
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNature's hydrides: rapid reduction of halocarbons by folate model compounds
Michael K.
Denk
*a,
Nicholas S.
Milutinović
a,
Katherine M.
Marczenko
a,
Natalie M.
Sadowski
a and
Athanasios
Paschos
bc
aDepartment of Chemistry, University of Guelph, 50 Stone Road East, Guelph, Ontario N1G 2W1, Canada
bDepartment of Biology, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4K1, Canada
cMohawk College of Applied Arts and Technology, Department of Chemical and Environmental Technology, 135 Fennell Ave West, Hamilton, Ontario L9C 1E9, Canada
First published on 17th November 2016
Abstract
Halocarbons R–X are reduced to hydrocarbons R–H by folate model compounds under biomimetic conditions. The reactions correspond to a halide–hydride exchange with the methylenetetrahydrofolate (MTHF) models acting as hydride donors. The MTHF models are also functional equivalents of dehalohydrogenases but, unlike these enzymes, do not require a metal cofactor. The reactions suggest that halocarbons have the potential to act as endocrinological disruptors of biochemical pathways involving MTHF. As a case in point, we observe the rapid reaction of the MTHF models with the inhalation anaesthetic halothane. The ready synthetic accessibility of the MTHF models as well as their dehalogenation activity in the presence of air and moisture allow for the remediation of toxic, halogenated hydrocarbons.
Introduction
The structural similarity between methylenetetrahydrofolate (MTHF) 2H1–5 and Thauer's hydrogenase 3H6 has led us to investigate imidazolidines 1H as model compounds for the reactivity of these fascinating biomolecules (Fig. 1).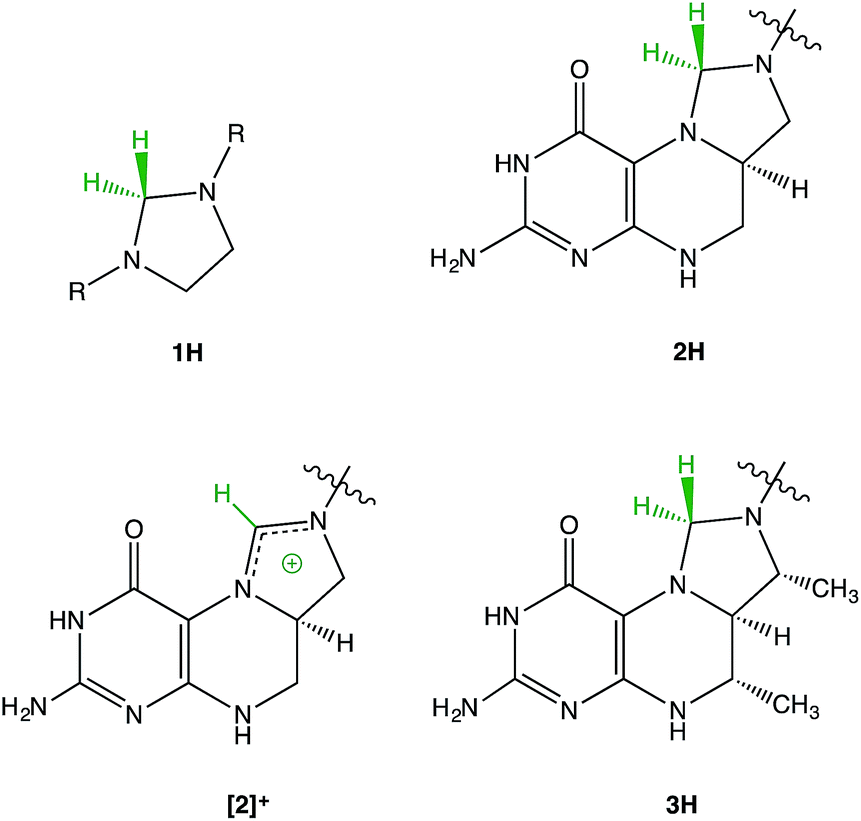 | ||
| Fig. 1 Structural relationship between imidazolidines 1H, methylenetetrahydrofolate (MTHF) 2H, and Thauer's hydrogenase 3H. Different structural formulae resulting from different N-protonation positions can be found in the literature for 2H. The structure presented in this figure is the one most commonly used.1 | ||
A type of reactivity shared by 2H and 3H is the transfer of negatively charged “hydridic” hydrogen leading to the very stable cationic species [2]+ and [3]+.
