
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkene hydrogenation activity of enoate reductases for an environmentally benign biosynthesis of adipic acid†
Jeong Chan
Joo‡
 *a,
Anna N.
Khusnutdinova‡
b,
Robert
Flick
b,
Taeho
Kim
b,
Uwe T.
Bornscheuer
c,
Alexander F.
Yakunin
*b and
Radhakrishnan
Mahadevan
*b
*a,
Anna N.
Khusnutdinova‡
b,
Robert
Flick
b,
Taeho
Kim
b,
Uwe T.
Bornscheuer
c,
Alexander F.
Yakunin
*b and
Radhakrishnan
Mahadevan
*b
aCenter for Bio-based Chemistry, Division of Convergence Chemistry, Korea Research Institute of Chemical Technology, 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, Republic of Korea. E-mail: jcjoo@krict.re.kr
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, ON M5S 3E5, Canada. E-mail: a.iakounine@utoronto.ca; krishna.mahadevan@utoronto.ca
cInstitute of Biochemistry, Department of Biotechnology & Enzyme Catalysis, Greifswald University, Felix-Hausdorff-Strasse 4, 17487 Greifswald, Germany
First published on 11th October 2016
Abstract
Adipic acid, a precursor for Nylon-6,6 polymer, is one of the most important commodity chemicals, which is currently produced from petroleum. The biosynthesis of adipic acid from glucose still remains challenging due to the absence of biocatalysts required for the hydrogenation of unsaturated six-carbon dicarboxylic acids to adipic acid. Here, we demonstrate the first enzymatic hydrogenation of 2-hexenedioic acid and muconic acid to adipic acid using enoate reductases (ERs). ERs can hydrogenate 2-hexenedioic acid and muconic acid producing adipic acid with a high conversion rate and yield in vivo and in vitro. Purified ERs exhibit a broad substrate spectrum including aromatic and aliphatic 2-enoates and a significant oxygen tolerance. The discovery of the hydrogenation activity of ERs contributes to an understanding of the catalytic mechanism of these poorly characterized enzymes and enables the environmentally benign biosynthesis of adipic acid and other chemicals from renewable resources.
Introduction
The vast majority of commodity chemicals are currently produced from fossil fuels.1 Due to growing concerns over petroleum shortages, climate change, energy security, and human health, there have been tremendous efforts to produce commodity chemicals from renewable resources including plant biomass.2–4 Multi-enzyme-catalyzed cascade reactions have been proposed for the synthesis of value-added chemicals including long-chain dicarboxylic acids, chiral lactones, alcohols, amines, and amino acids.5–8 In addition, several C2–C6 platform chemicals have been successfully produced from glucose using natural or engineered microbial strains.9Adipic acid is one of the most important aliphatic dicarboxylic acids, which is used for the synthesis of Nylon-6,6 polyamide (∼$ 6 billion global market). 2.6 million metric tonnes per year of adipic acid is produced from petroleum-derived benzene but current chemical processes operate under harsh conditions (e.g. extreme temperature or pressure) and produce toxic by-products such as nitrous oxide (N2O).10–12 Recently, the chemical company Rennovia Inc. demonstrated the chemical synthesis of adipic acid from glucose via oxidation of glucose to glucaric acid and the hydro-oxygenation of glucaric acid into adipic acid, but this process requires both high temperatures and pressures, as well as large amounts of organic solvents.13
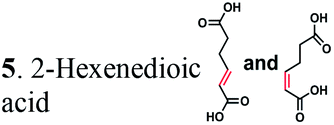
Due to the absence of natural pathways for the production of adipic acid, the bio-based production of adipic acid has been attempted using enzyme discovery and metabolic engineering approaches. A number of non-natural synthetic pathways have been proposed including muconic acid, α-aminoadipate, or α,ω-hydrocarbon dicarboxylic acid pathways.10–12,14,15 In particular, Escherichia coli cells with an engineered aromatic amino acid biosynthesis pathway can produce up to 59.2 g L−1 of cis,cis-muconic acid from glucose, which has to be hydrogenated to adipic acid using chemical catalysts.11 In addition, there have been several recent studies on the production of cis,cis-muconic acid in Saccharomyces cerevisiae, Klebsiella pneumoniae from glucose and in Pseudomonas putida from lignin derivatives.16–19 Recent patents proposed novel pathways to produce adipic acid from 2-hexenedioic acid instead of cis,cis-muconic acid.20 However, most of the proposed pathways require chemical hydrogenation to adipic acid due to the absence of known hydrogenation biocatalysts.10,21 Recently, a biocompatible palladium catalyst was developed for use with microbial growth media.22 But this chemical catalyst still has limitations such as low conversion yields (∼80%) and the inaccessibility of the catalyst to intracellular metabolites, and thus proposed bio- and chemo-catalysis is only suitable for small scale synthesis (Scheme 1).
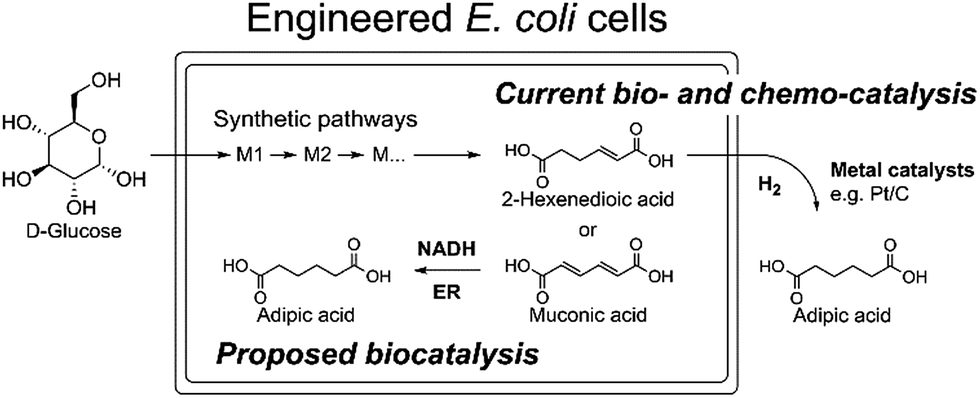 | ||
| Scheme 1 The current biocatalytic synthesis of adipic acid from glucose requires chemical hydrogenation of unsaturated six-carbon dicarboxylic acids e.g. cis,cis-muconic acid or 2-hexenedioic acid. | ||
In contrast to chemical alkene hydrogenation, there is no known enzyme with which biocatalytic hydrogenation to adipic acid has been shown experimentally.10,19,22–25 Thus, it is crucial to identify a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation biocatalyst to replace the chemical hydrogenation step in biochemical routes for the production of adipic acid from renewable feedstocks.
C hydrogenation biocatalyst to replace the chemical hydrogenation step in biochemical routes for the production of adipic acid from renewable feedstocks.
Previously, two families of flavoenzymes have been studied for the biocatalytic hydrogenation of alkenes: Old Yellow Enzymes (OYEs; EC 1.6.99.1) and enoate reductases (ERs; EC 1.3.1.31).26–28 These flavoenzymes exhibited a broad substrate spectrum toward unsaturated substrates bearing an electron-withdrawing group such as an aldehyde, ketone, or carboxylic acid.26,27 The redox reaction of OYEs is facilitated by a noncovalently bound flavin mononucleotide (FMN) cofactor, which is oxidized by the alkene substrate and regenerated via hydride transfer from NADPH. The structural and biochemical characterization of OYEs revealed their high potential in biocatalysis, because they allow the simultaneous introduction of up to two new stereocentres.29 OYEs have been predominantly investigated for the asymmetric reduction of activated alkenes, such as conjugated enals, enones, α,β-dicarboxylic acids, imides, nitroalkenes, and ynones.27,30 It has been shown that the substrate range and catalytic performance of OYEs can be improved using various protein engineering approaches.31–33 In addition, the potential for recombinant ene-reductases to catalyze the reduction of a wide variety of substrates including unsaturated acids has been shown even though no activity for adipic acid production has been reported.34 Compared with OYEs, ERs are less characterized, because these enzymes were found to be oxygen-sensitive.35–37 Clostridial ERs use NADH as an electron donor and contain FAD, FMN, and an [4Fe–4S] iron–sulfur cluster.37,38 ERs can reduce CC double bonds in a variety of monoesters and monoacids, including non-activated 2-enoates, a reaction not catalyzed by OYEs.36,39 Although both OYEs and ERs have the potential to catalyze the hydrogenation of unsaturated six-carbon dicarboxylic acids to adipic acid, these studies have not yet been reported.
In the present work, the CC hydrogenation activity of OYEs and ERs from different microorganisms was analyzed with the aim of identifying an enzyme for the biocatalytic production of adipic acid from unsaturated six-carbon dicarboxylic acids. We found that 25 purified OYEs from various microorganisms showed no hydrogenation activity against 2-hexenedioic acid or muconic acid. In contrast, four ERs from several Clostridia and Bacillus coagulans hydrogenated 2-hexenedioic acid and muconic acid both in vitro and in vivo, producing adipic acid with high yields. Thus, microbial ERs can be used for development of the biocatalytic hydrogenation of unsaturated six-carbon dicarboxylic acids and for the production of adipic acid from renewable resources.
Results and discussion
Screening of purified OYEs for alkene hydrogenation
It has been shown that the OYEs from B. subtilis or Solanum lycoperiscum can hydrogenate unsaturated branched dicarboxylic acids e.g. 2-methylmaleic acid (citraconic acid) and 2-methylfumaric acid (mesaconic acid).27,40 To verify if OYEs can be used for the biocatalytic production of adipic acid, we cloned 25 genes encoding putative OYEs from different microorganisms (Table S1 in the ESI†), which belong to the FMN oxidoreductase family PF00724.41 25 putative OYEs were over-expressed in E. coli and purified to near homogeneity using affinity chromatography on a Ni-NTA resin (data not shown). 21 purified proteins exhibited NADPH-dependent reductase activity against two common OYE substrates: 2-cyclohexen-1-one, but none of them were active in the hydrogenation of 2-hexenedioic acid or muconic acid (Table S1†). In addition, in vivo (whole-cell) biotransformation experiments using three E. coli strains over-expressing OYEs from E. coli (UniProt P77258), Saccharomyces cerevisiae (Q03558), or Bacillus subtilis (P54550) revealed no hydrogenation of 2-hexenedioic acid to adipic acid (data not shown).The analyzed microbial OYEs showed no hydrogenation activity toward unsaturated six-carbon dicarboxylic acids (5, 6, and 7). This is consistent with previous reports that OYEs cannot easily reduce α,β-unsaturated carboxylic acids without an activating group or additional electron-withdrawing groups such as a second acid- or ester group, a halogen or a nitrile,28 suggesting that OYEs may not be suitable for the hydrogenation of unsaturated six-carbon dicarboxylic acids. Although certain OYEs are capable of the asymmetric hydrogenation of methyl-branched dicarboxylic acids, i.e. 2-methylmaleic acid and 2-methylfumaric acid,40 activated α,β-unsaturated aldehydes or ketones are known to serve as preferred substrates42 and thus, there has been no report for the OYE-catalyzed hydrogenation of non-activated 2-enoates e.g. trans-cinnamic acid.
Whole-cell anaerobic hydrogenation of unsaturated six-carbon dicarboxylic acid using ERs
Next, we characterized the alkene hydrogenation activity of bacterial ERs. The first known ERs have been found mainly in Clostridia.37 It has been shown that the ER from Clostridium tyrobutyricum (ER-CT) exhibited a broad substrate specificity toward α,β-unsaturated carboxylates in vivo.39 ER-CT has a 50–82% sequence identity to ERs from B. coagulans (ER-BC), C. acetobutylicum (ER-CA), C. kluyveri (ER-CK), C. ljungdahlii (ER-CL), and Moorella thermoacetica (ER-MT) (Fig. S1 and Table S2†). We also selected the Bcoa0725 gene (GenBank CP003056.1, location: 769909-771897) from a thermosphilic lactic acid producer, B. coagulans 36D1, which is annotated as an uncharacterized NADH:flavin oxidoreductase/NADH oxidase.43 The Bcoa0725 protein sequence (ER-BC, G2TQU6) shows the presence of several putative cofactor binding domains, which are highly conserved in the ER family and a 2,4-dienoyl CoA reductase (DCR) from E. coli38 (Fig. S1 and S2†). Therefore, we conducted a series of in vivo biotransformations using E. coli cells expressing the six recombinant ERs.The whole-cell anaerobic biotransformation of 2-hexenedioic acid (20 mM) using the six ERs expressed in the E. coli strain revealed the production of adipic acid by the five ERs (except for ER-CT) (Fig. 1). Adipic acid was purified from the biotransformation medium using HPLC with an Aminex HPX-87H column, and its structural identity was confirmed by LC-MS and NMR (Fig. S3†). ER-BC, ER-CA, and ER-CK catalyzed the complete conversion of 2-hexenedioic acid (20 mM) to adipic acid within 3 h, while ER-CL and ER-MT showed complete hydrogenation of 2-hexenedioic acid after 6 h and 48 h, respectively (Table 1, Fig. S4†). These results indicate that the biotransformation of 2-hexenedioic acid can yield up to 2.5–3.0 g L−1 of adipic acid. The fast bioconversion of 2-hexenedioic acid to adipic acid by the ER-BC, ER-CA, and ER-CK strains suggests that E. coli cells can support high rates of substrate (2-hexenedioic acid) uptake and product (adipic acid) secretion, making this system useful for industrial applications. The complete transformation of 2-hexenedioic acid to adipic acid also implies that E. coli cells have no enzymes metabolizing these chemicals. The rates of in vivo biotransformation of 2-hexenedioic acid correlated with the expression level and solubility of recombinant ERs in E. coli cells (Fig. S4 and Table S3†). In particular, the inactivity of ER-CT in the conversion of 2-hexenedioic acid might be due to its low/insoluble expression in E. coli.
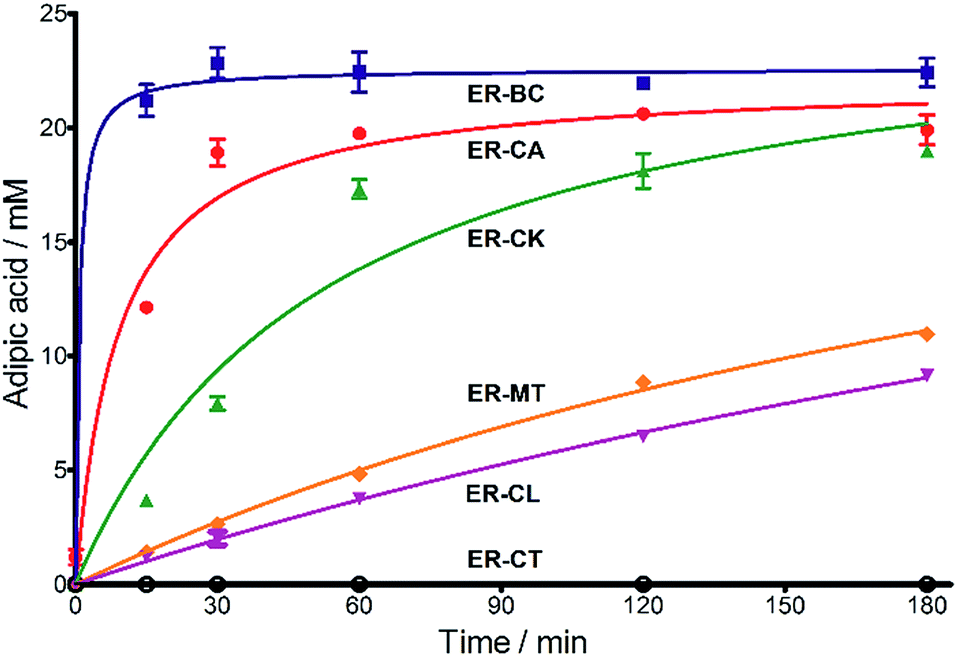 | ||
Fig. 1 Time course for the biotransformation of 2-hexenedioic acid into adipic acid by E. coli cells expressing ERs. A mixture of cis- and trans-isomers of 2-hexenedioic acid (20 mM) was used as a substrate. The formation of adipic acid was analyzed using HPLC. ( ) = ER-BC, ( ) = ER-BC, ( ) = ER-CA, ( ) = ER-CA, ( ) = ER-CK, ( ) = ER-CK, ( ) = ER-MT, ( ) = ER-MT, ( ) = ER-CL, and ( ) = ER-CL, and ( ) = ER-CT. ) = ER-CT. | ||
| Substratesa | Activity [U mg−1 protein] | Substrate conversion% in vitrob (12 h) | Substrate conversion% in vivo (24 h) | |||
|---|---|---|---|---|---|---|
| ER-BC | ER-CA | ER-BC | ER-CA | ER-BC | ER-CA | |
| a Substrate concentrations used for specific activity detection: 1 = 200 mM, 2 = 200 mM, 3 = 50 mM, 4 = 1 mM, 5 = 35 mM, 6 = 0.7 mM, 7 = 0.7 mM. Substrate concentrations used for conversion calculations in vivo and in vitro: 1 = 5 mM, 2 = 20 mM, 3 = 20 mM, 4 = 3 mM, 5 = 20 mM, 6 = 0.7 mM, 7 = 0.7 mM. b P33160 formate dehydrogenase was used to regenerate NADH in the reaction mixture. c E. coli cells without ERs can hydrogenate 2-cyclohexen-1-one due to the presence of the endogenous N-ethylmaleimide reductase NemA (P77258) (Table S1). This background activity was subtracted from the experimental data. d BDL – below detection limit; ND – not determined; MC – metabolized by cells. | ||||||
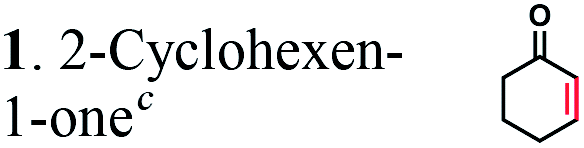
|
0.08 ± 0.03 | 0.028 ± 0.001 | BDL | ND | 21.3 ± 0.15 | 18.2 ± 1.8 |
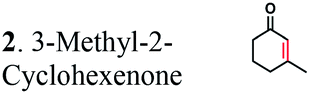
|
0.13 ± 0.03 | 0.091 ± 0.005 | BDL | BDL | BDL | BDL |
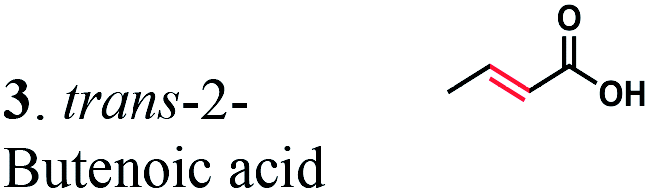
|
0.037 ± 0.01 | 0.062 ± 0.003 | BDL | 1.5 ± 1.05 | MC | MC |
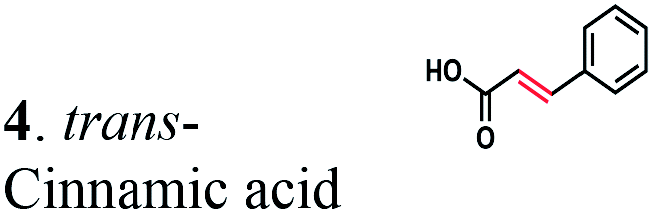
|
0.39 ± 0.001 | 3.2 ± 0.1 | 72 ± 1.8 | 85.3 ± 31.1 | MC | MC |
|
2.3 ± 0.04 | 0.056 ± 0.003 | 17.9 ± 0.01 | 5.74 ± 1.8 | 99.6 ± 3.5 | 93.4 ± 3.7 |
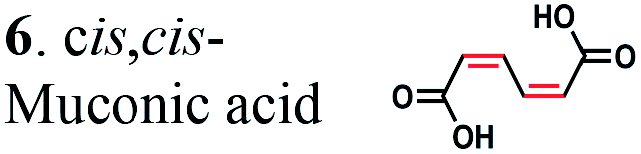
|
BDL | 0.056 ± 0.003 | BDL | BDL | 94.3 ± 0.23 | 64.3 ± 0.0 |
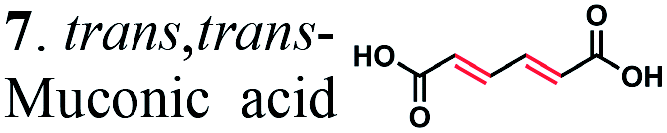
|
BDL | 0.059 ± 0.001 | BDL | BDL | 91.1 ± 0.79 | 81.0 ± 1.2 |
To further investigate the suitability of the ER strains for the biocatalytic production of adipic acid, we tested them for the in vivo bioconversion of muconic acid, which is the final bioproduct of the combined bio- and chemo-catalytic processes for the production of adipic acid from biomass.11,19 Whole-cell biotransformation experiments with cis,cis- and trans,trans-isomers of muconic acid (0.7 mM) revealed that the ER-BC, ER-CA and ER-MT strains hydrogenated both isomers to adipic acid (Fig. 2). After a 24 h incubation, no substrate (cis,cis- or trans,trans-isomers of muconic acid) or intermediate (2-hexenedioic acid) were detected in the culture, and a 99% yield of adipic acid was obtained with these strains (Fig. 2a–c). The ER-CK and ER-CL strains also exhibited a 99% yield of adipic acid from the trans,trans-isomer, but a lower yield (<29%) from the cis,cis-isomer of muconic acid (Fig. 2d and e). Interestingly, with the latter substrate the ER-CK and ER-CL strains produced three additional products identified as 2-hexenedioic, trans,trans-muconic acid, and a 3-hexenedioic-like compound with m/z 143.0319 (Fig. 2d and e) suggesting the presence of cis–trans isomerase activity in these ERs. The formation of a 3-hexenedioic acid-like compound from cis,cis-muconic acid by these enzymes is similar to the reductase activity of the E. coli DCR, which is a functional homolog of ERs and catalyzes the hydrogenation of enoyl-CoA substrates. Mutated E. coli DCR can catalyze the hydrogenation of 2,4-dienoyl CoA into 3-enoyl CoA instead of 2-enoyl CoA via a cryptic alternate proton donor in the absence of the primary donor.44 ER-CK and ER-CL may have a similar reaction mechanism. Our results indicate that the characterized microbial ERs can catalyze the sequential hydrogenation of the two CC bonds of the six-carbon dicarboxylic acids, but appear to have different isomeric preferences. Thus, whole-cell transformations of 2-hexenedioic, cis,cis-muconic and trans,trans-muconic acid revealed that ER-CA and ER-BC out of the six ERs tested in this work produced adipic acid from unsaturated six-carbon dicarboxylic acids with a high conversion rate and yield, and formed no by-products, indicating that these two ERs can be good candidates for metabolic engineering.
 | ||
| Fig. 2 Biotransformation of cis,cis- and trans,trans-isomers of muconic acid (0.70 mM) by E. coli cells expressing (a) ER-BC, (b) ER-CA, (c) ER-MT, (d) ER-CK, and (e) ER-CL. Substrates and products were detected by LC-MS. (1) cis,cis-muconic acid, (2) trans,trans-muconic acid, (3) 2-hexenedioic acid, (4) adipic acid, and (5) 3-hexenedioic acid-like compound with m/z 143.0319. | ||
Anaerobic over-expression and purification of bacterial ERs
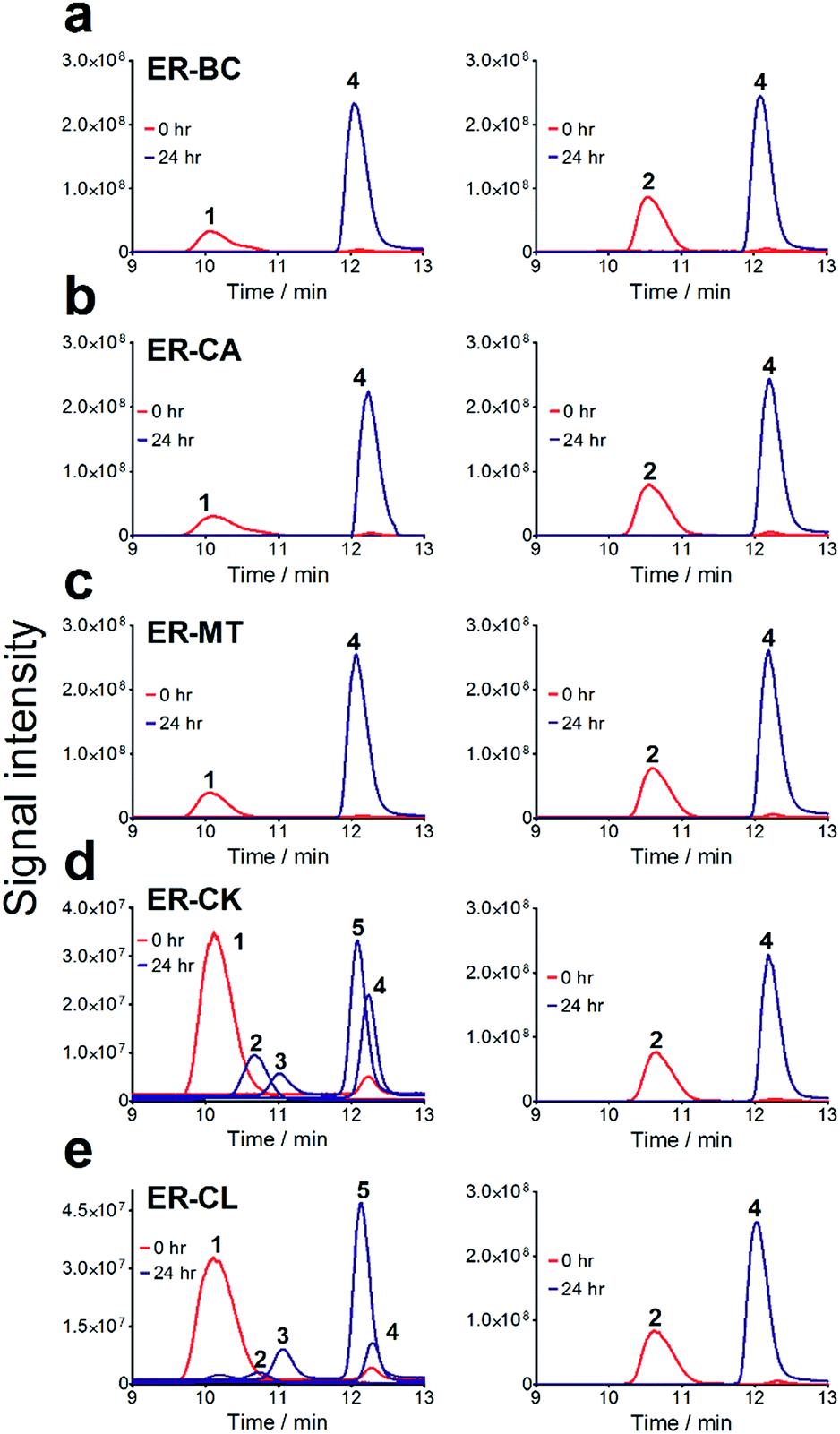
For the in vitro biochemical characterization of ERs, six ERs were recombinantly expressed and purified under anaerobic conditions. For the improved synthesis of Fe–S proteins, we used the E. coli BL21 (DE3) ΔiscR strain with a deletion of the iscR gene encoding a transcriptional repressor of proteins involved in the biosynthesis of Fe–S clusters.45 Anaerobic over-expression in E. coli and affinity purification of the six cloned ERs produced significant amounts of soluble protein for ER-CA, ER-CK, and ER-BC (>5 mg L−1), whereas the other three ERs showed lower expression (Fig. S5†). Purified ER-CA and ER-BC exhibited a brown colour in solution and an absorption spectrum with a shoulder at 380 nm and a flavin-like maximum at 450 nm (Fig. 3a and b), whereas preparations of the other four ERs were less coloured (data not shown). Both the brown colour of purified ER-CA and ER-BC and the 380 nm shoulder in their absorption spectra suggest the presence of a functional [4Fe–4S] cluster. Similar absorption spectra were also reported for ERs purified from the cells of C. kluyveri and C. tyrobutyricum.46,47 Moreover, anaerobically purified ER-CA and ER-BC are partially reduced and can be completely oxidized by incubation in air as indicated by the absorbance decrease at 380 nm and 450 nm. Based on the absorption spectra, the addition of NADH in excess (3 mM) resulted in complete reduction of both proteins, which can be partially re-oxidized by the subsequent addition of trans-cinnamic acid (6 mM) under anaerobic conditions. These results suggest that both ER-CA and ER-BC exhibit hydrogenation activity toward trans-cinnamic acid.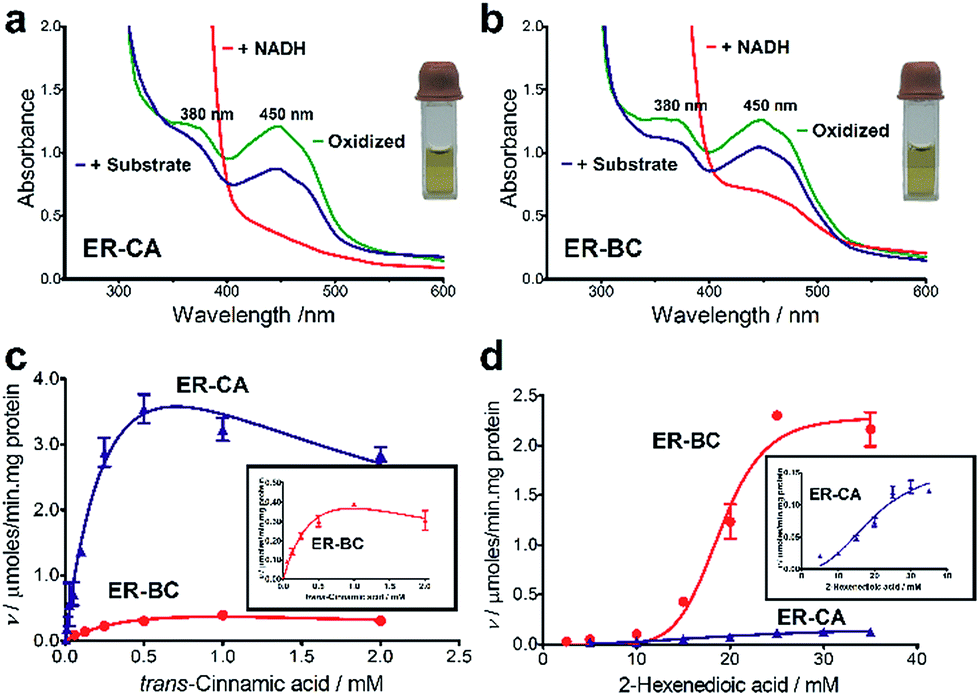 | ||
| Fig. 3 Absorption spectra and enoate reductase activity of purified ERs. (a and b) UV-visible absorption spectra of (a) ER-CA and (b) ER-BC. Green curves: ERs in the oxidized state (after overnight incubation in air). Red curves: ERs reduced by the addition of 3 mM NADH to the protein sample in the anaerobic cuvette (under argon). Blue curves: ERs partially oxidized by the addition of the substrate (6 mM trans-cinnamic acid) to the NADH-reduced protein (under argon). Inserts are photos of the sealed spectrophotometric cuvettes showing the brown colour of the purified ERs. (c and d) NADH-dependent anaerobic hydrogenation of (c) trans-cinnamic acid or (d) 2-hexenedioic acid by purified ER-CA (blue lines) and ER-BC (red lines). Enzyme reactions contained the indicated concentration of the substrates trans-cinnamic acid titrated with 1 N NaOH (c) or 2-hexenedioic acid dissolved in isopropanol (d). | ||
In vitro and in vivo hydrogenation activity of ER-CA and ER-BC
The substrate range of purified ERs was characterized using a set of unsaturated aliphatic and aromatic substrates including 2-cyclohexen-1-one, 3-methyl-2-cyclohexenone, trans-2-butenoic acid, trans-cinnamic acid, 2-hexenedioic acid, cis,cis-muconic acid, and trans,trans-muconic acid (Table 1). ER-BC exhibited both NADPH- and NADH-dependent hydrogenation activity against 2-hexenedioic acid with NADH supporting higher activity (Fig. S6†). Anaerobic assays revealed that purified ER-CA had high hydrogenation activity against cinnamic acid (3.5 U mg−1 protein) and low activity with 2-hexenedioic acid (0.05 U mg−1 protein in aqueous buffer solution, Fig. S7† and 0.13 U mg−1 protein in 14% isopropanol, Fig. 3d) and trans-butenoic acid (0.008 U mg−1 protein). In addition, ER-BC exhibited significant reductase activity against trans-cinnamic acid (0.39 U mg−1 protein), and 2-hexenedioic acid (0.09 U mg−1 protein in aqueous buffer solution, Fig. S7† and 2.3 U mg−1 protein in 14% isopropanol, Table 1). Although ER-BC showed >91% in vivo substrate conversion with cis,cis- and trans–trans isomers of muconic acid, no detectable in vitro hydrogenation activity of this enzyme against muconic acid could be demonstrated suggesting that it might have a low affinity for this substrate.Kinetic studies with purified ER-CA and ER-BC were performed using trans-cinnamic and 2-hexenedioic acid as substrates (Fig. 3c and d). Both ERs could efficiently produce 3-phenylpropanoic acid from trans-cinnamic acid (Fig. 3c). 3-Phenylpropanoic acid is an important intermediate metabolite in the phenylpropanoid pathway for the synthesis of flavonoids, which are valuable natural antioxidant products.35,48 The kinetic parameters (kcat and Km) of ER-CA for trans-cinnamic acid were 9.7 s−1 and 0.40 mM, and those of ER-BC were 1.1 s−1 and 0.68 mM, resulting in a 14.8-fold higher catalytic efficiency of ER-CA than that of ER-BC (kcat/Km, 24.0 vs. 1.6 s−1 mM−1). Both ERs exhibited moderate substrate inhibition with trans-cinnamic acid (Ki, 1.2 vs. 1.4 mM, respectively) (Fig. 3c). In contrast to trans-cinnamic acid, ER-CA and ER-BC showed no saturation for 2-hexenedioic acid dissolved in aqueous buffer solution despite their significant in vitro activity (Fig. S7†), which might be due to the limited solubility of the substrate in aqueous solutions.
To further increase the concentration of dissolved 2-hexenedioic acid in the assays, it was dissolved in a reaction mixture containing water-miscible organic solvents i.e., isopropanol, methanol, and dimethyl sulfoxide (DMSO) (Fig. 3 and S8†). Both ERs showed sigmoidal kinetics in the concentration range from 2.5 to 35 mM in the presence of 14% isopropanol (Fig. 3d). Compared with ER-CA, ER-BC had a higher turnover rate (kcat, 2.86 vs. 0.214 s−1) and a similar substrate binding affinity (Km, 18.9 vs. 20.5 mM) for 2-hexenedioic acid, resulting in a 14.5-fold higher catalytic efficiency of ER-BC compared with that of ER-CA (kcat/Km, 0.151 vs. 0.0104 s−1 mM−1). ER-BC also showed similar sigmoidal kinetics in the other two co-solvents (Fig. S8†). It is known that multimeric enzymes exhibit cooperative kinetics.49,50 The ER family can form a homo-multimeric protein37,46 and size exclusion chromatography revealed that ER-BC and ER-CA purified in this work also form a homo-dimer and trimer, respectively (data not shown). In addition, the presence of water-miscible organic solvents can affect the cooperative kinetics of enzymes by changing the solubility and the binding pattern of substrates.51 Therefore, the unusual sigmoidal kinetics with purified ER-CA and ER-BC for 2-hexenedioic acid might be due to their multimeric state or the presence of water-miscible solvents but further experiments will be required to elucidate the mechanism of enzyme catalysis. In vitro biochemical characterization of ERs indicate that ER-CA and ER-BC are the preferred reductases for bio-hydrogenation of aromatic and aliphatic alkenes, respectively.
Thermostability and oxygen tolerance of ER-CA and ER-BC
Both thermostability and oxygen tolerance are important performance parameters for enzyme applications in biocatalysis. The thermostability of purified ER-CA and ER-BC was evaluated by comparing their thermal aggregation curves over a broad range of temperatures (25 to 80 °C) and their temperatures of aggregation Tagg (Fig. 4a). Although ER-CA and ER-BC belong to the same protein family and have a similar structural fold (Fig. S2†), they had different thermal aggregation profiles. As shown in Fig. 4a, ER-BC from a thermosphilic strain exhibited a significantly higher thermostability (Tagg 49.9 °C) with a Tagg almost 11 °C higher than that of ER-CA (39.0 °C). This is consistent with a rather low thermostability of most proteins from mesophilic Clostridia.52–54 The addition of trans-cinnamic acid had no effect on the thermostability of both proteins (ER-CA, 39.0 vs. 39.3 °C and ER-BC, 49.9 vs. 49.3 °C), which is consistent with the low affinity of these enzymes to this substrate. | ||
| Fig. 4 Stability of purified ER-CA and ER-BC. (a) Thermostability: differential static light scattering analysis of ERs in the absence (blue lines) or presence (red lines) of 1 mM trans-cinnamic acid. (b) Oxygen tolerance: the initial activities (100%) of ER-CA (3.5 U mg−1 protein) and ER-BC (2.3 U mg−1 protein) were measured with trans-cinnamic acid and 2-hexenedioic acid, respectively. | ||
It is known that ERs are oxygen-sensitive enzymes due to the presence of an oxygen-labile [4Fe–4S] cluster coordinated by the strictly conserved motif C-2X-C-3X-C-11X-C (Fig. S1†).35 Despite the high biocatalytic potential of ERs for alkene hydrogenation, the oxygen sensitivity of these enzymes has impeded their application in industrial processes. However, our experiments revealed the significant tolerance of purified ER-CA and ER-BC to inactivation by oxygen (Fig. 4b). In the presence of air (21% oxygen), ER-CA exhibited 30% residual activity after one day of storage and was completely inactivated by oxygen after two additional days of storage. Purified ER-BC showed even higher oxygen tolerance with 33% remaining activity after three days of incubation with air, and with the same inactivation dynamics under aerobic and anaerobic conditions. The high oxygen tolerance of ER-CA and especially ER-BC may be associated with the high stability of their [4Fe–4S] cluster or with the restricted access of oxygen to the clusters.55–57 Future work using site-directed mutagenesis and protein crystallization is required to reveal the mechanism of the thermostability and oxygen tolerance of these enzymes.
Conclusions
In summary, we have demonstrated the biocatalytic hydrogenation of unsaturated six-carbon dicarboxylic acids to adipic acid using ERs. The substrate specificity of ERs is comparable to that of the biocompatible palladium catalyst, whereas conversion yields of ERs for 2-hexenedioic acid and muconic acid are higher than those of the palladium catalyst (91% vs. ∼80%).22 Given the high conversion yield of ERs and literature reports on muconic acid production of up to 59 g L−1,11,16–19 one can expect that an ER based process could lead to similarly high titers. In particular, ER-BC exhibits high oxygen tolerance and thermostability making it useful for in vivo and in vitro applications to overcome the limitations of chemical catalysts. Thus, the CC hydrogenation activity of ERs demonstrated in our work can potentially replace the chemical hydrogenation step in current synthetic protocols creating a completely bio-based pathway for the greener production of adipic acid or other biochemicals e.g. 3-phenylpropanoic acid from renewable feedstocks.
Acknowledgements
The authors thank all members of the BioZone Centre for Applied Science and Bioengineering and Christine Achampong for help in conducting the experiments. Dr K. T. Shanmugam (University of Florida) and Dr P. Hallenbeck (University of Montreal) are thanked for providing the B. coagulans 36D1 genomic DNA and the E. coli BL21 (DE3) ΔiscR strain, respectively. This work was supported by the Government of Canada through Genome Canada and the Ontario Genomics Institute (2009-OGI-ABC-1405), Ontario Research Fund (ORF-GL2-01-004), NSERC Strategic Project Grant, and BiofuelsNet, by Industrial Strategic Technology Development Program (10047910, Production of biobased cadaverine and polymerization of Bio-polyamide 510) funded by the Ministry of Trade, Industry & Energy (MOTEI, Korea).Notes and references
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- M. Kalim Akhtara, N. J. Turner and P. R. Jones, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 87–92 CrossRef PubMed.
- J. Nielsen, M. Fussenegger, J. Keasling, S. Y. Lee, J. C. Liao, K. Prather and B. Palsson, Nat. Chem. Biol., 2014, 10, 319–322 CrossRef CAS PubMed.
- H. C. Tseng and K. L. J. Prather, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17925–17930 CrossRef CAS PubMed.
- J. W. Song, E. Y. Jeon, D. H. Song, H. Y. Jang, U. T. Bornscheuer, D. K. Oh and J. B. Park, Angew. Chem., Int. Ed., 2013, 52, 2534–2537 CrossRef CAS PubMed.
- N. Oberleitner, C. Peters, J. Muschiol, M. Kadow, S. Saß, T. Bayer, P. Schaaf, N. Iqbal, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2013, 5, 3524–3528 CrossRef CAS.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- I. Oroz-Guinea and E. García-Junceda, Curr. Opin. Chem. Biol., 2013, 17, 236–249 CrossRef CAS PubMed.
- Y. S. Jang, B. Kim, J. H. Shin, Y. J. Choi, S. Choi, C. W. Song, J. Lee, H. G. Park and S. Y. Lee, Biotechnol. Bioeng., 2012, 109, 2437–2459 CrossRef CAS PubMed.
- T. Polen, M. Spelberg and M. Bott, J. Biotechnol., 2013, 167, 75–84 CrossRef CAS PubMed.
- N. Z. Xie, H. Liang, R. B. Huang and P. Xu, Biotechnol. Adv., 2014, 32, 615–622 CrossRef CAS PubMed.
- J.-L. Yu, X.-X. Xia, J.-J. Zhong and Z.-G. Qian, Biotechnol. Bioeng., 2014, 111, 2580–2586 CrossRef CAS PubMed.
- G. M. Diamond, V. Murphy and T. R. Boussie, in Modern Applications of High Throughput R&D in Heterogeneous Catalysis, ed. A. Hagemeyer and A. F. Volpe, Bentham Science, 2014, ch. 8, pp. 288–309, DOI:10.2174/97816080587231140101.
- J. M. Clomburg, M. D. Blankschien, J. E. Vick, A. Chou, S. Kim and R. Gonzalez, Metab. Eng., 2015, 28, 202–212 CrossRef CAS PubMed.
- S. Cheong, J. M. Clomburg and R. Gonzalez, Nat. Biotech., 2016, 34, 556–561 CrossRef CAS PubMed.
- K. A. Curran, J. M. Leavitt, A. S. Karim and H. S. Alper, Metab. Eng., 2013, 15, 55–66 CrossRef CAS PubMed.
- H.-M. Jung, M.-Y. Jung and M.-K. Oh, Appl. Microbiol. Biotechnol., 2015, 99, 5217–5225 CrossRef CAS PubMed.
- D. R. Vardon, N. A. Rorrer, D. Salvachua, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- A. P. Burgard, P. Pharkya and R. E. Osterhout, US Pat., US8592189 B2, 2013.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 567–576 CrossRef CAS.
- G. Sirasani, L. Tong and E. P. Balskus, Angew. Chem., Int. Ed., 2014, 53, 7785–7788 CrossRef CAS PubMed.
- A. J. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- Y. Deng, L. Ma and Y. Mao, Biochem. Eng. J., 2016, 105, 16–26 CrossRef CAS.
- X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed.
- H. Simon, J. Bader, H. Guenther, S. Neumann and J. Thanos, Angew. Chem., Int. Ed., 1985, 24, 539–553 CrossRef.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 CrossRef CAS.
- C. K. Winkler, G. Tasnádi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381–389 CrossRef CAS PubMed.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS PubMed.
- N. Iqbal, F. Rudroff, A. Brigé, J. Van Beeumen and M. D. Mihovilovic, Tetrahedron, 2012, 68, 7619–7623 CrossRef CAS PubMed.
- A. Z. Walton, B. Sullivan, A. C. Patterson-Orazem and J. D. Stewart, ACS Catal., 2014, 4, 2307–2318 CrossRef CAS PubMed.
- G. Steinkellner, C. C. Gruber, T. Pavkov-Keller, A. Binter, K. Steiner, C. Winkler, A. Łyskowski, O. Schwamberger, M. Oberer, H. Schwab, K. Faber, P. MacHeroux and K. Gruber, Nat. Commun., 2014, 5, 4150 CAS.
- A. B. Daugherty, S. Govindarajan and S. Lutz, J. Am. Chem. Soc., 2013, 135, 14425–14432 CrossRef CAS PubMed.
- T. Reß, W. Hummel, S. P. Hanlon, H. Iding and H. Gröger, ChemCatChem, 2015, 7, 1302–1311 CrossRef.
- M. Gall, M. Thomsen, C. Peters, I. V. Pavlidis, P. Jonczyk, P. P. Grunert, S. Beutel, T. Scheper, E. Gross, M. Backes, T. Geißler, J. P. Ley, J. M. Hilmer, G. Krammer, G. J. Palm, W. Hinrichs and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 1439–1442 CrossRef CAS PubMed.
- H. S. Toogood and N. S. Scrutton, Catal. Sci. Technol., 2013, 3, 2182–2194 CAS.
- F. Rohdich, A. Wiese, R. Feicht, H. Simon and A. Bacher, J. Biol. Chem., 2001, 276, 5779–5787 CrossRef CAS PubMed.
- P. A. Hubbard, X. Liang, H. Schulz and J. J. P. Kim, J. Biol. Chem., 2003, 278, 37553–37560 CrossRef CAS PubMed.
- H. Simon, J. Bader, H. Guenther, S. Neumann and J. Thanos, Angew. Chem., Int. Ed. Engl., 1985, 24, 539–553 CrossRef.
- C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409–5411 CrossRef CAS PubMed.
- R. D. Finn, A. Bateman, J. Clements, P. Coggill, R. Y. Eberhardt, S. R. Eddy, A. Heger, K. Hetherington, L. Holm, J. Mistry, E. L. L. Sonnhammer, J. Tate and M. Punta, Nucleic Acids Res., 2014, 42, D222–D230 CrossRef CAS PubMed.
- A. D. N. Vaz, Biochemistry, 1995, 34, 4246–4256 CrossRef CAS PubMed.
- M. S. Rhee, B. E. Moritz, G. Xie, T. Glavina Del Rio, E. Dalin, H. Tice, D. Bruce, L. Goodwin, O. Chertkov, T. Brettin, C. Han, C. Detter, S. Pitluck, M. Land, M. Patel, M. Ou, R. Harbrucker, L. O. Ingram and K. T. Shanmugam, Stand. Genomic Sci., 2011, 5, 331–340 CrossRef CAS PubMed.
- X. Tu, P. A. Hubbard, J. J. P. Kim and H. Schulz, Biochemistry, 2008, 47, 1167–1175 CrossRef CAS PubMed.
- M. K. Akhtar and P. R. Jones, Appl. Microbiol. Biotechnol., 2008, 78, 853–862 CrossRef CAS PubMed.
- W. Tischer, J. Bader and H. Simon, Eur. J. Biochem., 1979, 97, 103–112 CrossRef CAS PubMed.
- J. Caldeira, R. Feicht, H. White, M. Teixeira, J. J. G. Moura, H. Simon and I. Moura, J. Biol. Chem., 1996, 271, 18743–18748 CrossRef CAS PubMed.
- Y. Lin, R. Jain and Y. Yan, Curr. Opin. Biotechnol., 2014, 26, 71–78 CrossRef CAS PubMed.
- G. B. Birrell, T. O. Zaikova, A. V. Rukavishnikov, J. F. W. Keana and O. H. Griffith, Biophys. J., 2003, 84, 3264–3275 CrossRef CAS PubMed.
- D. Cubrilovic, W. Haap, K. Barylyuk, A. Ruf, M. Badertscher, M. Gubler, T. Tetaz, C. Joseph, J. Benz and R. Zenobi, ACS Chem. Biol., 2014, 9, 218–226 CrossRef CAS PubMed.
- E. Torres and J. Aburto, Arch. Biochem. Biophys., 2005, 437, 224–232 CrossRef CAS PubMed.
- F. Mingardon, J. D. Bagert, C. Maisonnier, D. L. Trudeau and F. H. Arnold, Appl. Environ. Microbiol., 2011, 77, 1436–1442 CrossRef CAS PubMed.
- C. Y. Lee, K. O. Yu, S. W. Kim and S. O. Han, J. Biosci. Bioeng., 2010, 109, 331–336 CrossRef CAS PubMed.
- E. Goihberg, O. Dym, S. Tel-Or, I. Levin, M. Peretz and Y. Burstein, Proteins: Struct., Funct., Genet., 2007, 66, 196–204 CrossRef CAS PubMed.
- A. J. Jervis, J. C. Crack, G. White, P. J. Artymiuk, M. R. Cheesman, A. J. Thomson, N. E. L. Brun and J. Green, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4659–4664 CrossRef CAS PubMed.
- T. Imai, K. Tagucbi, Y. Ogawara, D. Ohmori, F. Yamakura, H. Ikezawa and A. Urushiyama, J. Biochem., 2001, 130, 649–655 CrossRef CAS PubMed.
- T. Goris, A. F. Wait, M. Saggu, J. Fritsch, N. Heidary, M. Stein, I. Zebger, F. Lendzian, F. A. Armstrong, B. Friedrich and O. Lenz, Nat. Chem. Biol., 2011, 7, 310–318 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02842j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |