DOI:
10.1039/C6SC03196J
(Edge Article)
Chem. Sci., 2017,
8, 361-365
Photochromic Torsional Switch (PTS): a light-driven actuator for the dynamic tuning of π-conjugation extension†
Received
19th July 2016
, Accepted 15th August 2016
First published on 16th August 2016
Abstract
Here we present a molecular architecture that can reversibly change the geometric conformation of its π-system backbone via irradiation with two different wavelengths. The proposed ‘molecular actuator’ consists of a photoswitchable azobenzene orthogonally connected to a π-conjugated bithiophene by both direct and aliphatic linker-assisted bonding. Upon exposure to 350 nm light, the trans azobenzene moiety isomerizes to its cis form, causing the bithiophene to assume a semiplanar anti conformation (extended π-conjugation). Exposure to 254 nm light promotes the isomerization of the azobenzene unit back to its initial extended trans conformation, thus forcing the bithiophene fragment to twist out of coplanarity (restricted π-conjugation). The molecular conformation of the bithiophene was characterized using steady-state UV-vis and nuclear magnetic resonance spectroscopy, as well as ab initio computations. The proposed molecular design could be envisaged as a π-conjugation modulator, which has potential to be incorporated into extended linear π-systems, i.e. via the terminal α-thiophene positions, and used to tune their optical and electronic properties.
Introduction
The ability to control mechanical motion at the molecular level is pivotal for the development of novel responsive materials able to translate external stimuli into work. In the last 20 years, a large variety of ‘molecular actuators’ able to convert thermal, chemical, and photochemical energy into operating motion have been successfully employed to perform tasks at the meso- and macroscopic levels.1–12 The restrained mechanistic action of these molecular actuators is commonly correlated with thoughtful design of their dynamic molecular structure. In order to provide different functions, various molecular architectures have been proposed, e.g. shuttles,13,14 rotors,15–19 scissors,20,21 cars,22,23 chemical valves,24,25 and artificial molecular-based muscles.26–29 Despite the large variety of synthetic responsive architectures reported in the literature, examples of designs conceived to exploit the light-mediated molecular motion as a means to tune the conjugation length of linear π-systems are few. Molecular actuators proposed for such a scope commonly rely on their ability to mechanically modulate the π-bond geometry of a conjugated structure either by the cleavage-formation of π-bonds,30–33 or by the torsion-planarization of π-orbitals.34–36 In the first case, molecular actuators, e.g. diarylethenes, are usually incorporated as monomeric unit into the π-conjugated backbone of oligomers and polymers, however their operation reversibility efficiency decreases inversely with the enhancement of the π-system. In contrast, the second class of actuators are usually incorporated as pendant functionality along π-conjugated structures, thus their operation is not directly affected by the extension of π-system. The function of the latter type of actuators, however, is usually limited by the possibility to switch the orientation of their constituents between only two semiplanar conformations (e.g. syn and anti)34–36 that do not differ significantly in their electronic structure. The restricted operation of these molecular architectures does not offer the possibility to obtain highly twisted π-orbital geometries, which if achieved would allow for the full exploitation of the physical properties of π-conjugated systems.37–43 Molecular actuators able to modulate the π-bond geometry beyond the usual semiplanar conformation are thus extremely desirable as they can allow for preparation of novel responsive semiconductor materials, e.g. π-conjugated oligomers and polymers, that may allow for the fabrication of optical and electronic devices with unprecedented properties and tunability. Herein we report on a novel molecular design, referred to as a ‘Photochromic Torsional Switch’ (PTS), able to reversibly change the conformation of its π-conjugated backbone from twisted to planar in response to light. The proposed PTS actuator can be envisaged as a chemical motif that can be potentially incorporated into the backbone of linear π-systems to reversibly modulate their conjugation length.
Results and discussion
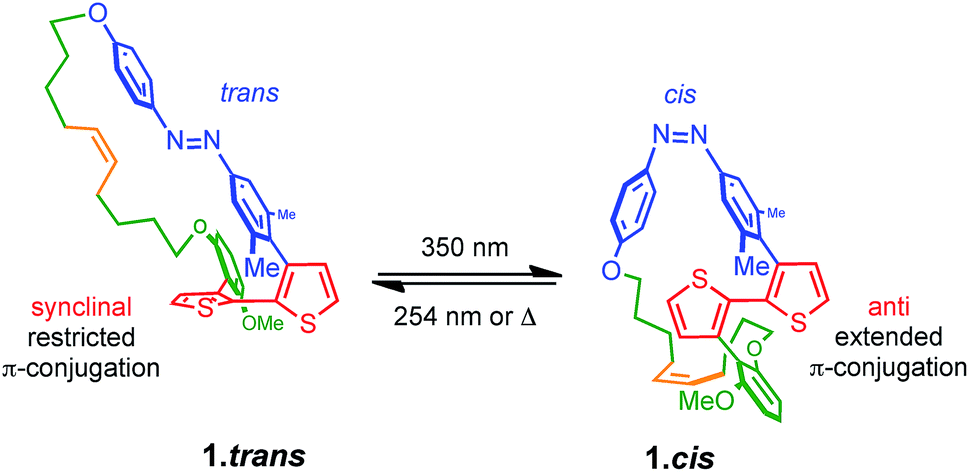
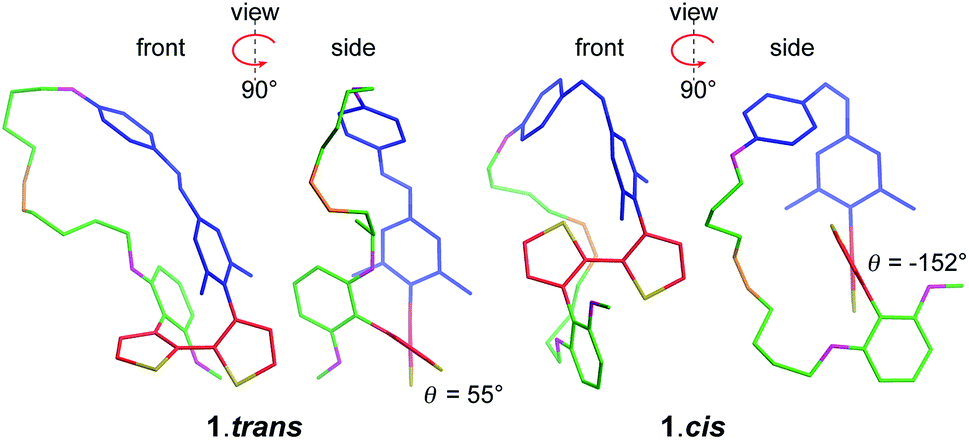
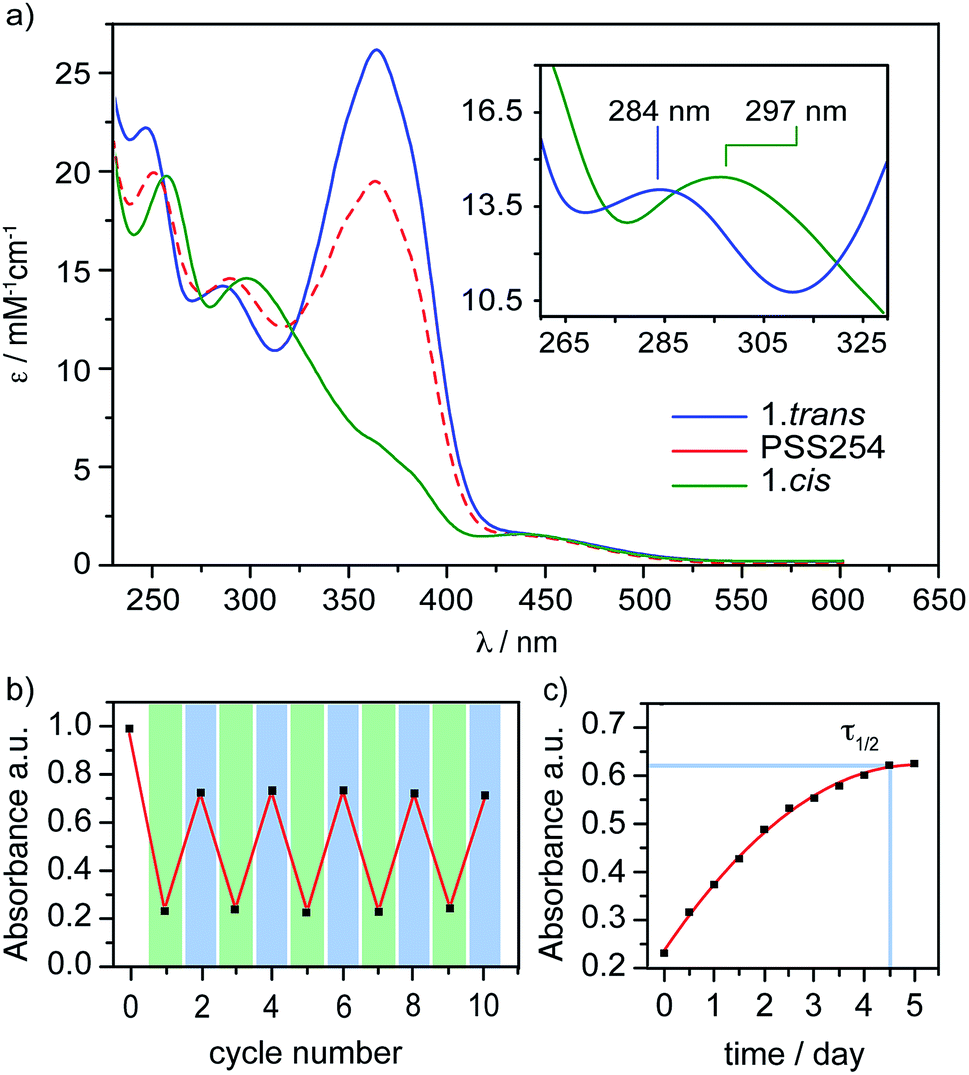
The design of PTS 1 is illustrated in Scheme 1. The PTS actuator consists of an azobenzene-switch laterally connected to a bithiophene unit by both direct and aliphatic linker-assisted bonding. Two methyl units in the meta position of the azobenzene guarantees its orientation in an orthogonal arrangement to the bithiophene fragment, thus reducing the communication between the π-orbitals of the two constituents. A ten-carbon alkene chain connected with an alkoxy benzene unit mechanically transfers the isomerization motion of the azobenzene to the bithiophene backbone. PTS actuator 1 was synthesized via a convergent route involving the attachment of the azobenzene and the resorcinol monomethyl ether groups to bithiophenic unit, followed by the introduction of terminal olefin side chains for the final macrocyclization. The desired PTS compound 1 was obtained as the pure trans conformer by intra-molecular cross metathesis of the two terminal alkenes of its open precursor (for details about the synthesis, see ESI†). The geometrical conformations of both 1.trans and 1.cis, were determined by replica-exchange molecular dynamic (REMD) computations performed at the density functional tight binding (DFTB) level (REMD@DFTB).44 Several minima were identified and optimized statically at the density functional theory level (see ESI for details†). The lowest-lying structures revealed that when the azobenzene is in its extended trans conformation (1.trans), the bithiophene unit is forced to twist out of coplanarity with a dihedral angle (θ) of 55° (Fig. 1). Contrarily, when the azobenzene assumes its cis form (1.cis) the bithiophene assumes a more planar and π-conjugated conformation with θ = −152°. The latter dihedral angle is in good agreement with the literature values reported for unsubstituted bithiophenes in their anti-conformation (θ = 146–152°).45,46 On the other hand, the bithiophene configuration in 1.trans has a dihedral angle that significantly differs from the values commonly observed for either the syn or the anti conformers (θ = 35–37°).45,46 The synclinal arrangement assumed by the thiophenes in 1.trans is the result of a suppressed rotation along the thiophene–thiophene bond derived by the stretching of the aryloxy alkene linker. Thus, the bithiophene is mechanically arrested in a conformation that would be energetically unfavourable for the corresponding unmodified counterparts. In order to probe any structural variations of the bithiophenes following azobenzene isomerization, we used UV-vis and nuclear magnetic resonance spectroscopy, and electronic structure computations. The absorption spectra of the 1.trans and 1.cis are the superposition of individual constituting components, namely the azobenzene, in its trans and cis form, and the phenylene–bithiophene segment (see ESI†). The absorption spectrum of 1.trans displays four distinguishable peaks: 454 nm, 363 nm, 284 nm and 245 nm (Fig. 2a). According to TDDFT computation performed on top of the lowest-energy structures as extracted by REMD@DFTB, the first two bands correspond to the typical S0 → S1 and S0 → S2 excitations represented respectively by the n–π* and π–π* transitions localized on the azobenzene. The peak centred at 284 nm is mainly the result of the S0 → S4 excitation of the phenyl-functionalized bithiophene. In the framework of the molecular orbitals, this excitation can be described as a HOMO → LUMO+1 single particle transition involving the π and π* orbitals prevalently localized on the bithiophene with partial delocalization on the phenylene segment and azobenzene unit (see ESI for details†). Notably, this peak is not observed for the open precursor of 1, which exhibits a broader band centred at 363 nm with a shoulder at 270 nm (see ESI†). The resulting spectral broadening of the PTS-open precursor can be ascribed to the retention of full rotational freedom along the thiophene–thiophene bond. The last blue-shifted band of 1 at 245 nm is the result of a high-energy electronic transition involving the π–π* orbitals localized on the phenyl rings of the azobenzene. Upon irradiation at 350 nm approximately 86% of the trans isomer of azobenzene is converted to the cis form (Fig. 2a). The resulting absorption spectrum, 1.cis, exhibits the typical signature of the azobenzene in its cis conformation, with a reduction in the oscillation strength for the π–π* transition (∼23% absorption intensity at 363 nm). However, no significant changes in the absorption profile were observed for the S0 → S1 (n–π*) transition. Conversely, bands at 284 nm and 245 nm exhibit a bathochromic shift of 13 nm and 11 nm, respectively (Fig. 2a). Since the π-conjugation extension of the bithiophene is directly correlated with the peak at 284 nm, any conformational change in its π-bonds geometry will result in a spectral shift of this band. According to the spectral changes and TDDFT assignments, the red-shift observed for this band is ascribable to the planarization of the bithiophenic segment which results in a more effective delocalization of its molecular orbitals along the two thiophene units. After the trans-to-cis isomerization of 1, it is possible to recover about ∼55% of the initial trans isomer by irradiating at 254 nm (photostationary state, Fig. 2a).47 The latter wavelength was selected as an alternative excitation to the usual 430 nm due to the similar oscillator strengths of the n–π* band for the two conformers. Monitoring the UV-vis absorptions of 1 after many repeated alternating irradiation cycles at λ = 350 nm and at λ = 254 nm, respectively, did not result in any noticeable degradation of the compound, highlighting the robustness of the PTS architecture (Fig. 2b). The recovery of the initial 1.trans conformer can also be obtained by thermal relaxation of the azobenzene moiety with a half-life (τ1/2) of ca. 4.5 days at 25 °C (Fig. 2c). It is noteworthy that, such long thermal stability of the cis isomer is not observed for the non-PTS alkoxy-azobenzene analogue (see ESI†). Slow thermal relaxation has been usually reported for azobenzenes that are functionalized in the ortho position with electron donating48 and withdrawing49 groups. The lack of electron directing functionality on the PTS azobenzene, along with the faster thermal relaxation of its open precursor and non-PTS azobenzene analogue suggest that the slow thermal relaxation of 1.cis can be associated to a reduced degree of freedom in the isomerization motion (see ESI†).
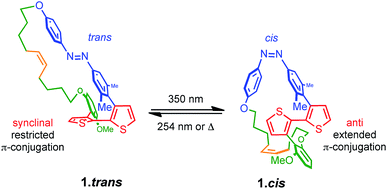 |
| | Scheme 1 Structures of the studied Photochromic Torsional Switch (PTS) unit. | |
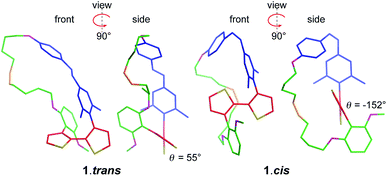 |
| | Fig. 1 Optimized geometries of 1.trans (left) and 1.cis (right) identified from REMD computation. | |
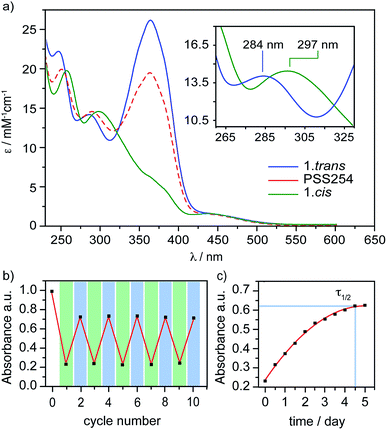 |
| | Fig. 2 (a) Absorption spectrum of 1.trans (blue), 1.cis (green), and the photostationary state PSS254 (red) in tetrahydrofuran (THF) solution. The inset shows the S0 → S4 band of the phenylene–bithiophene segment on an enlarged scale. (b) Measured absorbance at λ = 363 nm of 1 in THF solution alternating irradiation at λ = 350 nm (green) and λ = 254 nm (blue) in repeated switching cycles. (c) Measured absorbance at λ = 363 nm of 1.cis in THF solution during thermal relaxation. | |
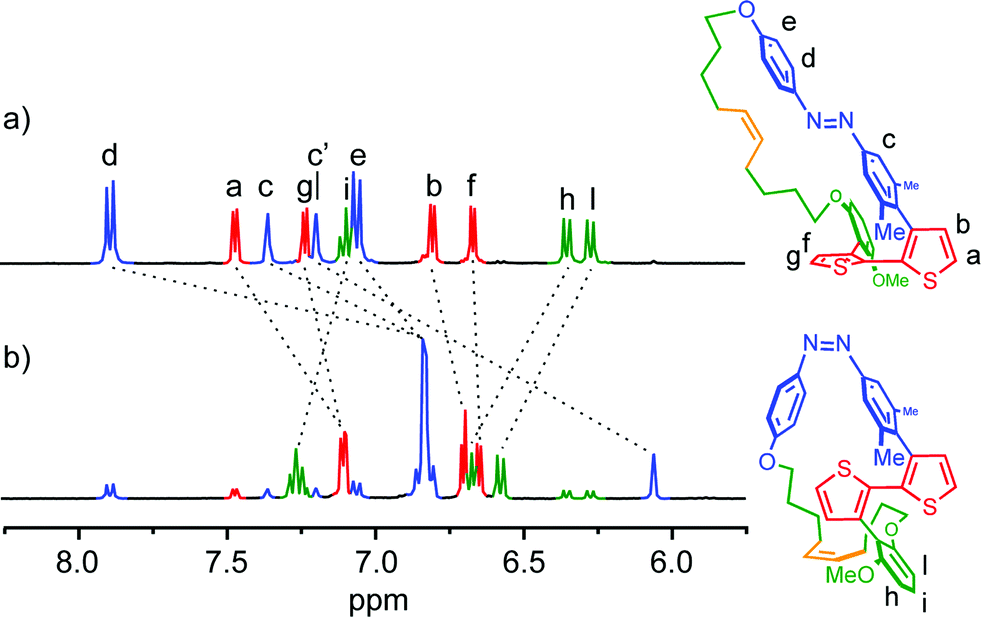
Finally, 1H-NMR spectroscopy was conducted to investigate the rotation along the thiophene–thiophene bond upon the isomerization of the azobenzene moiety. After the trans-to-cis isomerization of 1, the aromatic protons of thiophene Ha (δ 7.47 ppm) and Hb (δ 6.81 ppm) showed a significant up-field shifts of 0.36 and 0.11 ppm, respectively (Fig. 3). On the other hand, the aromatic signals of the linker's phenylene, Hi, Hh and Hl, observed respectively at 7.10, 6.36, and 6.28 ppm for 1.trans, showed downfield shifts of 0.17, 0.33, and 0.30 ppm, respectively. These spectral changes are reasonable given the different molecular geometry assumed by the two PTS conformers. In 1.trans, the thiophene units assume a synclinal arrangement, orienting the two phenyl fragments of the linker and the azobenzene in a parallel-displaced conformation. Such arrangement leads to the lowering of the magnetic shielding effect in the thiophenes (downfield shift) while increasing that of the phenylenes due to π–π stack interactions (upfield shift). When the azobenzene isomerizes to its cis form, the thiophenes assume a more planar conformation with concomitant edge-to-face arrangement with the corresponding pseudo-orthogonal phenyl rings of the linker and azobenzene. This conformation results in an increase in the magnetic shielding of the thiophene protons (upfield shift), and a decrease in the shielding of the phenylene counterpart (downfield shift). The resulting 1H-NMR spectral profile of 1.cis is in good agreement with bithiophene signature of the open analogue as well as with ‘planarly constrained’ bithiophenes reported by Sugiyasu et al.50 Hence, it can be concluded that compound 1 undergoes a rotation-like (twisted-planar) motion along the thiophene–thiophene bond upon trans–cis isomerization of the azobenzene switch.
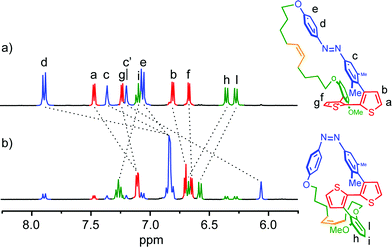 |
| | Fig. 3 Aromatic region of the 1H-NMR spectrum of compound 1 in tetrahydrofuran-d8. Before (a) and after 1 h of irradiation at 350 nm (b). | |
Conclusions
In conclusion, a novel molecular actuator capable of modulating the extension of its π-conjugated backbone in response to light has been designed. The mechanical motion associated with the trans–cis isomerization of an azobenzene has been translated to a change in the planarity of the connected bithiophene, thus allowing for the dynamic tuning of its π-conjugation. This work provides a basis for the development of novel light-driven molecular actuators that can be used to tune the physical properties of extended π-conjugated system. The possibility to integrate the proposed PTS structure into conjugated oligomers and polymers, i.e. via the terminal α-thiophene positions, can potentially lead to the next-generation of photochromic molecular materials with both photochromic and photoconductive behaviour, and allow for the fabrication of novel light responsive optoelectronic devices.
Acknowledgements
We would like to thank Prof. Holger Frauenrath for his helpful discussions, and the Swiss National Science Foundation (SNSF) Ambizione program (PZ00P2_148050) for the financial support. D. F. acknowledges the Alexander von Humboldt foundation for a postdoctoral research fellowship. C. C. acknowledges funding from the European Research Council (ERC StG Grant 306528, “COMPOREL”).
Notes and references
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- D. Bléger, Z. Yu and S. Hecht, Chem. Commun., 2011, 47, 12260–12266 RSC.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247–2279 CrossRef.
- V. Ferri, M. Elbing, G. Pace, M. D. Dickey, M. Zharnikov, P. Samorì, M. Mayor and M. A. Rampi, Angew. Chem., Int. Ed., 2008, 47, 3407–3409 CrossRef CAS PubMed.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- S. Iamsaard, S. J. Aßhoff, B. Matt, T. Kudernac, J. J. L. M. Cornelissen, S. P. Fletcher and N. Katsonis, Nat. Chem., 2014, 6, 229–235 CrossRef CAS PubMed.
- L. Zhang, H. Liang, J. Jacob and P. Naumov, Nat. Commun., 2015, 6, 7429–7440 CrossRef CAS PubMed.
- K. Ichimura, S. Oh and M. Nakagawa, Science, 2000, 288, 1624–1626 CrossRef CAS PubMed.
- J. Bernà,
et al.
, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed.
- S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- J. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS PubMed.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 150–155 CrossRef PubMed.
- T. V. Khuong, H. Dang, P. D. Jarowski, E. F. Maverick and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2007, 129, 839–845 CrossRef CAS PubMed.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612–5613 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Macià, N. NaKatsonis, S. R. Harutyunyan, K. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- J. Morin, Y. Shirai and J. M. Tour, Org. Lett., 2006, 8, 1713–1716 CrossRef CAS PubMed.
- T. D. Nguyen, H. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS PubMed.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375–4379 CrossRef CAS PubMed.
- Y. Liu,
et al.
, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- J. Wu, K. C. Leung, D. Benítez, J. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270–1278 CrossRef PubMed.
- R. H. Baughman, Synth. Met., 1996, 78, 339–353 CrossRef CAS.
- F. Stellacci, C. Bertarelli, F. Toscano, M. C. Gallazzi, G. Zotti and G. Zerbi, Adv. Mater., 1999, 11, 292–295 CrossRef CAS.
- N. Tanifuji, M. Irie and K. Matsuda, Chem. Lett., 2005, 34, 1580–1581 CrossRef CAS.
- T. Kawai, T. Kunitake and M. Irie, Chem. Lett., 1999, 28, 905–906 CrossRef.
- H. Cho and E. Kim, Macromolecules, 2002, 35, 8684–8687 CrossRef CAS.
- B. Jousselme, P. Blanchard, N. Gallego-Planas, J. Delaunay, M. Alain, P. Richomme, E. Levillain and J. Roncali, J. Am. Chem. Soc., 2003, 125, 2888–2889 CrossRef CAS PubMed.
- B. Jousselme, P. Blanchard, M. Alain, E. Levillain, M. Dias and J. Roncali, J. Phys. Chem. A, 2006, 110, 3488–3494 CrossRef CAS PubMed.
- V. A. Azov, J. Cordes, D. Schlüter, T. Dülcks, M. Böckmann and N. L. Doltsinis, J. Org. Chem., 2014, 79, 11714–11721 CrossRef CAS PubMed.
- M. J. Marsella and T. M. Swager, J. Am. Chem. Soc., 1993, 115, 12214–12215 CrossRef CAS.
- A. Giovannitti,
et al.
, Adv. Funct. Mater., 2016, 26, 514–523 CrossRef CAS.
- R. D. Mc Cullough, S. Tristram-Nagle, S. P. Wiliams, R. D. Lowe and M. Jayaraman, J. Am. Chem. Soc., 1993, 115, 4910–4911 CrossRef CAS.
- A. Fin, A. Vargas Jentzsch, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2012, 51, 12736–12739 CrossRef CAS PubMed.
- D. Alonso Doval, M. Dal Molin, S. Ward, A. Fin, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 2819–2825 RSC.
- D. Vonlanthen, A. Mishchenko, M. Elbing, M. Neuburger, T. Wandlowski and M. Mayor, Angew. Chem., Int. Ed., 2009, 48, 8886–8890 CrossRef CAS PubMed.
- D. Vonlanthen, A. Rudnev, A. Mishchenko, A. Käslin, J. Rotzler, M. Neuburger, T. Wandlowski and M. Mayor, Chem.–Eur. J., 2011, 17, 7236–7250 CrossRef CAS PubMed.
- R. Petraglia, A. Nicolaï, M. D. Wodrich, M. Ceriotti and C. Corminboeuf, J. Comput. Chem., 2016, 37, 83–92 CrossRef CAS PubMed.
- V. Hernandez and J. T. L. Navarrete, J. Chem. Phys., 1994, 101, 1369–1377 CrossRef CAS.
- T. Lin and S. Lin, Phys. Chem. Chem. Phys., 2015, 17, 4127–4136 RSC.
- Note: ∼55% of azobenzene trans isomer corresponds to ∼72% absorption intensity at 363 nm. Irradiation at 254 nm does not result in any isomerization of olefin double bond (see ESI†).
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, TDDFT calculations and excited states assignments. See DOI: 10.1039/c6sc03196j |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence