixture of GlcNAc 1, Et3N, and thioacetic acid. However if the order was changed then acetylation was not the outcome; for example it was found that if the thioacetic acid was added last (i.e. after the DMC 1) then either a 1,6-anhydro sugar was formed, or, in the case of a 2-acetamido sugar, the corresponding 1,2-trans glycosyl thioacetate was formed. We reasoned that a plausible mechanism for the formation of 3 may involve attack of thioacetate 4 on DMC 1 to first generate a thiouronium ester intermediate 5. Intermediate 5 may then subsequently be attacked by the de-protonated anomeric hydroxyl group of 6 to generate the glycosyl acetate 7 and the thiourea 8 (Fig. 1), which was isolated and characterized as a by-product from these reactions.17
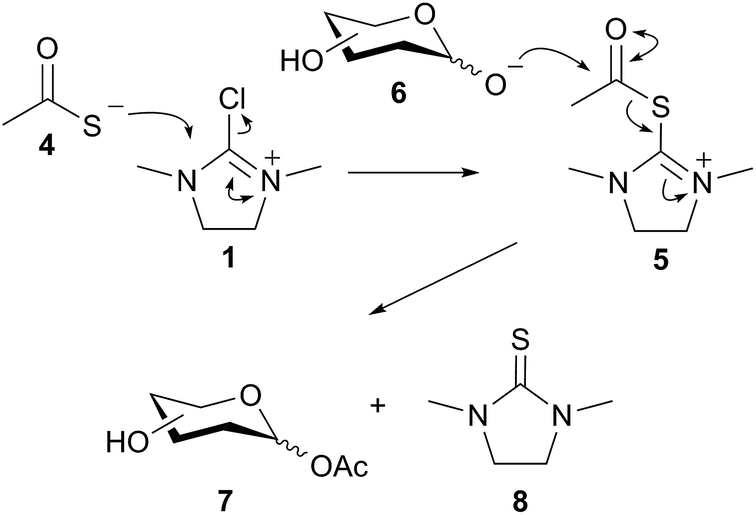 | ||
| Fig. 1 Plausible reaction mechanism. | ||
Further study revealed the process to be quite general; application to a variety of unprotected monosaccharides gave the corresponding glycosyl acetates in good yield (Table 1, entries 1–4, 78–88% yield). Interestingly reactions of sugars with a free hydroxyl group at position-2 were invariably found to be stereoselective, producing only the 1,2-trans products. Contrastingly reactions of 2-acetamido-sugars gave the corresponding glycosyl acetates as a mixture of anomers.18 Although the process was itself highly efficient, as evidenced by 1H NMR and t.l.c. monitoring of reaction progress, a problem was encountered during product isolation. The removal of the large excess of triethylammonium salts formed during the reaction from the highly polar reaction products proved tedious, and, in certain cases, acetate migration and/or hydrolysis was observed during this process. The use of an alternative base and the pH dependence19 of the efficiency of the process were investigated. Initial trials using sodium carbonate, previously applied to 2-chloro-1,3-dimethyl-1H-benzimidazol-3-ium chloride (CDMBI) mediated reactions as a replacement for triethylamine by Shoda,20 only gave very low yields of glycosyl acetate products (<20%). However the use of an iterative reagent addition procedure, similar to that reported by Winssinger et al.14 produced the desired glycosyl acetates in good to high yields.
| Entry | Sugar | Product | Yield/[%] | α/β |
|---|---|---|---|---|
| a Reagents and conditions: sugar, Et3N (10 equiv.), AcSH (5 equiv.), H2O, 0 °C, then 1 (3 equiv.), 30 min. b Reagents and conditions: sugar, Na2CO3 (1.6 equiv.), AcSH (1.6 equiv.), 0 °C, then 1 (1 equiv.); five iterations of reagent addition in total. | ||||
| 1 | GlcNAc 2 |

|
86a | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
| 92b | ||||
| 2 | GalNAc |
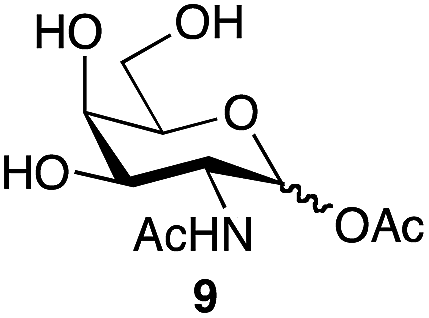
|
88a | 1:1.1 |
| 86b | ||||
| 3 | Galactose |
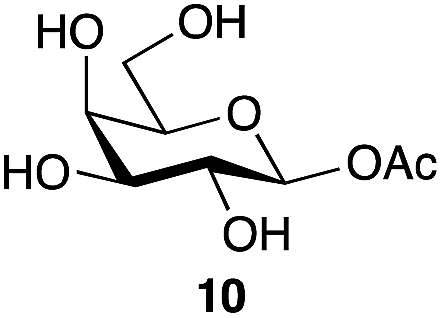
|
78a | β only |
| 81b | ||||
| 4 | Mannose |
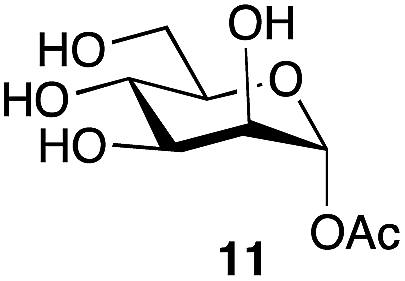
|
82a | α only |
| 79b | ||||
| 5 | Lactose |
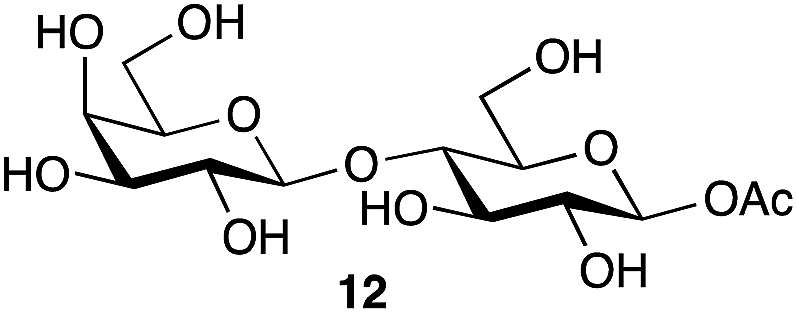
|
77b | β only |
| 6 | Galβ1,3-GalNAc |
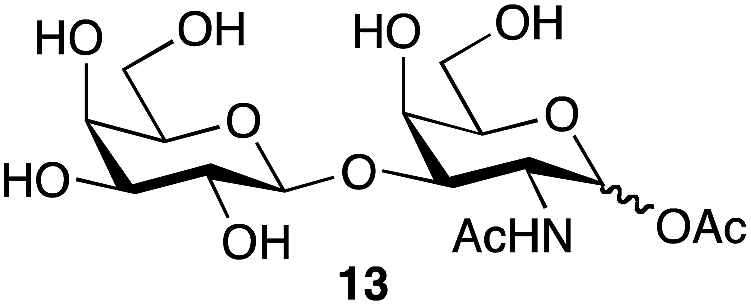
|
87b | 1:1.1 |
This optimized procedure,§ using sodium carbonate as the base, was then applied to number of mono-, and disaccharides (Table 1, entries 1–6). The ready scalability of the process was demonstrated by selective acetylation of mannose on a 5 g scale, to produce the α-acetate 11 in 79% yield (Table 1, entry 4).
With a rapid one-step method for the production of glycosyl acetates in hand, the potential utility of these materials, previously only accessible by multi-step reaction sequences involving selective protecting group manipulations in the presence of a labile glycosyl acetate, was investigated. Shoda and co-workers have previously reported21 the direct production of triazinyl glycosides in water using DMC, and found them to be useful donors for enzyme-catalyzed glycosylation. Mindful of that work, and also the structural similarities between a glycosyl acetate and the enzyme-bound intermediate of hydrolytic reactions catalyzed by retaining glycosidases,22 one area of application appeared to be as possible donor substrates for glycosidase-mediated synthesis. Indeed, Neustroev and co-workers had previously reported the use of the galactose β-acetate 10 as a donor substrate in glycosylations catalyzed by the β-galactosidase from Penicillium sp.23 Interestingly they reported that the galactose β-acetate 10 was hydrolyzed 100-times faster by this β-galactosidase than the corresponding p-nitrophenyl-β-D-galactopyranoside. That finding suggests that glycosyl acetates may in some cases be more active substrates for enzyme catalyzed processes than the currently very widely used p-nitrophenyl glycosides. However, their further study and any potential applications have up to now been limited by the difficulties associated with their synthesis.
We tested the ability of retaining endo-α-N-acetylgalactoaminidases (EC 3.2.1.97) from family GH101 (ref. 24) to use glycosyl acetates as donor substrates. Two enzymes, engBL25 from Bifidobacterium longum and engEF26 from Enterococcus faecalis, were expressed and purified following reported procedures. The pure α-acetates of GalNAc 9α and the O-glycan core 1 disaccharide Galβ1,3-GalNAc 13α, each of which was each separated from the corresponding β-acetate by RP-HPLC, were treated with buffered aqueous methanol (15% v/v) in the presence of both engBL and engEF. In each case, an enzyme-catalyzed reaction27 led to the stereoselective formation of the corresponding α-methyl glycoside, in addition to some hydrolysis (Scheme 2). The ability of engBL to use acetate 9α as a donor substrate contrasts with its reported inability to process the corresponding α-p-nitrophenyl glycoside.25
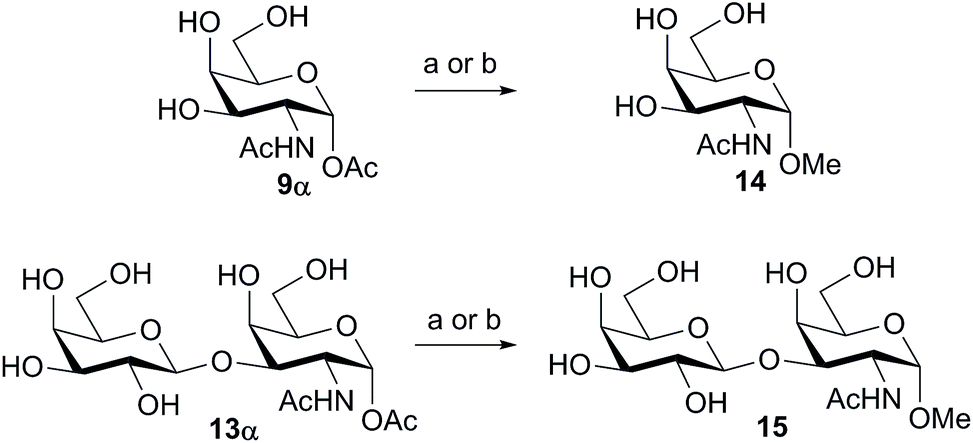 | ||
| Scheme 2 Glycosylation of the α-acetates of GalNAc and the core 1 disaccharide Gal-β1,3-GalNAc with methanol using family GH101 endo-α-N-acetylgalactosaminidases. Reagents and conditions: (a) engBL, 15% (v/v) aqueous MeOH, 25 mM NaOAc, pH 6.0, 37 °C, 16 h; 14, 15%; 15, 38%; (b) engEF, 15% (v/v) aqueous MeOH, 25 mM NaOAc, pH 6.0, 37 °C, 16 h; 14, 40%; 15, 37%. Yields determined by 1H NMR integration of the anomeric and/or OMe protons. | ||
Interestingly the corresponding β-acetates, although of the same anomeric configuration as the enzyme-bound intermediate of these retaining α-galactosidases, were not found to be effective donors for the WT enzymes, presumably due to steric clashes within the enzyme active sites. Glycosyl acetates may however prove to be useful substrates for mutant glycosynthase enzymes in which the catalytic nucleophile has been replaced by a smaller residue.28
The above reactions, catalyzed by family GH101 α-glycosidases, used the α-acetates as the donor species. In the case of 2-acetamido sugars this involved purification and separation of the anomeric mixture that had been produced by the acetylation reaction. However, since the anomeric acetylation of 2-hydroxy sugars was stereoselective, we considered the possibility of directly using the aqueous reaction mixtures containing these crude anomeric acetates for glycosidase-catalyzed reactions. Thus D-glucose 16 was acetylated in aqueous solution using DMC 1, thioacetic acid, and Na2CO3 as the base (Scheme 3) using the iterative addition procedure. The crude reaction mixture containing the intermediate β-glucosyl acetate 17 was then treated with the retaining family GH1 β-glucosidase from almonds29 (EC 3.2.1.21) and methanol (15% v/v) as the acceptor. After 16 h, 1H NMR indicated that the glucosyl acetate 17 had completely disappeared, and the desired methyl β-D-glucopyranoside 18 had been formed, together with some glucose as the result of competitive hydrolysis. This result demonstrates the potential for the direct application of aqueous solutions of glycosyl acetates made in situ as substrates for enzymatic catalyzed glycosylations, obviating the need to handle or purify these highly polar and quite labile materials.
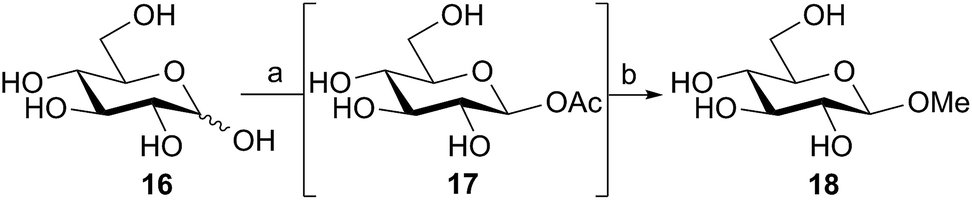 | ||
| Scheme 3 Direct use of β-glucosyl acetate as a substrate for almond β-glucosidase. Reagents and conditions: (a) Na2CO3 (1.6 equiv.), AcSH (1.6 equiv.), 0 °C, then dropwise addition of 1 (1.0 equiv.) dissolved in water, 100% conversion with five iterations; then (b) β-glucosidase from almonds (100 U), 15% (v/v) MeOH, 37 °C, 16 h, 26%. Yields determined by 1H NMR integration of the anomeric and/or OMe protons. | ||
Conclusions
In summary the combination of a suitable base, thioacetic acid, and DMC 1, added in that order, provides a remarkable and unprecedented method for the selective acetylation of the anomeric centre of a variety of sugars in aqueous solution. The procedure is simple, efficient, and may be performed on a multi-gram scale. In addition to providing a novel method for differential protection of carbohydrates, the glycosyl acetate products, now available in a single step from the unprotected parent sugar, are suitable donor substrates for glycosidases. Furthermore, the crude aqueous acetylation reaction mixture may be used for subsequent enzyme-catalyzed processes. The one step synthetic availability of glycosyl acetates by this method may increase their utility as donors for glycosidase-catalyzed synthesis, in addition to opening up their applications as readily accessible materials for other synthetic transformations.Acknowledgements
We thank Prof. Shinya Fushinobu (University of Tokyo, Japan) and Prof. Makoto Ito (Kyushu University, Japan) for their donation of the plasmids for engBL and engEF, respectively, the University of Canterbury (Ph.D Scholarship to D. L.) and the Bimolecular Interaction Centre for financial support.Notes and references
- (a) M. J. X. Jäger and A. J. Minnaard, Chem. Commun., 2015, 52, 656–664 Search PubMed; (b) T. Saloranta and R. Leino, Synlett, 2015, 26, 421–425 CrossRef CAS; (c) D. Lee and M. Taylor, Synthesis, 2012, 3421–3431 Search PubMed.
- (a) E. Fischer, Ber. Dtsch. Chem. Ges., 1893, 26, 2400–2412 Search PubMed; (b) B. Capon, Chem. Rev., 1969, 69, 407–498 Search PubMed.
- (a) S. Feng, C. Bagia and G. Mpourmpakis, J. Phys. Chem. A, 2013, 117, 5211–5219 Search PubMed; (b) E. P. Sergeant and B. Dempsey, Ionisation Constants of Organic Acids in Aqueous Solution; IUPAC Chemical Data Series No. 23, Pergamon Press, New York, 1979, pp. 175–177 Search PubMed; (c) J. J. Christensen, J. H. Rytting and R. M. Izatt, J. Chem. Soc. B, 1970, 1646–1648 Search PubMed; (d) R. M. Izatt, J. H. Rytting, L. D. Hansen and J. J. Christensen, J. Am. Chem. Soc., 1966, 88, 2641–2643 Search PubMed.
- I. F. Pelyvás, T. K. Lindhorst, H. Streicher and J. Thiem, Synthesis, 1991, 1991, 1015–1018 Search PubMed.
- For some examples see (a) D. Zhu, K. N. Baryal, S. Adhikari and J. Zhu, J. Am. Chem. Soc., 2014, 136, 3172–3175 Search PubMed; (b) D. A. Ryan and D. Y. Gin, J. Am. Chem. Soc., 2008, 130, 15228–15229 Search PubMed; (c) R. R. Schmidt, Angew. Chem., Int. Ed., 1986, 25, 212–235 Search PubMed; (d) R. R. Schmidt and J. Michel, Tetrahedron Lett., 1984, 25, 821–824 CrossRef CAS.
- (a) R. R. Schmidt and W. Klotz, Synlett, 1991, 168–170 Search PubMed; (b) Y. E. Tsvetkov, W. Klotz and R. R. Schmidt, Liebigs Ann. Chem., 1992, 1992, 371–375 Search PubMed.
- (a) H. Pfander and F. Witter, Helv. Chim. Acta, 1979, 62, 1944–1951 Search PubMed; (b) H. Pfander, M. Läderach and F. Witter, Helv. Chim. Acta, 1980, 63, 277–283 Search PubMed; (c) H. Pfander and M. Läderach, Carbohydr. Res., 1982, 99, 175–179 CrossRef CAS.
- D. Plusquellec, F. Roulleau, F. Bertho, M. Lefeuvre and E. Brown, Tetrahedron, 1986, 42, 2457–2467 Search PubMed.
- (a) T. Isobe and T. Ichikawa, J. Org. Chem., 1999, 64, 6984–6988 Search PubMed; (b) T. Isobe and T. Ichikawa, J. Org. Chem., 1999, 64, 6989–6992 Search PubMed.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S.-I. Shoda, J. Org. Chem., 2009, 74, 2210–2212 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. I. Kobayashi and S.-I. Shoda, Chem. Commun., 2009, 3378–3379 Search PubMed.
- (a) D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907–11911 Search PubMed; (b) D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., 2014, 126, 12101–12105 CrossRef.
- N. Yoshida, M. Noguchi, T. Tanaka, T. Matsumoto, N. Aida, M. Ishihara, A. Kobayashi and S.-I. Shoda, Chem.–Asian J., 2011, 6, 1876–1885 Search PubMed.
- A. Novoa, S. Barluenga, C. Serba and N. Winssinger, Chem. Commun., 2013, 49, 7608–7610 Search PubMed.
- DMC activation of unprotected mono- and oligosaccharides may also be used to access 1,6-anhydro sugars in a process that is often observed as a side reaction to the attack of external nucleophiles at the anomeric centre. See: T. Tanaka, W. C. Huang, M. Noguchi, A. Kobayashi and S.-I. Shoda, Tetrahedron Lett., 2009, 50, 2154–2157 Search PubMed.
- S. R. Alexander and A. J. Fairbanks, Org. Biomol. Chem., 2016, 14, 6679–6682 Search PubMed.
- Spectral data for thiourea 8 matched that previously reported see: M. K. Denk, S. Gupta, J. Brownie, S. Tajammul and A. J. Lough, Chem.–Eur. J., 2001, 7, 4477–4486 Search PubMed.
- The reason for the difference in stereochemical outcome is not yet clear, particularly since mutarotation of 2-acetamido and 2-hydroxy sugars is expected to proceed at similar rates under the reaction conditions. See: D. M. L. Morgan and A. Neuberger, Proc. R. Soc. London, Ser. A, 1974, 337, 317–332 Search PubMed.
- See ESI† (Fig. 4 and 5).
- M. Noguchi, T. Fujieda, W. C. Huang, M. Ishihara, A. Kobayashi and S.-I. Shoda, Helv. Chim. Acta, 2012, 95, 1928–1936 Search PubMed.
- (a) T. Tanaka, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Commun., 2008, 2016–2018 Search PubMed; (b) M. Noguchi, M. Nakamura, A. Ohno, T. Tanaka, A. Kobayashi, M. Ishihara, M. Fujita, A. Tsuchida, M. Mizuno and S.-I. Shoda, Chem. Commun., 2012, 48, 5560–5562 RSC.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 Search PubMed.
- A. Zinin, E. Eneyskaya, K. Shabalin, A. Kulminskaya, S. Shishlyannikov and K. Neustroev, Carbohydr. Res., 2002, 337, 635–642 Search PubMed.
- http://www.cazy.org/GH101.html .
- K. Fujita, F. Oura, N. Nagamine, T. Katayama, J. Hiratake, K. Sakata, H. Kumagai and K. Yamamoto, J. Biol. Chem., 2005, 280, 37415–37422 Search PubMed.
- H. M. Goda, K. Ushigusa, H. Ito, N. Okino, H. Narimatsu and M. Ito, Biochem. Biophys. Res. Commun., 2008, 375, 541–546 Search PubMed.
- Control reactions performed in the absence of enzyme did not result in the formation of any methyl glycosides.
- P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784–1794 Search PubMed.
- S. He and S. G. Withers, J. Biol. Chem., 1997, 272, 24864–24867 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of all compounds and spectra. See DOI: 10.1039/c6sc04667c |
| ‡ Typical experimental procedure using triethylamine as base: N-acetyl-D-glucosamine 2 (500 mg, 2.26 mmol), thioacetic acid (0.8 mL, 11.3 mmol, 5 equiv.), and triethylamine (3.1 mL, 22.6 mmol, 10 equiv.) were stirred in H2O (10 mL), and the mixture was cooled to 0 °C. DMC 1 (1.2 g, 6.78 mmol, 3 equiv.) was then added portion-wise. After 30 min, t.l.c. (CHCl3/MeOH, 2:1) indicated complete consumption of starting material (Rf 0.2) and the formation of a major product (Rf 0.5). The reaction mixture was then diluted with water (20 mL), washed with DCM (5 × 20 mL), concentrated to 1 mL, filtered through a column of Amberlite® IR120 (H+ form, previously converted to the Na+ form by washing with 1 M aqueous NaOH), and concentrated in vacuo. Purification by flash column chromatography (CHCl3:MeOH, 5:1) gave 1-acetyl-2-acetamido-2-deoxy-D-glucopyranoside 3 (510 mg, 86%) as a white solid (α/β, 3:2). |
| § Typical experimental procedure using Na2CO3 as base: D-mannose (5 g, 27.8 mmol) and Na2CO3 (4.70 g, 44.4 mmol, 1.6 equiv.) were dissolved in water (100 mL), the reaction mixture stirred, and cooled to 0 °C. Thioacetic acid (3.1 mL, 44.4 mmol, 2 equiv.) was then added, followed by the drop-wise addition of DMC 1 (4.70 g, 27.8 mmol, 1 equiv., dissolved in water [10 mL]). The reaction mixture was stirred for 15 min, before Na2CO3 (4.70 g, 44.4 mmol, 1.6 equiv.) and thioacetic acid (0.31 mL, 44.4 mmol, 1.6 equiv.) were added, followed by the drop-wise addition of DMC 1 (4.70 g, 27.8 mmol, 1 equiv., dissolved in water [10 mL]). This stepwise addition was repeated another three times (i.e. a total of 5 iterations), before the reaction mixture was diluted with water, washed with DCM, and lyophilized. Purification by flash column chromatography (10:1 CHCl3:MeOH → 5:1 CHCl3:MeOH) afforded acetyl α-D-mannopyranoside 11 (4.5 g, 79%) as a white foam. |
| This journal is © The Royal Society of Chemistry 2017 |