
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free O–H/C–H difunctionalization of phenols by o-hydroxyarylsulfonium salts in water†
Dengfeng
Chen
,
Qingyuan
Feng
,
Yunqin
Yang
,
Xu-Min
Cai
,
Fei
Wang
and
Shenlin
Huang
*
College of Chemical Engineering, Jiangsu Key Lab of Biomass-Based Green Fuels and Chemicals, Nanjing Forestry University, Nanjing, 210037, P. R. China. E-mail: shuang@njfu.edu.cn
First published on 4th November 2016
Abstract
An environmentally benign method for C–H/O–H difunctionalization of phenols with sulfoxides under mild conditions has been developed. The reaction process is mediated by an electrophilic aromatic substitution and subsequent selective aryl or alkyl migration, involving C–S and C–O bond formations with broad substrate scope.
Introduction
Carbon–heteroatom bond forming reactions are one of the most important tools used in pharmaceutical chemistry, medicinal chemistry, and material sciences.1 In particular, transition metal catalyzed C–O or C–S cross-couplings,2 have been widely used to construct important families of target molecules, aryl ethers, sulfides and sulfoxides.3 Traditionally, these reactions are carried out in organic solvents (e.g., THF, CH3CN, DMSO, and DMF), which may cause potential health, safety, and waste disposal issues.4 Therefore, transition-metal-free C–O and C–S coupling reactions5 under greener conditions are desirable, especially in the pharmaceutical industry.6 Herein, we report our efforts toward a novel environmentally benign O–H/C–H difunctionalization of phenols by o-hydroxyarylsulfonium salts in water (Scheme 1).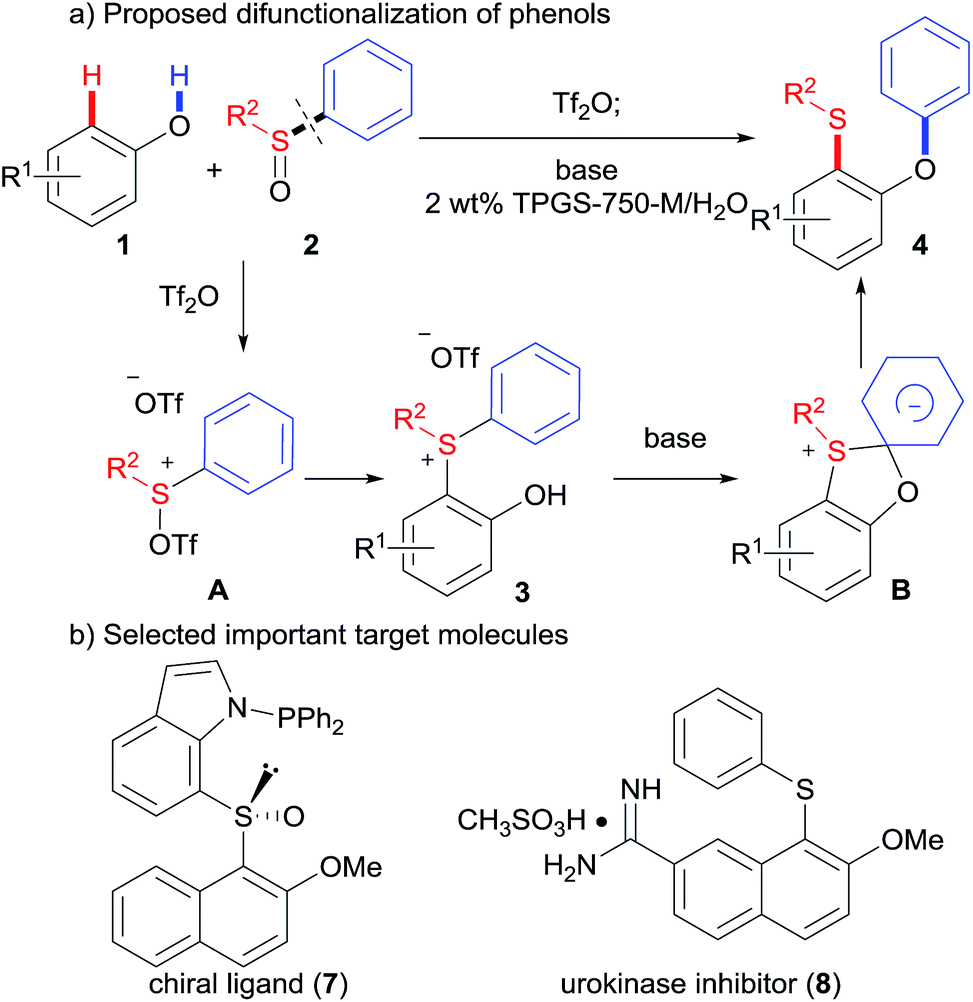 | ||
| Scheme 1 Proposed difunctionalization of phenols. | ||
Our reaction design is detailed in Scheme 1. It is reasonable to hypothesize that sulfoxides 2 may be activated using a sulfonylating agent like trifluoromethanesulfonic anhydride (Tf2O), inspired by the Pummerer reaction.7 The activated sulfoxide A could be attacked by phenols 1 at sulfur, giving rise to o-hydroxyarylsulfonium intermediates 3. Such intermediates might then undergo a Smiles-like rearrangement,8 thus affording versatile products o-(phenoxy)aryl sulfides, 4, which could be readily converted to many important ligands9 and biologically active molecules10 (Scheme 1b). However, the following challenges in this strategy were anticipated: (1) the activated sulfoxide can be attacked by the phenolic hydroxyl group at the cationic sulfur, according to previous studies;11 (2) it may require one electron-withdrawing group (e.g., NO2) to activate the migration of the aromatic ring in the Smiles rearrangement; (3) such rearrangement of sulfonium salts has been seldom reported.12
Results and discussion
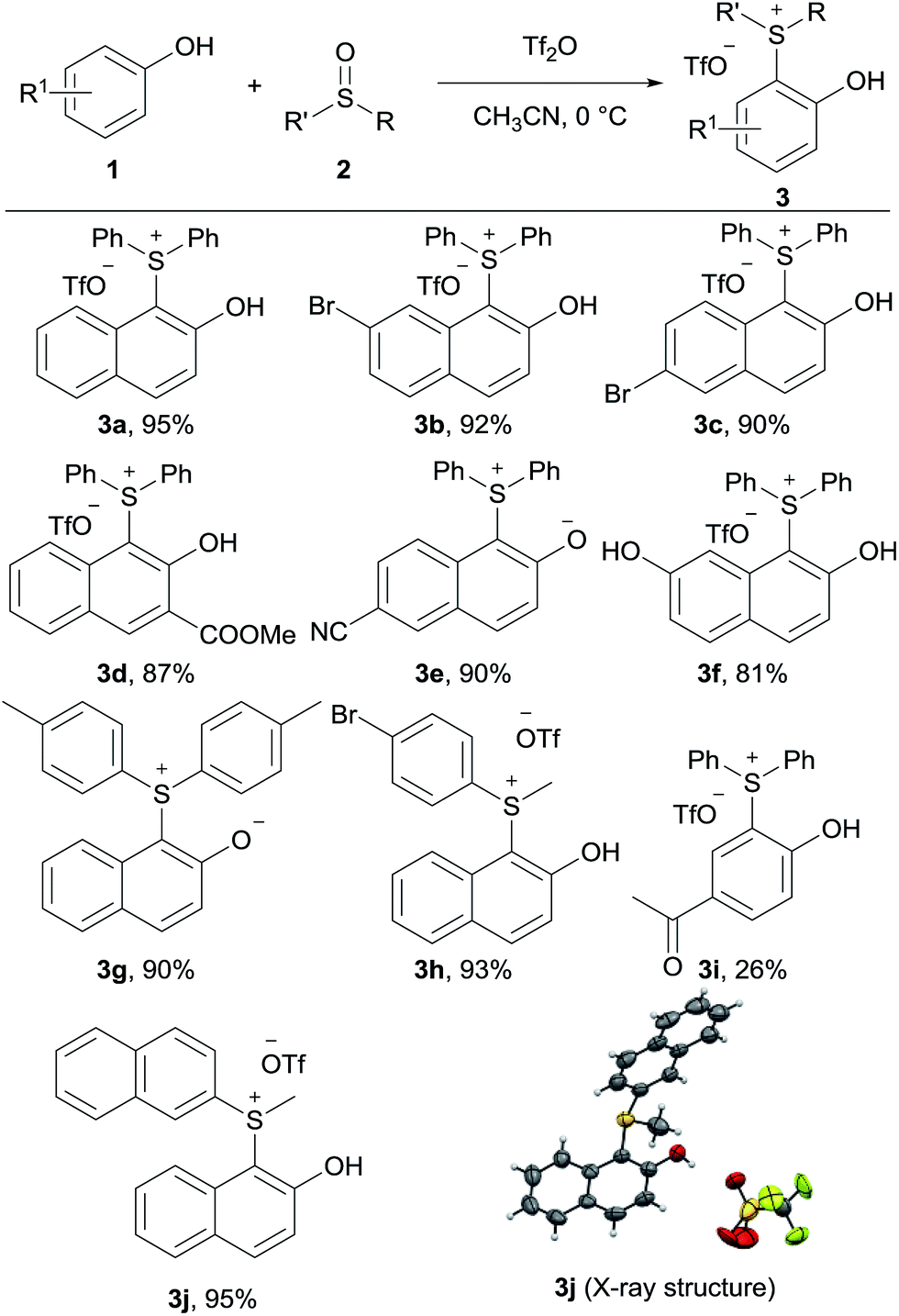
Our initial investigation began with 2-naphthol 1a and diphenyl sulfoxide 2a in the presence of Tf2O in acetonitrile at 0 °C. To our delight, the new sulfonium salt 3a was isolated in 95% yield. Somewhat surprisingly, this type of o-hydroxyarylsulfonium salt has never been prepared by this electrophilic aromatic substitution approach. The efficiency of this process was briefly examined with various phenols and sulfoxides, as shown in Table 1. Naphthols with electron-withdrawing substituents (3b–3e) worked equally well as those with electron-donating groups (3f). In addition, both diaryl sulfoxides and alkyl aryl sulfoxides led to the corresponding products in excellent yields (3g, 3h, and 3j). Interestingly, 3e and 3g were isolated as zwitterions. Unfortunately, though not entirely unexpected, a poor yield was observed when 4-hydroxyacetophenone was treated with diphenyl sulfoxide 2a.| a Reaction conditions: phenol 1 (0.2 mmol), sulfoxide 2 (0.22 mmol), Tf2O (0.24 mmol), MeCN (0.1 M) at 0 °C for 3 h. |
|---|
|
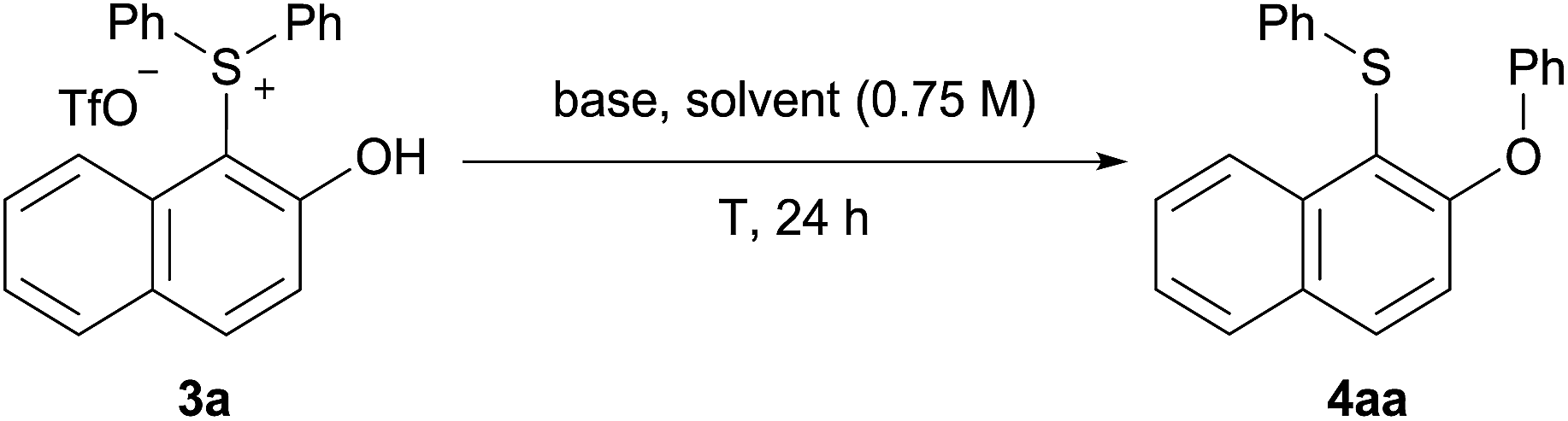
With isolated salt 3a in hand, we sought to determine its reactivity in a Smiles-like rearrangement, as a stepwise and general methodology to o-(phenoxy)naphthyl sulfide 4aa (Table 2). We first explored the choice of base under aqueous micellar conditions13 at 70 °C (entries 1–3), utilizing TPGS-750-M, a “benign-by-design” surfactant developed by Lipshutz.14 To our delight, in this medium, K3PO4 was identified to be the most effective base for this rearrangement (entry 4). Lowering the reaction temperature led to incomplete conversion and, therefore, a decreased yield (entry 5). No increase in yield was observed at higher temperature (entry 6). Other surfactants including Triton X-100 and SDS were screened, as were organic solvents (CH3CN, THF, and DMF). All afforded inferior results (entries 7–11). A control reaction “on water” (i.e., in the absence of TPGS-750-M) was conducted and a significantly lower yield was observed (entry 12), confirming the importance of micellar catalysis in facilitating this transformation in aqueous media.
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | T | Yieldb |
| a Conditions: 3a (0.375 mmol), base (1.125 mmol), solvent (0.75 M) for 24 h. b Yields were determined by HPLC analysis with nitrobenzene as an internal standard. c Using 2 wt% surfactant in water. SDS = sodium dodecyl sulfate. | ||||
| 1 | NaHCO3 | TPGS-750-M/H2Oc | 70 °C | 78% |
| 2 | CsF | TPGS-750-M/H2Oc | 70 °C | 60% |
| 3 | Et3N | TPGS-750-M/H2Oc | 70 °C | 88% |
| 4 | K3PO4 | TPGS-750-M/H2Oc | 70 °C | 93% |
| 5 | K3PO4 | TPGS-750-M/H2Oc | 60 °C | 70% |
| 6 | K3PO4 | TPGS-750-M/H2Oc | 80 °C | 90% |
| 7 | K3PO4 | Triton X-100/H2Oc | 70 °C | 76% |
| 8 | K3PO4 | SDS/H2O | 70 °C | 58% |
| 9 | K3PO4 | CH3CN | 70 °C | 75% |
| 10 | K3PO4 | THF | 70 °C | 78% |
| 11 | K3PO4 | DMF | 70 °C | 86% |
| 12 | K3PO4 | H2O | 70 °C | 70% |
At this stage, we sought to develop a one-pot sequence. Unfortunately, synthesis of the sulfonium salts either in aqueous media or under solvent-free conditions was low yielding. Hence, sulfonium salts were prepared in situ and used directly for the rearrangement without chromatography. Gratifyingly, the desired sulfide 4aa was produced in 86% yield from the reaction of 2-naphthol 1a and diphenyl sulfoxide 2a (Table 3).
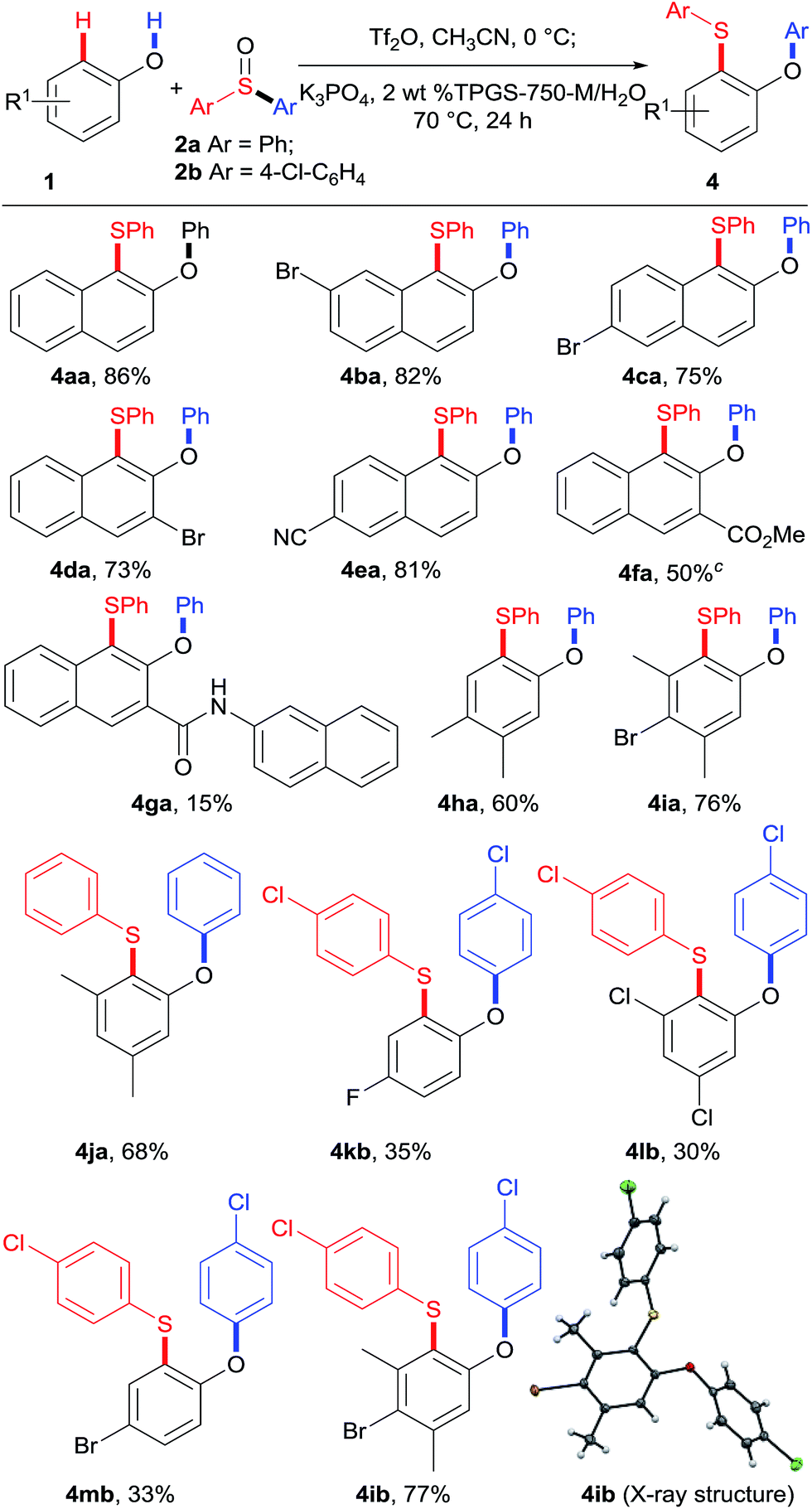
With the optimized conditions in hand, we proceeded to explore the substrate scope of phenols 1, as shown in Table 3. Naphthols with different substituents proceeded smoothly under the standard reaction conditions, leading to products in good to high yields (4aa–4fa). Importantly, these products (4ba–4ea) bearing Br and CN substituents could be potentially further functionalized. Unfortunately, naphthol 1g with an amide functional group gave the corresponding product 4ga in only 15% yield. Besides naphthols, the substrate scope was further expanded to phenol derivatives. Phenols 1h–1j containing electron-donating groups reacted smoothly with sulfoxides 2a and 2b generating the corresponding sulfides 4ha–4ja and 4ib (X-ray) in good yields. Phenols bearing electron-withdrawing groups showed lower reactivity, leading to products 4kb–4mb in moderate yields. Interestingly, for p-unsubstituted phenols 1j and 1l, the desired products 4ja and 4lb were obtained selectively.
The scope of sulfoxides was then evaluated in reactions with 2-naphthol 1a (Table 4). Adducts 4ab and 4ac were obtained in good yields from symmetrical diaryl sulfoxides 2b and 2c with different electronic properties. Furthermore, unsymmetrical diaryl sulfoxides 2d–2h were also investigated. Remarkably, the use of sulfoxide 2d, simultaneously bearing p-tolyl and phenyl residues resulted in phenyl-migrated product 4ad, while the p-tolyl-migrated isomer was not detected. A similar trend was observed for sulfoxides 2e–2h, and the more electron-deficient aryl groups were selectively migrated delivering the corresponding sulfides 4ae–4ah. These results suggest that the rearrangement takes place by ipso-attack of a negatively charged oxygen atom onto the more electron-deficient aryl group.
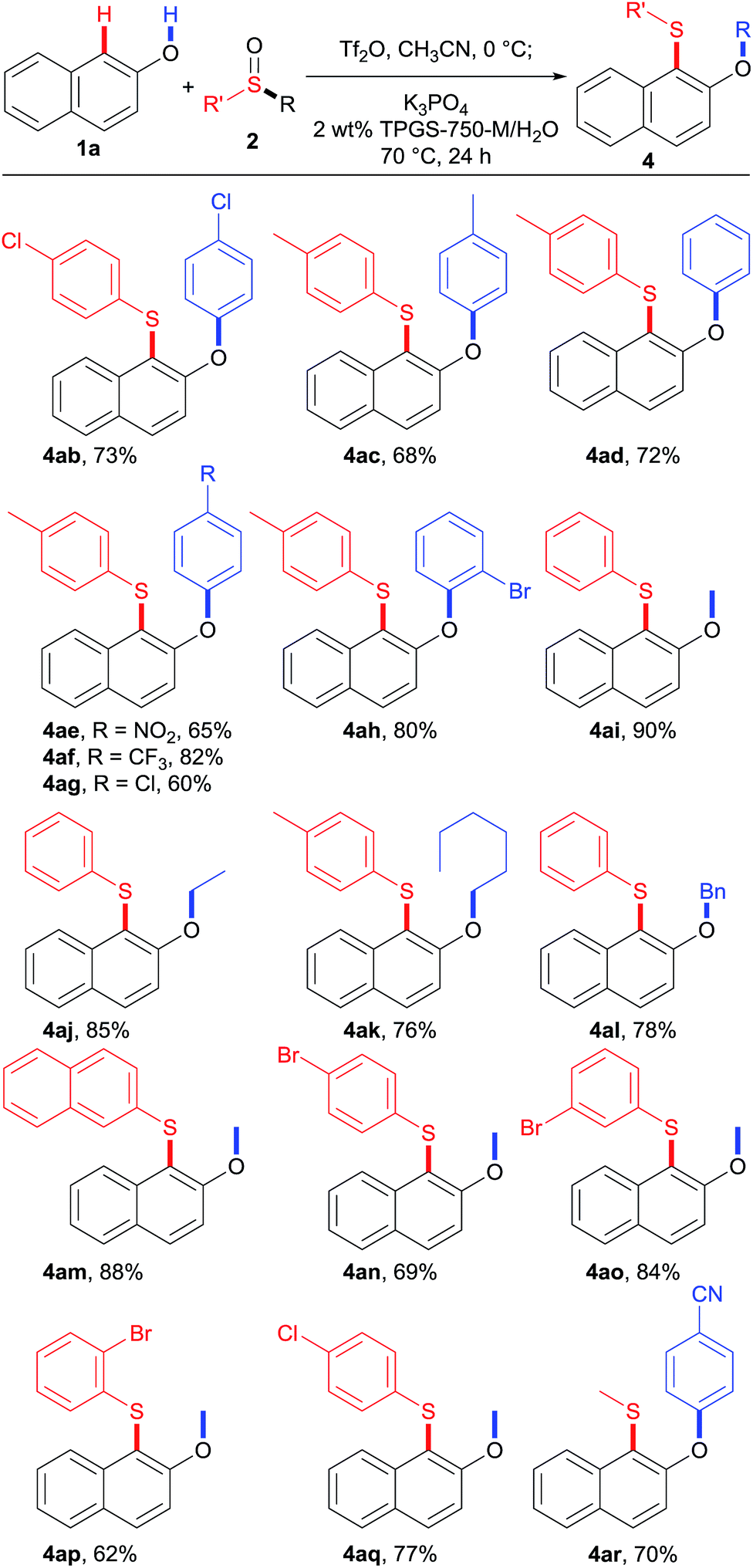
Besides diaryl sulfoxides, alkyl aryl sulfoxides also participated in this transformation, further expanding the range of participating substrates. Increasing the length of the alkyl chain slightly decreased the reaction efficiency, as demonstrated by products 4ai–4ak. In addition, benzyl phenyl sulfoxide 2l is also a competent reaction partner, providing 4al in 78% yield. Moreover, the aryl group could also be modified with different electronic properties, and the reactions furnished the corresponding sulfides in good to high yields (4ak and 4am–4ar). Importantly, there was no significant effect of substituents in the para, meta, or ortho positions. For example, p-bromophenyl methyl sulfoxide (2n), m-bromophenyl methyl sulfoxide (2o), and o-bromophenyl methyl sulfoxide (2p) all reacted uneventfully and gave methyl-migrated sulfides 4an–4ap in useful yields. Notably, p-cyanophenyl methyl sulfoxide (2r) afforded cyanophenyl-migrated product 4ar exclusively, whereas aryl methyl sulfoxides 2m–2q all provided methyl-migrated products 4am–4aq. These results suggest that stronger electron-withdrawing groups on the aryl moiety can suppress alkyl migration, leading to aryl migration instead.
To further explore the applicability of this approach, we tested this new transformation on estrone 1n containing a complex architecture. Accordingly, difunctionalization of estrone proceeded smoothly, affording 4ni or 4nb in 68% yield (Scheme 2a), demonstrating the mild and selective nature of this overall transformation. In addition, a formal synthesis of urokinase inhibitor 8 was accomplished efficiently as shown in Scheme 2b.
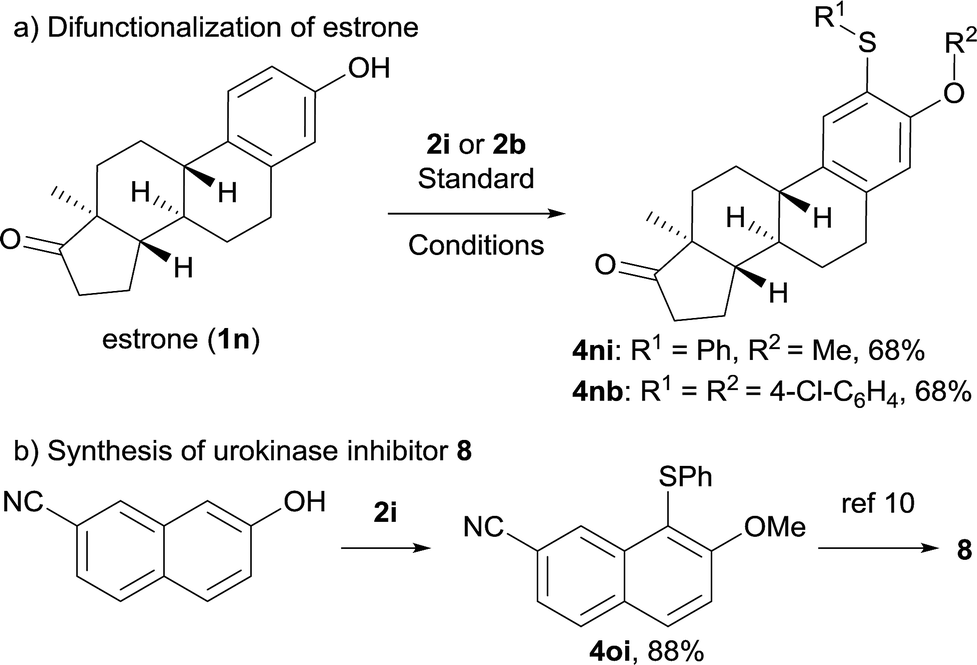 | ||
| Scheme 2 Application in late-stage construction and scaffold construction. | ||
We next sought to prepare sulfoxides and sulfones in one-pot (Scheme 3). Starting from 2-naphthol 1a and diphenyl sulfoxide 2a, sulfonium salt formation and then rearrangement in water gave sulfide 4aa. Without isolation, the resulting sulfide could then be oxidized to sulfoxide 5aa and sulfone 6aa in good overall yields, respectively (Scheme 3a). Sulfone 6eg was also obtained in high yield under similar reaction conditions. The selectivity of the Smiles-like rearrangement was unambiguously confirmed by single crystal X-ray analysis of 6eg (Scheme 3b).
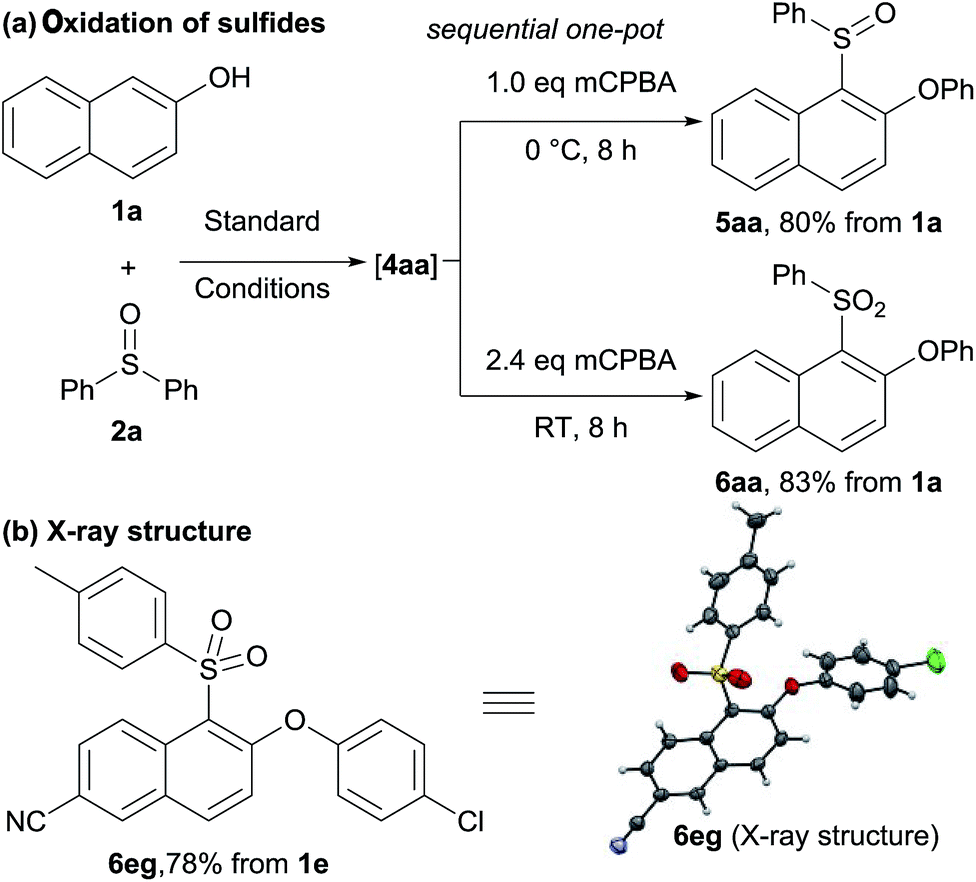 | ||
| Scheme 3 Oxidation of the products in water. | ||
To understand whether the rearrangement step is intramolecular or intermolecular, a crossover reaction of naphthol 1a with diaryl sulfoxides 2b and 2c was conducted under the standard reaction conditions (Scheme 4). It was found that no cross-product was detected, which suggests that this aryl migration is intramolecular. Furthermore, 4ab was formed in 78% yield while 4ac was isolated in only 39% yield, indicating that sulfoxide 2b was more reactive than sulfoxide 2c.
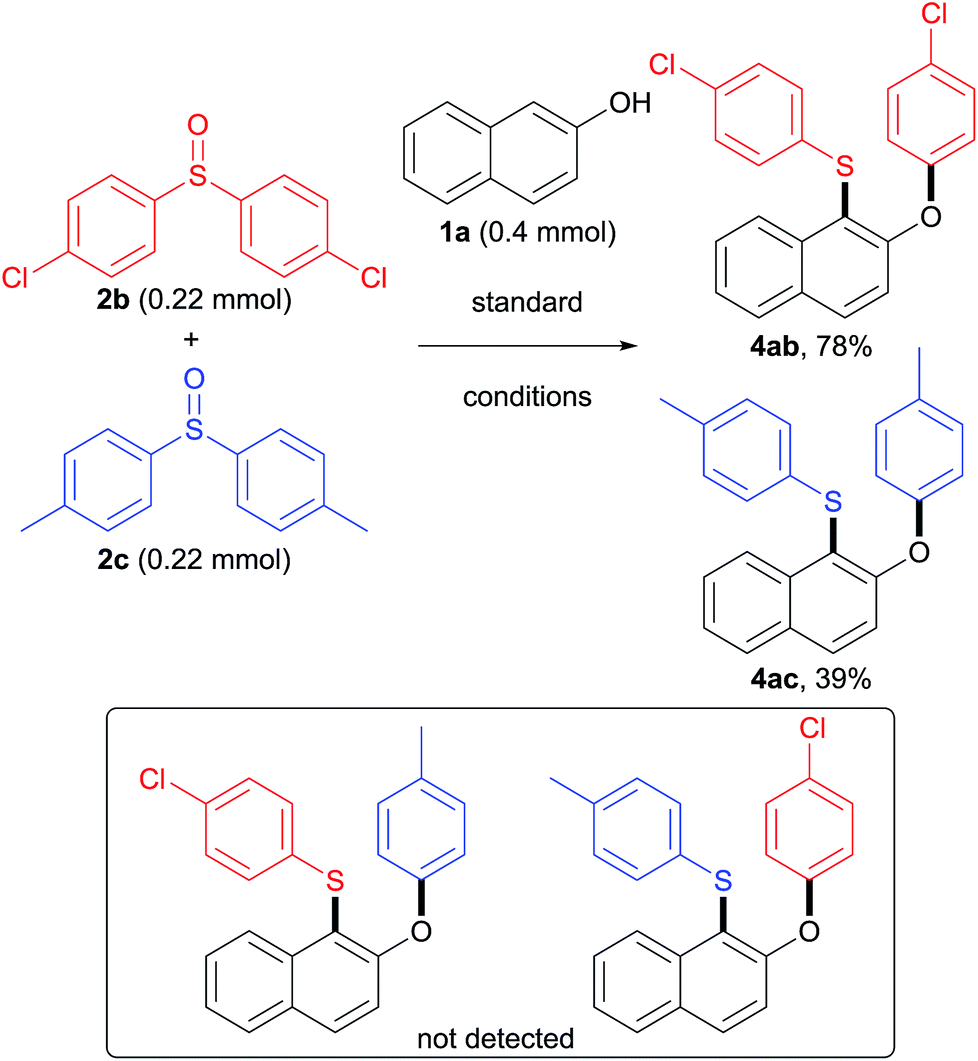 | ||
| Scheme 4 Application in late-stage construction and scaffold construction. | ||
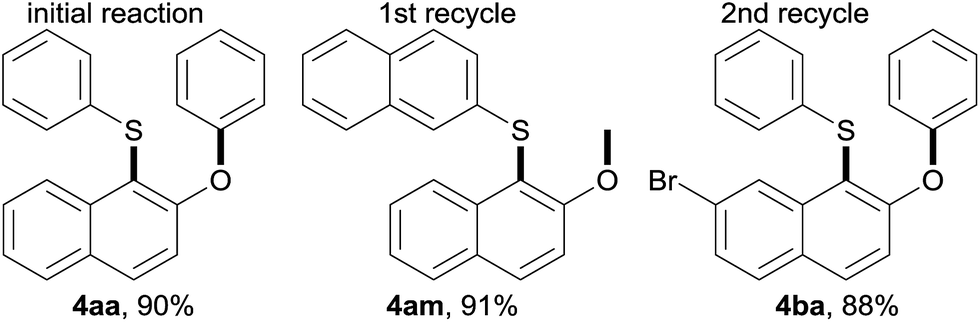
Lastly, E Factors4d,15 were calculated for the rearrangement under aqueous micellar conditions, to assess the level of waste generation. Initially, an E Factor of only 4 was obtained based on organic solvent used, as illustrated in Table 5. An E Factor of only 8.6, was realized even with water included in the calculation. Moreover, the aqueous medium containing TPGS-750-M can be recycled to generate a different sulfide 4am or 4ba efficiently. Since no additional water needs to be used in each cycle, the associated E Factors dropped significantly.
Conclusions
In conclusion, we have developed an unprecedented approach for C–H/O–H difunctionalization of phenols under mild conditions. The process is mediated by an electrophilic aromatic substitution and subsequent Smiles-like rearrangement, involving C–S and C–O bond formations. Notably, this rearrangement proceeded under aqueous micellar conditions, successfully addressing several environmental issues. Further studies on related rearrangements featuring multiple bond-formations in water are currently in progress in our laboratories.Acknowledgements
Financial support provided by the National Key Research and Development Program (2016YFD0600801), the Natural Science Foundation of China (21502094), the Jiangsu Specially-Appointed Professor Plan, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) is warmly acknowledged with thanks.Notes and references
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere, and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-I. Negishi, Wiley-Interscience, New York, 2002 Search PubMed; (c) Cross-Coupling Reactions: A Practical Guide, Topics in Current Chemistry Series 219, ed. N. Miyaura, Springer, New York, 2002 Search PubMed; (d) Catalyzed Carbon–Heteroatom Bond Formation, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2011 Search PubMed; (e) J. Yin, in Applications of Transition Metal Catalysis in Drug Discovery and Development, ed. M. L. Crawley and B. M. Trost, John Wiley and Sons, Hoboken, 2012, pp. 97–163 Search PubMed.
- For selected reviews and examples, see: (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (b) P. Bichler and J. A. Love, Top. Organomet. Chem., 2010, 31, 39 CrossRef CAS; (c) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (d) M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397 CrossRef CAS; (e) M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180 CrossRef PubMed; (f) K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694 CrossRef CAS PubMed; (g) S. Gowrisankar, A. G. Sergeev, P. Anbarasan, A. Spannenberg, H. Neumann and M. Beller, J. Am. Chem. Soc., 2010, 132, 11592 CrossRef CAS PubMed; (h) G. Bastug and S. P. Nolan, J. Org. Chem., 2013, 78, 9303 CrossRef CAS PubMed; (i) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS PubMed; (j) Y. Tan, J. M. Muñoz-Molina, G. C. Fu and J. C. Peters, Chem. Sci., 2014, 5, 2831 RSC; (k) J. Mao, T. Jia, G. Frensch and P. J. Walsh, Org. Lett., 2014, 16, 5304 CrossRef CAS PubMed; (l) Z. Qiao, J. Wei and X. Jiang, Org. Lett., 2014, 16, 1212 CrossRef CAS PubMed; (m) M. W. Johnson, K. I. Hannoun, Y. Tan, G. C. Fu and J. C. Peters, Chem. Sci., 2016, 7, 4091 RSC; (n) F.-G. Sun, M. Li, C.-F. He, B. Wang, B. Li, X.-W. Sui and Z.-H. Gu, J. Am. Chem. Soc., 2016, 138, 7456 CrossRef CAS PubMed.
- (a) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133 CrossRef CAS PubMed; (b) D. A. Evans, K. R. Campos, J. S. Tedrow, F. E. Michael and M. R. Gagne, J. Am. Chem. Soc., 2000, 122, 7905 CrossRef CAS; (c) D. A. Evans, F. E. Michael, J. S. Tedrow and K. R. Campos, J. Am. Chem. Soc., 2003, 125, 3534 CrossRef CAS PubMed; (d) D.-H. Bao, H.-L. Wu, C.-L. Liu, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2015, 54, 8791 CrossRef CAS PubMed; (e) Y. Wei, L.-Q. Lu, T.-R. Li, B. Feng, Q. Wang, W.-J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2016, 55, 2200 CrossRef CAS PubMed; (f) L. A. Damani, Sulphur-Containing Drugs and Related Organic Compounds, Wiley, 1989 Search PubMed; (g) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- (a) T. Laird, Org. Process Res. Dev., 2012, 16, 1 CAS; (b) P. J. Dunn, Pharmaceutical Process Development, ed. J. A. Blacker and M. T. Williams, Royal Society of Chemistry, London, 2011, ch. 6 Search PubMed; (c) C. Jimenez-Gonzales and D. J. Constable, Green Chemistry and Engineering: A Practical Approach, Wiley, New York, 2011 Search PubMed; (d) R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) T. Hostier, V. Ferey, G. Ricci, P. D. Gomez and J. Cossy, Org. Lett., 2015, 17, 3898 CrossRef CAS PubMed; (b) A. M. Wagner and M. S. Sanford, J. Org. Chem., 2014, 79, 2263 CrossRef CAS PubMed; (c) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed; (d) A. S. Henderson, S. Medina, J. F. Bower and M. C. Galan, Org. Lett., 2015, 17, 4846 CrossRef CAS PubMed; (e) Y.-Y. Dong, M. I. Lipschutz and T. D. Tilley, Org. Lett., 2016, 18, 1530 CrossRef CAS PubMed.
- (a) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS; (b) V. W. Rosso, D. A. Lust, P. J. Bernot, J. A. Grosso, S. P. Modi, A. Rusowicz, T. C. Sedergran, J. H. Simpson, S. K. Srivastava, M. J. Humora and N. G. Anderson, Org. Process Res. Dev., 1997, 1, 311 CrossRef CAS.
- For selected reviews and examples, see: (a) A. Shafir, Tetrahedron Lett., 2016, 57, 2673 CrossRef CAS; (b) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842 CrossRef CAS PubMed; (c) L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832 CrossRef CAS PubMed; (d) S. K. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401 CrossRef CAS PubMed; (e) J. A. Fernández-Salas, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 790 CrossRef PubMed; (f) J. A. Fernández-Salas, A. P. Pulis and D. J. Procter, Chem. Commun., 2016, 52, 12364 RSC.
- (a) J. F. Burnett and R. E. Zalher, Chem. Rev., 1951, 49, 273 CrossRef; (b) W. E. Truce, E. M. Kreider and W. W. Brand, Org. React., 1970, 18, 99 CAS.
- (a) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834 RSC, and references cited therein; (b) Y. Tang, Y. Sun, J. Liu and S. Duttwyler, Org. Biomol. Chem., 2016, 14, 5580 RSC.
- (a) A. G. Geyer, W. J. McClellan, T. W. Rochway, K. D. Stewart, M. Weitzberg and M. D. Wendt, US Pat. 6,258,822, 2001; (b) A. G. Geyer, W. J. McClellan, T. W. Rochway, K. D. Stewart, M. Weitzberg and M. D. Wendt, US Pat. 6,284,796, 2001; (c) T. Nakazawa, J. Xu, T. Nishikawa, T. Oda, A. Fujita, K. Ukai, R. E. P. Mangindaan, H. Rotinsulu, H. Kobayashi and M. Namikoshi, J. Nat. Prod., 2007, 70, 439 CrossRef CAS PubMed; (d) M. P. Crump, T. A. Ceska, L. Spyracopoulos, A. Henry, S. C. Archibald, R. Alexander, R. J. Taylor, S. C. Findlow, J. O'Connell, M. K. Robinson and A. Shock, Biochemistry, 2004, 43, 2394 CrossRef CAS PubMed; (e) B. Wang, L. Li, T. D. Hurley and S. O. Meroueh, J. Chem. Inf. Model., 2013, 53, 2659 CrossRef CAS PubMed; (f) T. A. Blizzard, F. DiNinno, J. D. Morgan II, H. Y. Chen, J. Y. Wu, S. Kim, W. Chan, E. T. Birzin, Y. T. Yang, L.-Y. Pai, P. M. D. Fitzgerald, N. Sharma, Y. Li, Z. Zhang, E. C. Hayes, C. A. DaSilva, W. Tang, S. P. Rohrer, J. M. Schaeffer and M. L. Hammond, Bioorg. Med. Chem. Lett., 2005, 15, 107 CrossRef CAS PubMed; (g) T. A. Blizzard, F. DiNinno, H. Y. Chen, S. Kim, J. Y. Wu, W. Chan, E. T. Birzin, Y. T. Yang, L.-Y. Pai, E. C. Hayes, C. A. DaSilva, S. P. Rohrer, J. M. Schaeffer and M. L. Hammond, Bioorg. Med. Chem. Lett., 2005, 15, 3912 CrossRef CAS PubMed; (h) T. A. Blizzard, F. DiNinno, H. Y. Chen, S. Kim, J. Y. Wu, W. Chan, E. T. Birzin, Y. T. Yang, L.-Y. Pai, E. C. Hayes, C. A. DaSilva, S. P. Rohrer, J. M. Schaeffer and M. L. Hammond, Bioorg. Med. Chem. Lett., 2005, 15, 5124 CrossRef CAS PubMed; (i) S. Zhuang, J. Zhang, F. Zhang, Z. Zhang, Y. Wena and W. Liu, Bioorg. Med. Chem. Lett., 2011, 21, 7298 CrossRef CAS PubMed; (j) L. D. B. Salum, I. Polikarpov and A. D. Andricopulo, J. Mol. Graphics Modell., 2007, 26, 434 CrossRef CAS PubMed; (k) S. Park, I. Kufareva and R. Abagyan, J. Comput.-Aided Mol. Des., 2010, 24, 459 CrossRef CAS PubMed; (l) L. Gao, Y. Tu and L. A. Eriksson, J. Biophys. Chem., 2011, 2, 233 CrossRef CAS; (m) F. P. DiNinno, T. A. Blizzard and J. D. Morgan II, US Pat. 0,234,245, 2005.
- (a) K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 7510 CrossRef CAS PubMed; (b) X. Huang, M. Patil, C. Farès, W. Thiel and N. Maulide, J. Am. Chem. Soc., 2013, 135, 7312 CrossRef CAS PubMed; (c) X. Huang and N. Maulide, J. Am. Chem. Soc., 2011, 133, 8510 CrossRef CAS PubMed; (d) Y. Ookubo, A. Wakamiya, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2012, 18, 12690 CrossRef CAS PubMed; (e) T. Kobatake, D. Fujino, S. Yoshida, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2010, 132, 11838 CrossRef CAS PubMed; (f) T. Kobatake, S. Yoshida, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2010, 49, 2340 CrossRef CAS PubMed.
- (a) X. Huang, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 8979 CrossRef CAS PubMed; (b) M. N. Kim and K. Kim, J. Heterocycl. Chem., 1998, 35, 235 CrossRef CAS.
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660 RSC , and references cited therein.
- (a) B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed; (b) Aldrich catalog number 763918.
- (a) B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952 CrossRef CAS PubMed, and references cited therein; (b) F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1502491, 1502489 and 1502490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04504a |
| This journal is © The Royal Society of Chemistry 2017 |