In addition to its function as a hydride donor, MTHF is involved in several other biochemical pathways including folate-dependent DNA methylation, purine, pyrimidine, and amino acid syntheses, as well as the methylation of homocysteine to methionine. MTHF is also the precursor to 5-methyltetrahydrofolate, an important intermediate in its own right.1–5
The recent discovery that the hydride donor system 2H/[2]+ acts as a photobiological cofactor in the light-activated repair enzyme DNA photolyase5 demonstrates that MTHF offers significant surprises even after more than sixty years of extensive research. For comprehensive reviews on Thauer's hydrogenase 3H see ref. 6a–d.
Results
Reduction of halogenated hydrocarbons
During our initial studies,7 we were surprised to find that imidazolidines 1H (R: methyl, tert-butyl)8 are unstable in chloroform and resemble many organometallic compounds and hydrido complexes in this respect. A closer examination revealed that the reason for this instability was the reduction of chloroform to dichloromethane by 1H acting as a hydride donor. By-product of the reduction is the salt [1]+Cl− (Scheme 1). We now report that this type of reduction is remarkably general and allows for the dehalogenation of a wide variety of halogenated hydrocarbons including those of toxicological, environmental, and pharmacological significance.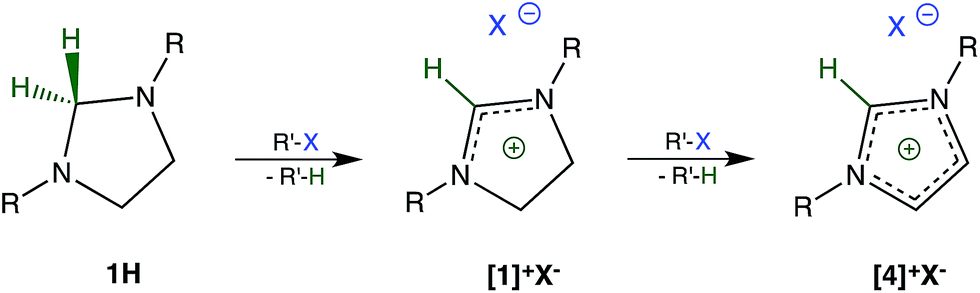 | ||
| Scheme 1 Reduction of carbon–halogen bonds R′–X by imidazolidine 1H (R: tert-butyl). | ||
To further explore the scope of the reductive dehalogenation, the reactivity of 1H towards a series of chloromethanes and bromomethanes was investigated. With the exceptions of dichloromethane, chloromethane, and bromomethane, all chlorine- and bromine-substituted methanes are reduced by 1H. The reductions occur at room temperature, but the time required for a complete conversion ranges from weeks for chloroform to only minutes for bromoform and carbon tetrabromide. Reactions of 1H with allyl bromide and ethyl 2-bromoacetate resulted in the formation of complex product mixtures. Details about the reaction as well as the compatibility of functional groups are the subject of on-going studies and will be reported in a separate publication.
The nature of the employed solvent (ethanol, hexanes), an excess of 1H, or an excess of the halogenated methane do not significantly influence the reaction rates. Remarkably, the dehalogenation reactions are not inhibited by air or moisture.
An excess of the halogenated hydrocarbon and prolonged reaction times lead to further oxidation of [1]+X− to [4]+X− (Scheme 1).
The unsaturated imidazoline analogues of 1H were previously found to be incompatible with biomimetic conditions due to their high sensitivity towards moisture.7c To investigate if the lack of reduction observed for some of the halogenated substrates is due to thermodynamic reasons or is the result of kinetic factors the reaction energies ΔG° for the halide–hydride exchange were determined by quantum chemical calculations (Table 1).9 A computational solvation model for water was added to simulate biological conditions (see Methods).10
| A | B | X:Cl | X:Br |
|---|---|---|---|
| CX4 | CHX3 | −48.3 | −37.9 |
| CHX3 | CH2X2 | −44.3 | −39.1 |
| CH2X2 | CH3X | −41.0 | −36.5 |
| CH3X | CH4 | −39.8 | −29.3 |
| PhX | PhH | −35.9 | −31.3 |
The calculated energies show that all reactions are thermodynamically favourable. The lack of reactivity observed for bromomethane, chloromethane, and dichloromethane is accordingly only the result of low reaction rates.
The observed reaction rates show no correspondence to the calculated reaction energies ΔG° but closely match the order of decreasing carbon–halogen bond energies (CBS-QB3 level, kcal mol−1): CH3Br (77.1), CHCl3 (77.0), CH2Br2 (71.7), CCl4 (71.9), CHBr3 (65.3), CBr4 (62.9).
To test for the ability of 1H to reduce polybrominated commercial flame retardants,12 1,3,5-tribromobenzene was investigated (Scheme 2). Dehalogenation to 1,3-dibromobenzene occurred within minutes, even at room temperature, although the total reduction to benzene requires prolonged heating at 150 °C.
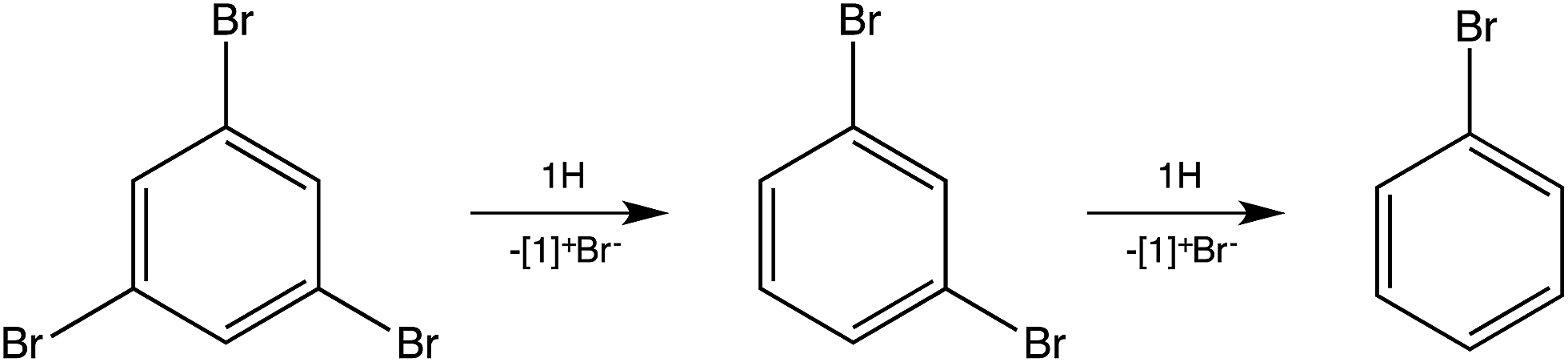 | ||
| Scheme 2 Reaction of imidazolidines 1H with 1,3,5-tribromobenzene. | ||
As carbon–halogen bonds are found in many medicinal drugs, the dehalogenation reactions observed for the MTHF model 1H may well occur in vivo with MTHF itself, with obvious implications for drug metabolism as well as toxicology.
As a test case, the reaction of the inhalation anaesthetic halothane, CF3CH(Br)Cl, with 1H was examined (Scheme 3). Dehalogenation was observed (NMR) at room temperature within minutes with 2-chloro-1,1,1-trifluoroethane, CF3CH2Cl,13 being formed as the sole reduction product.
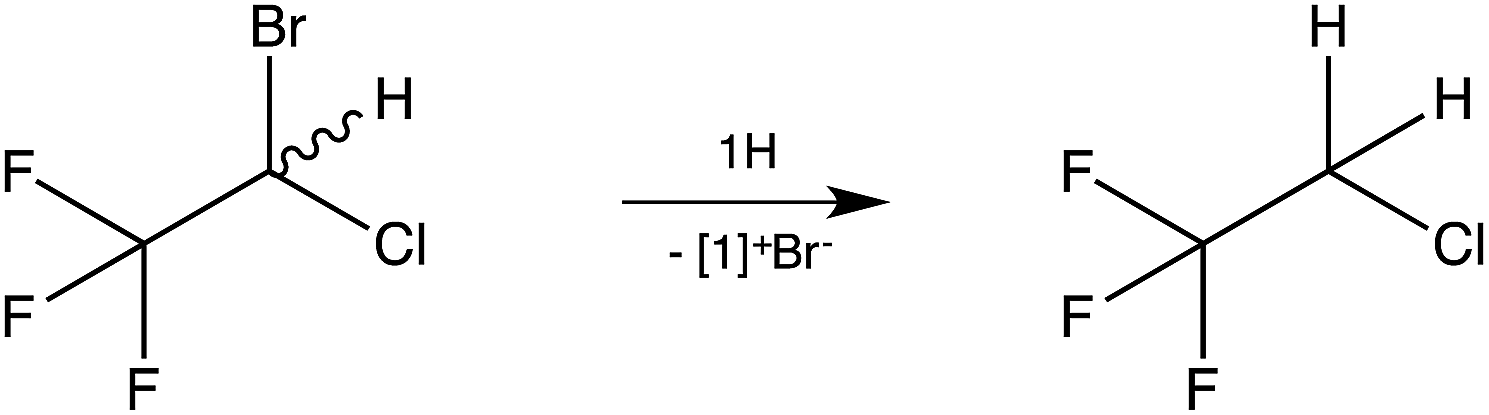 | ||
| Scheme 3 Reaction of imidazolidines 1H with 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane). | ||
The high reactivity and selectivity observed suggests that 1H may become a new substitute for the tin hydrides18 and hydrosilanes19 previously used for selective debrominations.
Discussion
While in vivo studies will inevitably be required to further study the biochemical implications of these findings, the reactivity of 1H does suggest that MTHF-dependent biochemical pathways are likely to be disrupted by halogenated hydrocarbons. It is of interest to note in this context that MTHF is the precursor to 5-methyltetrahydrofolate – a compound under current investigation for the treatment of depression and schizophrenia.4The reported reductive dehalogenation reactions are highly unusual for a simple, neutral, and stable organic molecule like 1H, but have precedence in the fascinating reactivity of dehalohydrogenases.15
Dehalohydrogenases have been isolated from a variety of microorganisms14–16 and have attracted great interest for their ability to reduce a number of highly toxic, halogenated hydrocarbons like the notorious 2,3,5,6-tetrachlorodibenzodioxin.16 A notable difference between our system 1H and dehalohydrogenases reported to date is that the latter require metal cofactors like iron-sulphur proteins or cobalamines.17 The fact that dehalohydrogenase-type activity is possible with metal-free folate models 1H suggests that metal-free dehalohydrogenases can exist as well.
Methods
Quantum chemical calculations
All calculations were carried out with Gaussian 09, Revision D.01.9 The energies were obtained from full optimizations with the CBS-QB3 method11 and the SMD solvent model10 for water. Minima were verified by the absence of virtual frequencies.Starting materials
The imidazolines 1H (R: tert-butyl, methyl, para-methyphenyl) were prepared from the respective 1,2-diaminoethanes as described elsewhere.7 Halothane and 1,3,5-tribromobenzene were purchased from Sigma-Aldrich Inc.Reduction of halocarbons
To the best of our knowledge the only previously reported reduction of a halogenated hydrocarbon by a MTHF analogue is that of CCl4.8 Reductions were carried out with a slight excess of the imidazolidines 1H (1.1 equivalent per reducible halogen). In nonpolar solvents like hexanes or diethyl ether, the onset of the reduction is indicated by the appearance of turbidity with subsequent precipitation of the salts [1]+X−. Control experiments in polar solvents or under inert gas (99.995% argon) with degassed materials did not show noticeably different reaction rates or products. For the less reactive aryl halides and chloromethanes, heating was required to drive the reaction to completion. The progress of the reductions was monitored by NMR and GC-MS. After completion of the reaction, the salts were isolated by filtration under inert gas, washed with diethyl ether, and dried in vacuo. Yield 85–95%. Traces of [4]+X− could be removed from [1]+X− by recrystallization from 95% ethanol.Reduction of halothane
To retain the highly volatile product 2-chloro-1,1,1-trifluoroethane (bp +6 °C)13a the reduction of halothane was monitored in a flame-sealed NMR tube.Spectroscopic data
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3)3], 45.3 [CH2CH2], 57.2 [C(H3)3], 154.1 [N2CH+].
H3)3], 45.5 [CH2CH2], 57.2 [(CH3)3], 153.1 [N2CH+].
H3)3], 45.3 [CH2CH2], 57.2 [C(H3)3], 154.1 [N2CH+].
H3)3], 45.5 [CH2CH2], 57.2 [(CH3)3], 153.1 [N2CH+].
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH], 10.54 [1H, s, N2CH+]. 13C-NMR (400 MHz, CDCl3): δ 30.3 [s, C(H3)3], 60.8 [(CH3)3], 119.7 [HCCH], 134.5 [N2CH+].
CH], 10.37 [1H, s, N2CH+]. 13C-NMR (400 MHz, CDCl3): δ 30.4 [C(H3)3], 61.2 [(CH3)3], 118.9 [HCCH], 135.2 [N2CH+].
H3], 46.8 [CH2CH2], 66.4 [N2CH2], 112.5, 126.7, 129.8, 144.4. GC-MS: tr = 17.9 min m/z (rel. int%): 252(72)[M+], 251(100), 133(39), 118(16), 105(81), 91(30), 65(10).
H3], 48.2 [CH2CH2], 118.1, 129.9, 133.6, 136.4, 150.9 [N2CH+].
CH], 10.54 [1H, s, N2CH+]. 13C-NMR (400 MHz, CDCl3): δ 30.3 [s, C(H3)3], 60.8 [(CH3)3], 119.7 [HCCH], 134.5 [N2CH+].
CH], 10.37 [1H, s, N2CH+]. 13C-NMR (400 MHz, CDCl3): δ 30.4 [C(H3)3], 61.2 [(CH3)3], 118.9 [HCCH], 135.2 [N2CH+].
H3], 46.8 [CH2CH2], 66.4 [N2CH2], 112.5, 126.7, 129.8, 144.4. GC-MS: tr = 17.9 min m/z (rel. int%): 252(72)[M+], 251(100), 133(39), 118(16), 105(81), 91(30), 65(10).
H3], 48.2 [CH2CH2], 118.1, 129.9, 133.6, 136.4, 150.9 [N2CH+].
Conclusions
Our observation that dehalohydrogenase activity is possible with a simple, metal-free MTHF model 1H suggests that metal-free dehalohydrogenases may likewise exist.The ready synthetic accessibility of the model compounds 1H as well as their dehalogenation activity in the presence of air and moisture suggest their use for the remediation of toxic, halogenated hydrocarbons. The reactions suggest that halocarbons are likely to act as endocrinological disruptors of folate dependent pathways.
Acknowledgements
The authors are grateful for editorial comments by professors Gordon Lange and Mario Monteiro at different stages of the manuscript. N. S. M. and M. K. D. wrote the paper and M. K. D. carried out the quantum chemical calculations. All authors contributed about equally to the experiments. We thank one of the referees for enquiring about the reactivity of allyl bromide and ethyl 2-bromoacetate. The compatibility of other functional groups with the reported reduction is under investigation.Notes and references
- R. G. Kallen and W. P. Jencks, J. Biol. Chem., 1966, 241, 5851–5863 Search PubMed.
- (a) L. Jaenicke, Angew. Chem., 1961, 73, 449–480 Search PubMed; (b) L. Jaenicke, Annu. Rev. Biochem., 1964, 33, 287–312 CrossRef CAS PubMed; (c) J. T. Fox and P. J. Stover, Folic Acid and Folates, ed. G. Litwack, Elsevier Academic Press Inc., San Diego, 1st edn, 2008, vol. 79, ch. 1, pp. 1–44 Search PubMed; (d) E. P. Quinlivan, A. D. Hanson and J. F. Gregory, Anal. Biochem., 2006, 348, 163–184 Search PubMed.
- (a) E. R. Werner, N. Blau and B. Thoöny, Biochem. J., 2011, 438, 397–414 Search PubMed; (b) M. Födinger, W. H. Hörl and G. Sunder-Plassmann, J. Nephrol., 2000, 13, 20–33 Search PubMed; (c) K. Robien and C. M. Ulrich, Am. J. Epidemiol., 2003, 157, 571–582 Search PubMed.
- (a) A. L. Miller, Altern. Med. Rev., 2008, 13, 216–226 Search PubMed; (b) M. Fava and D. Mischoulon, J. Clin. Psychiatry, 2009, 70(5), 12–17 Search PubMed; (c) S. M. Stahl, CNS Spectr., 2007, 12, 739–744 Search PubMed.
- (a) A. Sancar, Chem. Rev., 2003, 103, 2203–2237 Search PubMed; (b) R. F. Pauszek III, G. Kodali and R. J. Stanley, J. Phys. Chem. A, 2014, 118, 8320–8328 CrossRef PubMed.
- (a) S. Shima and R. K. Thauer, Chem. Rec., 2007, 7, 37–46 Search PubMed; (b) B. H. Geierstanger, T. Prasch, C. Griesinger, G. Hartmann, G. Buurman and R. K. Thauer, Angew. Chem., Int. Ed. Engl., 1998, 37, 3300–3303 Search PubMed; (c) S. Shima, E. J. Lyon, M. Sordel-Klippert, M. Kauß, J. Kahnt, R. K. Thauer, K. Steinbach, X. Xie, L. Verdier and C. Griesinger, Angew. Chem., Int. Ed. Engl., 2004, 43, 2547–2551 Search PubMed; (d) E. J. Lyon, S. Shima, G. Buurman, S. Chowdhuri, A. Batschauer, K. Steinbach and R. K. Thauer, Eur. J. Biochem., 2004, 271, 195–204 Search PubMed.
- (a) M. K. Denk, S. Gupta, J. Brownie, S. Tajammul and A. J. Lough, Chem.–Eur. J., 2001, 7, 4477–4486 Search PubMed; (b) M. K. Denk, A. Hezarkhani and F. Zheng, Eur. J. Inorg. Chem., 2007, 3527–3534 Search PubMed; (c) M. K. Denk, J. M. Rodezno, S. Gupta and A. J. Lough, J. Organomet. Chem., 2001, 617–618, 242–253 CrossRef CAS.
- E. Rabe and H. W. Wanzlick, Liebigs Ann. Chem., 1973, 40–44 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- A. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 Search PubMed.
- (a) C. A. de Wit, Chemosphere, 2002, 46, 583–624 Search PubMed; (b) The Stockholm convention (2001) and its annexes list polychlorinated and brominated hydrocarbons that have been phased out due to their persistence in the environment, their bioaccumulation, their toxicity and capability to act as endocrinological disruptors (http://chm.pops.int/). Excluding the list of newly proposed compounds, the list now (2015) comprises 23 chlorinated and brominated hydrocarbons.
- (a) J. Chapman, R. Hill, J. Muir, C. W. Suckling and D. J. Viney, J. Pharm. Pharmacol., 1967, 19, 231–239 Search PubMed; (b) F. J. Weigert and J. K. Karel, J. Fluorine Chem., 1987, 37, 125–149 Search PubMed.
- J. Dolfing, A. J. van den Wijngaard and D. B. Janssen, Biodegradation, 1993, 4, 261–282 Search PubMed.
- (a) P. Adriaens and D. Grbić-Galić, Chemosphere, 1994, 29, 2253–2259 Search PubMed; (b) A. Neumann, G. Wohlfarth and G. Diekert, J. Biol. Chem., 1996, 271, 16515–16519 Search PubMed; (c) X. Maymó-Gatell, Y. T. Chien, J. M. Gossett and S. H. Zinder, Science, 1997, 276, 1568–1571 Search PubMed; (d) W. Schumacher, C. Holliger, A. J. Zehnder and W. R. Hagen, FEBS Lett., 1997, 409, 421–425 Search PubMed; (e) G. Wohlfarth and G. Diekert, Curr. Opin. Biotechnol., 1997, 8, 290–295 Search PubMed; (f) C. Holliger, G. Wohlfarth and G. Diekert, FEMS Microbiol. Rev., 1998, 22, 383–398 Search PubMed; (g) A. Mägli, M. Messmer and T. Leisinger, Appl. Environ. Microbiol., 1998, 64, 646–650 Search PubMed; (h) L. Adrian, U. Szewzyk, J. Wecke and H. Görisch, Nature, 2000, 408, 580–583 Search PubMed.
- (a) M. Bunge, L. Adrian, A. Kraus, M. Opel, W. G. Lorenz, J. R. Andreesen, H. Görisch and U. Lechner, Nature, 2003, 421, 357–360 Search PubMed; (b) H. Smidt and W. M. de Vos, Annu. Rev. Microbiol., 2004, 58, 43–73 Search PubMed.
- (a) M. Bommer, C. Kunze, J. Fesseler, T. Schubert, G. Diekert and H. Dobbek, Science, 2014, 346, 455–458 Search PubMed; (b) K. A. Payne, C. P. Quezada, K. Fisher, M. S. Dunstan, F. A. Collins, H. Sjuts, C. Levy, S. Hay, S. E. Rigby and D. Leys, Nature, 2015, 517, 513–516 Search PubMed.
- (a) R. J. Strunk, P. M. DiGiacomo, K. Aso and H. G. Kuivila, J. Am. Chem. Soc., 1970, 92, 2849–2856 Search PubMed; (b) A. Sanders and W. P. Giering, J. Org. Chem., 1973, 38, 3055 Search PubMed; (c) T. Ando, F. Namigata, H. Yamanaka and W. Funasaka, J. Am. Chem. Soc., 1967, 89, 5719–5721 Search PubMed.
- (a) C. Chatgilialoglu, D. Griller and M. Lesage, J. Org. Chem., 1988, 53, 3641–3642 Search PubMed; (b) M. Ballestri, C. Chatgilialoglu, K. B. Clark, D. Griller, B. Geise and B. Kopping, J. Org. Chem., 1991, 56, 678–683 Search PubMed; (c) C. Chatgilialoglu, Chem.–Eur. J., 2008, 14, 2310–2320 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |