
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodriven hydrogen evolution by molecular catalysts using Al2O3-protected perylene-3,4-dicarboximide on NiO electrodes†
Rebecca J.
Kamire
,
Marek B.
Majewski
,
William L.
Hoffeditz
,
Brian T.
Phelan
,
Omar K.
Farha
,
Joseph T.
Hupp
and
Michael R.
Wasielewski
*
Department of Chemistry and Argonne-Northwestern Solar Energy Research (ANSER) Center, Northwestern University, Evanston, IL 60208-3113, USA. E-mail: m-wasielewski@northwestern.edu
First published on 30th August 2016
Abstract
The design of efficient hydrogen-evolving photocathodes for dye-sensitized photoelectrochemical cells (DSPECs) requires the incorporation of molecular light absorbing chromophores that are capable of delivering reducing equivalents to molecular proton reduction catalysts at rates exceeding those of charge recombination events. Here, we report the functionalization and kinetic analysis of a nanostructured NiO electrode with a modified perylene-3,4-dicarboximide chromophore (PMI) that is stabilized against degradation by atomic layer deposition (ALD) of thick insulating Al2O3 layers. Following photoinduced charge injection into NiO in high yield, films with Al2O3 layers demonstrate longer charge separated lifetimes as characterized via femtosecond transient absorption spectroscopy and photoelectrochemical techniques. The photoelectrochemical behavior of the electrodes in the presence of Co(II) and Ni(II) molecular proton reduction catalysts is examined, revealing reduction of both catalysts. Under prolonged irradiation, evolved H2 is directly observed by gas chromatography supporting the applicability of PMI embedded in Al2O3 as a photocathode architecture in DSPECs.
Introduction
The harvesting and storage of light energy in the chemical bonds of liquid and gaseous fuels has been of increasing interest in recent years.1–3 One of many promising approaches uses pairs of dye-sensitized electrodes within photoelectrochemical cells (DSPECs) to drive molecular catalysts to perform the two half reactions of water oxidation and hydrogen evolution.4–6 Incorporation of both light-driven reactions into one efficient system presents many challenges and opportunities to understand photodriven electron transfer events, charge accumulation steps, catalytic mechanisms, and avoidance of degradation pathways within the components of DSPECs. Several designs that are capable of photodriven (or photoassisted) hydrogen evolution combine molecular catalysts with photosensitized nanomaterials in solution,7–11 or photosensitized electrodes,12–18 or use semiconductors as light absorbers.19–27 Mechanistic investigation of dye-sensitized photocathode architectures remains particularly rare,28 and such work could lead to improvement in the performance of devices. Current device designs suffer from degradation of the molecular light absorbers over time and losses in efficiency due to charge recombination at rates competitive with charge accumulation and catalysis. New chromophores based on earth abundant elements are also desirable, since the molecular photosensitizers used in the aforementioned device architectures are almost exclusively ruthenium-based, with the exception of only a few organic chromophores.13,15,29We previously demonstrated that a perylene-3,4-dicarboximide (PMI) chromophore (PMI, Fig. 1) is capable of electron injection into nanostructured TiO2 following photoexcitation and further capable of oxidizing a molecular water oxidation precatalyst.30 Studies have also demonstrated the utility of PMI-based chromophores for photodriven hole injection into NiO in p-type dye-sensitized solar cells (DSCs),31–36 which is encouraging for future work on hydrogen evolving photocathodes. One report suggests that no molecular catalyst is necessary for photodriven hydrogen evolution by a chromophore that includes a PMI moiety on NiO at pH 7,37 and a similar PMI-based chromophore has driven hydrogen evolution by a cubane molybdenum-sulfide cluster in acidic conditions.29 However, these photosensitizer designs generally require complex synthesis in order to incorporate a donor–acceptor character that extends the lifetime of the charge separated state. PMI can be synthesized from commercially available perylene-3,4:9,10-tetracarboxydianhydride in only four steps, and the monoanhydride opens to form the dicarboxylate when exposed to metal oxide semiconductors for convenient electrode functionalization.30
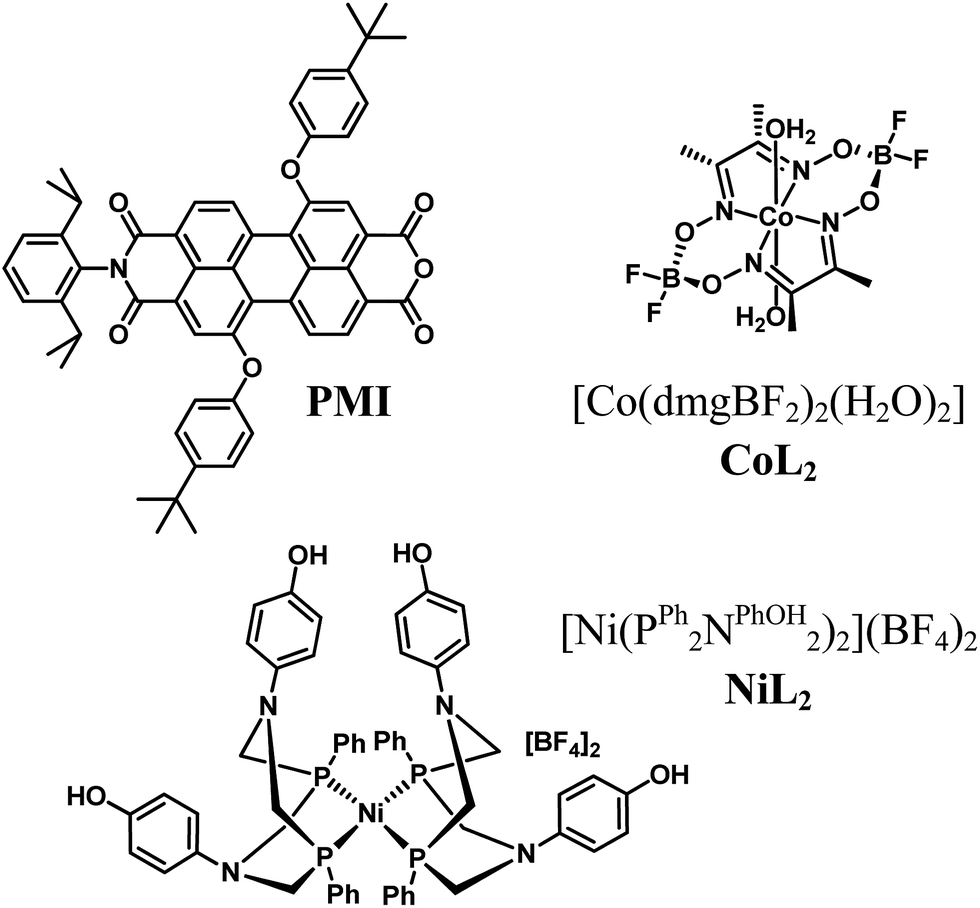 | ||
| Fig. 1 Chemical structures of the PMI photosensitizer and the H2 evolution catalysts. | ||
The application of an Al2O3 tunneling barrier by atomic layer deposition (ALD) before dye loading on a surface has been shown to act as an alternative to complex molecular synthesis by providing the important advantages of slowing back electron transfer between charges in semiconductors and on chromophores and redox shuttles in DSCs.16,38–46 In DSPECs, slowing charge recombination at the interface is similarly advantageous for efficient charge accumulation and catalysis. We investigate here the use of an ALD layer of varying thickness deposited after dye loading to slow charge recombination between charges in the dye and semiconductor with those on the catalyst.
However, ALD can also serve an additional purpose important in DSPECs. Dye desorption and degradation limit the performance of electrodes fabricated with PMI, since desorption in the acidic operating conditions and oxidative or reductive degradation of organic chromophores under catalytic conditions are some of the most significant limiting factors in the stability of DSPECs.3,5 We and others have demonstrated that ALD of either TiO2 or Al2O3 following dye absorption on nanostructured semiconductors can prevent desorption of bound molecules from surfaces in DSCs44,47–50 and electrodes for water oxidation.51,52 In addition, we hypothesize that a thicker layer of metal oxide surrounding the PMI molecules can protect the dyes from undesirable degradation side reactions. Meyer and coworkers have recently observed such a stabilization effect by Al2O3 ALD layers over Ru-based dyes and water oxidation catalysts on nanostructured semiconductors.53
Here we report the ability of PMI bound to nanostructured NiO electrodes and protected by Al2O3 to drive well-characterized cobaloxime54,55 and [Ni(PR2NR2)2]2+ (ref. 56 and 57) hydrogen evolution electrocatalysts (Fig. 1). Besides their efficacy in other photodriven systems, these catalysts were selected based on their electrocatalytic hydrogen production at comparable redox potentials (Table 1, Fig. S4†). We explore the charge transfer dynamics and find that the Al2O3 layers not only stabilize the organic chromophores against desorption and degradation but also favor longer charge separated state lifetimes and light-driven hydrogen evolution.
| E 00 (eV) | [H2SO4] (M) | E ox (V) | E red1 (V) | E red2 (V) | |
|---|---|---|---|---|---|
a Measurements on PMI diester in CH2Cl2 for optical expts; in 0.1 M TBAPF6 in CH3CN for redox expts.28
b 1.0 mM Fc, 0.1 M Na2SO4 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:MeCN, [H2SO4] listed (Fig. S4).
c Catalytic onset, as the intersection of the line of the catalytic slope with the cyclic voltammogram trace before acid addition. :1 H2O:MeCN, [H2SO4] listed (Fig. S4).
c Catalytic onset, as the intersection of the line of the catalytic slope with the cyclic voltammogram trace before acid addition.
|
|||||
| PMI | 2.30 | 0.00 | 1.34 | −0.79 | −1.05 |
| NiL2 | — | 0.00 | — | −0.47 | −0.74 |
| — | 0.07 | — | — | −0.45c | |
| CoL2 | — | 0.00 | — | −0.51 | — |
| — | 0.10 | — | −0.49 | −0.85c | |
Experimental
Synthesis and film preparation
The PMI dye and PMI diester (see structure in ESI†),30 cobaloxime catalyst,58 and nickel catalyst57 were prepared following reported procedures. Nanostructured NiO films on conductive fluorine doped tin oxide (FTO) slides were prepared based on a literature procedure59 and stored in a 110 °C oven until dye loading. To dye functionalize, films were gently shaken in a 0.45 mM solution of PMI in 1:3 toluene:methanol protected from light overnight, rinsed with CH2Cl2, and dried under a nitrogen stream. The NiO|PMI surface was then treated with 0–30 ALD cycles of dimethylaluminum isopropoxide and water to yield NiO|PMI|Al2O3 films, as described in the ESI.† Films for femtosecond transient absorption (fsTA) spectroscopy were either sealed under nitrogen with glass slides using UV-curable epoxy in a glove box, or placed in an acidic electrolyte solution of 0.1 M H2SO4 and 0.1 M Na2SO4 in 1:1 H2O:MeCN in gas-tight cuvettes and purged with argon. The films for photoelectrochemical experiments were attached to stranded conductive wire using conductive silver epoxy (CircuitWorks Chemtronics), which was then covered with non-conductive epoxy (LOCTITE 9340 Hysol) and cured at 110 °C for 10 min.
Optical spectroscopy
Femtosecond transient absorption (fsTA) spectroscopy experiments were conducted using a regeneratively amplified Ti:sapphire laser system with samples translated in two dimensions and irradiated at 495 nm as previously described.30,60 Further details and the fitting procedures are provided in the ESI.†
Electrochemical and photoelectrochemical experiments
Electrochemical measurements were performed using a CH Instruments Model 660A or 750E electrochemical workstation. Measurements on the dye-functionalized films employed the films as the working electrode, a 3.0 mm diameter glassy carbon counter electrode, and an Ag/AgCl (3 M NaCl) reference electrode. Light was applied from two white LEDs (100 mW cm−2 incident on the sample per LED) covered with 410 nm long-pass filters. Photocatalysis experiments were performed in a sealed cell under argon with a −0.40 V bias applied concurrently with illumination for two hours. The H2 in the headspace was identified following triplicate injections of 300 μL each into a gas chromatograph. Faradaic efficiencies were calculated from the amount of H2 in the headspace and the amount of current passed during the experiment.Results and discussion
Steady-state spectroscopy
The films were characterized by UV-Vis spectroscopy to gain information on the physical effect of ALD on the dye molecules (Fig. 2). The absorption spectrum for NiO|PMI films without ALD displays a (0,0) vibronic absorption maximum at 506 nm, a (0,1) band at 485 nm, and a small shoulder around 575 nm. The low energy shoulder is suggestive of dye aggregation45,61 but is absent after ALD is applied. Thus, we postulate that aggregates should not play a significant role in the photophysics of the ALD-treated films. As the number of cycles of ALD is increased, the signal broadens until by 30 cycles the vibronic structure is no longer distinct and the λmax is red-shifted to 522 nm. Increasing to 40 and 50 cycles produces no further spectral change (Fig. S1†). This spectrum is identical to that of the corresponding PMI diester spin coated onto glass but not to that of PMI diester encapsulated in a soft and flexible PMMA matrix (Fig. S2†), which suggests that the change in band shape after ALD could be due to the imposition of structural rigidity on PMI enforced by the surrounding Al2O3. The red-shift is likely due to an untwisting of the PMI core that is twisted in the free molecule by the presence of the bulky phenoxy groups.61 It is unlikely that the shift results from a change in dielectric environment, as we have not observed that effect in previous work.45,47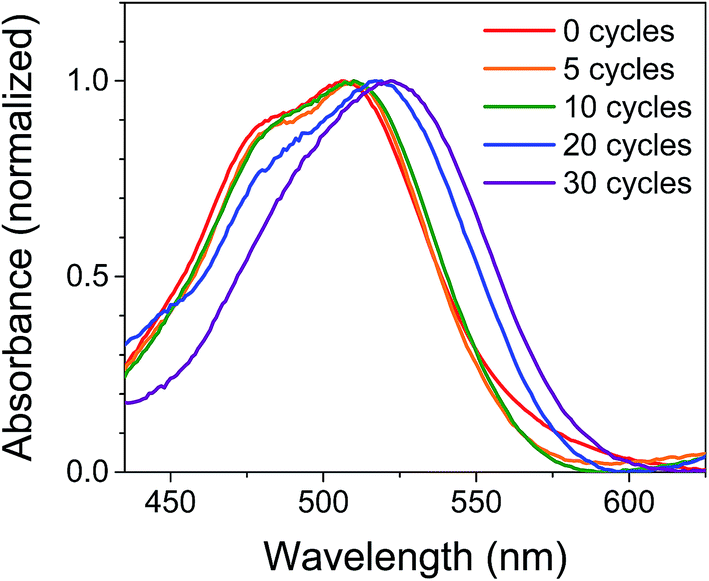 | ||
| Fig. 2 Normalized UV-Vis absorption spectra of NiO|PMI films treated with 0–30 cycles of Al2O3 ALD. The NiO background is subtracted. | ||
In an attempt to predict the Al2O3 thickness with respect to the PMI molecules, the geometry optimized PMI diester structure was obtained from DFT calculations (B3LYP/6-31G*) and shows that the maximum height of PMI from the NiO surface cannot be more than 17.5 Å (Fig. S23 and Table S3†). The ALD growth rate on flat surfaces is ca. 1 Å per cycle as determined by ellipsometry, so it was initially anticipated that the Al2O3 layer would fully encapsulate PMI by 20 cycles of ALD. However, we surmise based on the change in band shapes in the absorption spectra between 20 and 30 cycles of ALD, and no further changes thereafter, that only by 30 cycles of ALD is the dye molecule effectively encapsulated in Al2O3.
The stability of the dye molecules on the surface was also investigated by UV-Vis spectroscopy after the ALD process and after exposure to ambient light and air over 45 days (Fig. S3†). Minimal dye degradation was observed during the ALD treatment, based on the change in shape of the absorption curves, but following ALD, dye degradation does occur over time. Degradation decreases with increasing Al2O3 thickness so that by 30 cycles degradation is negligible over the 45 day period investigated, which is promising for use of ALD-treated NiO|dye films in devices and further supports the assertion that 30 cycles of ALD is sufficient to cover PMI.
Electrochemistry
In order to examine the energetic driving force for the desired photodriven charge transfer steps, we compare the redox properties of PMI as determined in our previous work with those of the catalysts and with the NiO valence band (Table 1).30 The NiO valence band is sufficiently low in energy that it is accessible for hole injection from PMI even at low pH (0.52 V vs. Ag/AgCl),12,62,63 and fast hole injection from similar PMI-based chromophores has been observed previously.64 The first reduction potentials, which are reversible in the absence of acid and semi-reversible in its presence, and the onsets of catalysis for NiL2 and CoL2 were measured by cyclic voltammetry and are reported in Fig. S4† and summarized in Table 1. The first reduction potential of PMI, Ered = −0.79 V vs. Ag/AgCl, is sufficiently negative to reduce either catalyst by one electron. In the presence of 0.1 M H2SO4, the catalytic onset for NiL2 is milder than the first reduction without acid, so PMI should be able to initiate catalysis. The CoL2 catalyst, however, has a catalytic onset of −0.85 V vs. Ag/AgCl, so catalysis without applied bias, if possible, is expected to be less efficient than for NiL2.Transient absorption spectroscopy
We have previously reported the fsTA spectroscopic characterization of the excited state decay of the singlet excited state of PMI (1*PMI) on Al2O3, where neither electron nor hole injection into the semiconductor is energetically accessible.30 The 1*PMI state that is formed upon excitation of Al2O3|PMI (dry) films is characterized by a ground state bleach and stimulated emission signal at 440 to 600 nm and an excited state absorption with a maximum centered at 675 nm (species A, Fig. S7A†). The subsequent decay of 1*PMI along a number of pathways including excimer formation65 is approximated using a species-associated model in which 1*PMI (species A) decays to species B, which decays to species C, which decays to the ground state (GS). While the spectra for “species B” and “species C” contain contributions from the several species formed along these decay pathways, excimer can be identified by a loss of stimulated emission and red-shift and broadening of the absorptive signal in species C (Fig. S7A†).NiO|PMI films were then studied by fsTA spectroscopy and, by comparison to the results for Al2O3|PMI, photodriven hole injection into NiO and subsequent charge recombination were identified for both dry films and films in acidic solution (Fig. 3 and S6–S9†). The lack of significant stimulated emission and excimer signal in the time-resolved spectra for all NiO|PMI samples indicates that nearly quantitative hole injection into NiO occurs very rapidly. The 1*PMI spectra (Fig. 3B, species A) and rate of hole injection were identified by global fitting to an A → B → C → D → GS model, where hole injection (A → B) fits to ∼0.5 ps for all samples and is likely multiexponential with an even faster component occurring within the instrument response time (Table S1†).
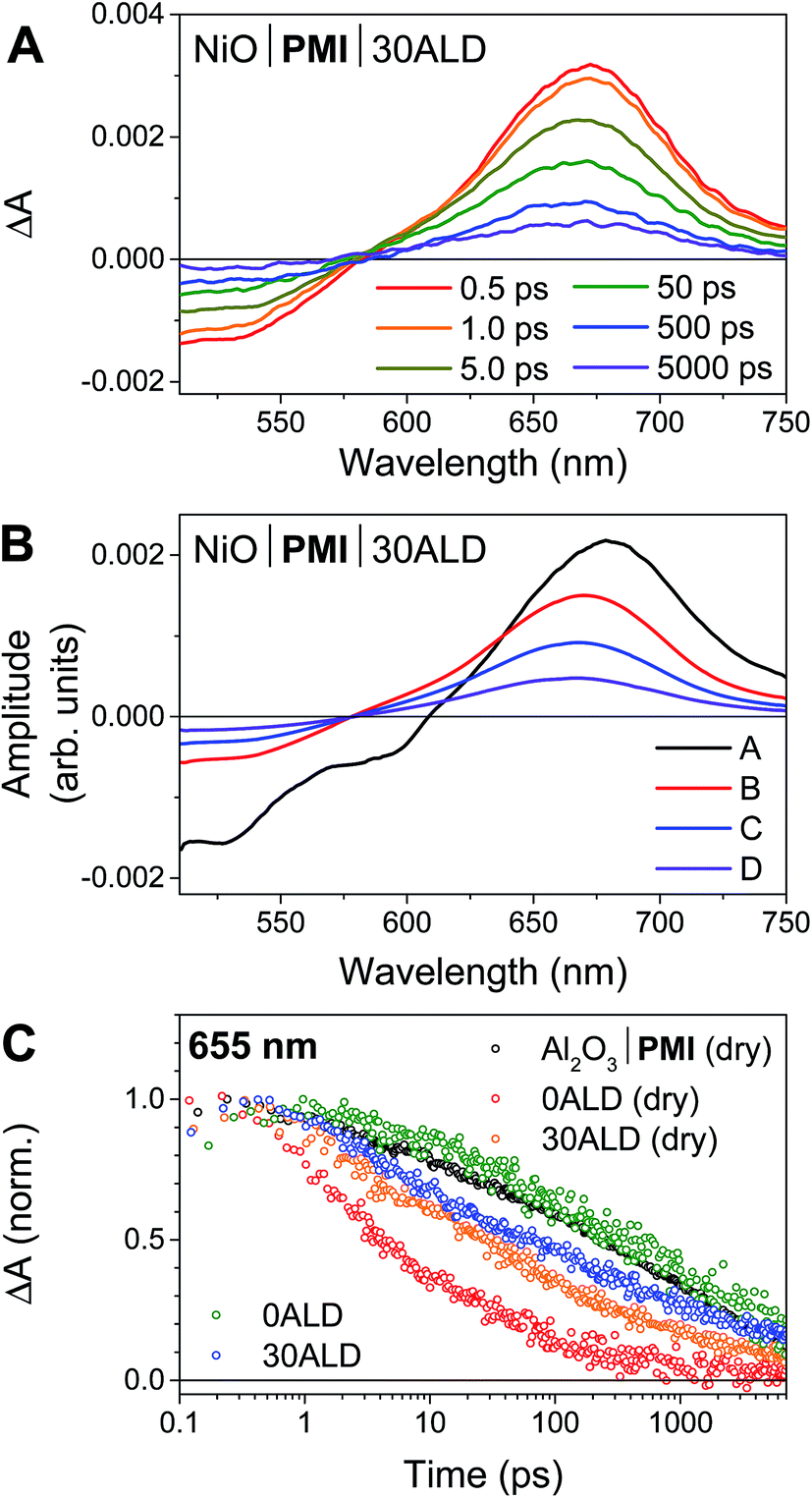 | ||
| Fig. 3 (A) fsTA spectra of NiO|PMI|30ALD in 0.1 M H2SO4 + 0.1 M Na2SO4 in 1:1 H2O:MeCN under argon following excitation with a 495 nm laser pulse. (B) Species-associated spectra obtained by a global fit of the fsTA spectra to an A → B → C → D → ground state (GS) model. (C) Normalized single wavelength traces at 655 nm from fsTA results of Al2O3|PMI (black), NiO|PMI (red), and NiO|PMI|30ALD (orange) dry under nitrogen and NiO|PMI (green) and NiO|PMI|30ALD (blue) in the electrolyte conditions as above. | ||
For the NiO|PMI (dry) film, an excimer population is observed spectrally as a shoulder at 560–600 nm in species B–D (Fig. S9†) and kinetically as a fast rate of decay at 655 nm (Fig. 3C). The shape of the 0.5 ps spectrum for the film without ALD, with a very blue-shifted x-intercept of about 575 nm, indicates that the excimer population is large and begins to form within the instrument response time. The A → B lifetime of 0.6 ± 0.3 ps is not significantly different from the A → B lifetimes for the other samples, so excimer formation likely occurs on a similar timescale and competitively with hole injection. Given the large driving force for hole injection from 1*PMI discussed above, it is likely that hole injection also occurs from the excimer state. A much smaller excimer population is also observed in the 500 ps and 5000 ps spectra at 550–600 nm for films without ALD under solvent conditions. The low excimer intensity in these samples likely results from dye interaction with solvent molecules weakening the interchromophore coupling. No excimer is observed for any of the films with 30 cycles of ALD, which indicates that the disaggregation induced by ALD eliminates excimer formation so that hole injection becomes the strongly dominant process.
Following hole injection, the resulting signal is characterized by a ground state bleach centered at 525 nm and an absorptive feature with a λmax around 665 nm and is consistent with PMI˙− (Fig. 3A).64,66 This signal decays with little change in shape as recombination with holes in NiO occurs (species B–D, Fig. 3B). Recombination is non-exponential, as expected for dye-sensitized semiconductors,64,67 and could not be adequately fit with fewer than three rates of decay. Consequently, the resulting fit carries a large degree of uncertainty, with values varying only slightly from those used as artificial starting values when initiating the fit. A comparison of the normalized kinetic traces at 665 nm provides more meaningful information (Fig. 3C). Recombination is slowed by the presence of the acidic solution for films with 0ALD, as the PMI molecules experience a more polar environment in solution and thus a lower energy PMI˙− state.68 This finding agrees with a previous study that found PMI-sensitized NiO to lie in the Marcus normal region.64 Thus, dye molecules encased in Al2O3, which experience an environment of intermediate polarity, display recombination rates between those for 0ALD films with and without solution present. Recombination is only minimally impeded by solution for films with 30ALD because the polarity of the dye environment remains unchanged. This finding indicates that the ALD layer should also shield PMI from catalyst molecules in solution, and slow charge recombination between the reduced catalyst and the surface, as long as the layer is thin enough for initial electron transfer through the layer to the catalyst to occur. However, catalyst reduction could not be directly identified from the fsTA experiments because samples with either NiL2 or CoL2 in solution had indistinguishable rates of decay from samples without catalyst (Fig. S10†). We conclude that, while PMI successfully injects holes into NiO in high yield and ALD favors injection over excimer formation, the majority of the electrons on PMI molecules recombine with holes in NiO before catalyst diffusion to the surface and subsequent catalyst reduction can occur. All reported photocathodes based on sensitized NiO, including those with surface-bound catalyst, are similarly limited by fast recombination.12–18,28 We turned to photoelectrochemical techniques to probe the fate of any charge separated states that are sufficiently long-lived to drive photocatalysis.
Photoelectrochemistry: evidence of hydrogen evolution
Following confirmation that the desired hole injection occurs from PMI into NiO, photoelectrochemical techniques were used to probe the ability of PMI to reduce NiL2 and CoL2 catalysts and drive hydrogen evolution. Fig. 4 displays results for linear sweep voltammetry (LSV) experiments on NiO|PMI with 30 cycles of Al2O3 ALD (NiO|PMI|30ALD) in an acidic 1:1 H2O:MeCN solution (5 mV s−1 from 0.00 to −0.55 V, 10 s light on/off cycles). In the absence of PMI, photocurrent is negligible (blue), but when PMI is present, the photocurrent is enhanced (black). When the light is turned on, an initial strong photocurrent that rapidly decays results from reduction of the PMI molecules on the surface and local capacitance effects53 and hole injection that is more rapid than dye regeneration.69 It is followed by a plateau where hole injection and dye regeneration are in equilibrium. With either NiL2 or CoL2 but without acid, the photocurrent is enhanced in a wave-like feature around the first reduction of each catalyst (−0.3 to −0.5 V) (Fig. S11 and S12†), which suggests that photoassisted catalyst reduction from PMI occurs. Upon addition of both acid and 0.5 mM NiL2 or 0.5 mM CoL2 to the solution (Fig. 4, red), the photocurrent traces are not simply the sum of those observed for the films with only H2SO4 and those with only catalyst. Instead, the films display a further enhanced photocurrent that increases at increasingly negative potentials and lacks capacitive features. The stronger photocurrent at more negative biases reflects the higher yield of reduced catalyst resulting from slowed hole recombination at these potentials. The lack of capacitive features indicates that PMI is rapidly regenerated by the catalyst following hole injection into NiO at any applied potential within the range. These changes together indicate that the second reduction of each catalyst and subsequent turnover to produce hydrogen occur.12,53,69
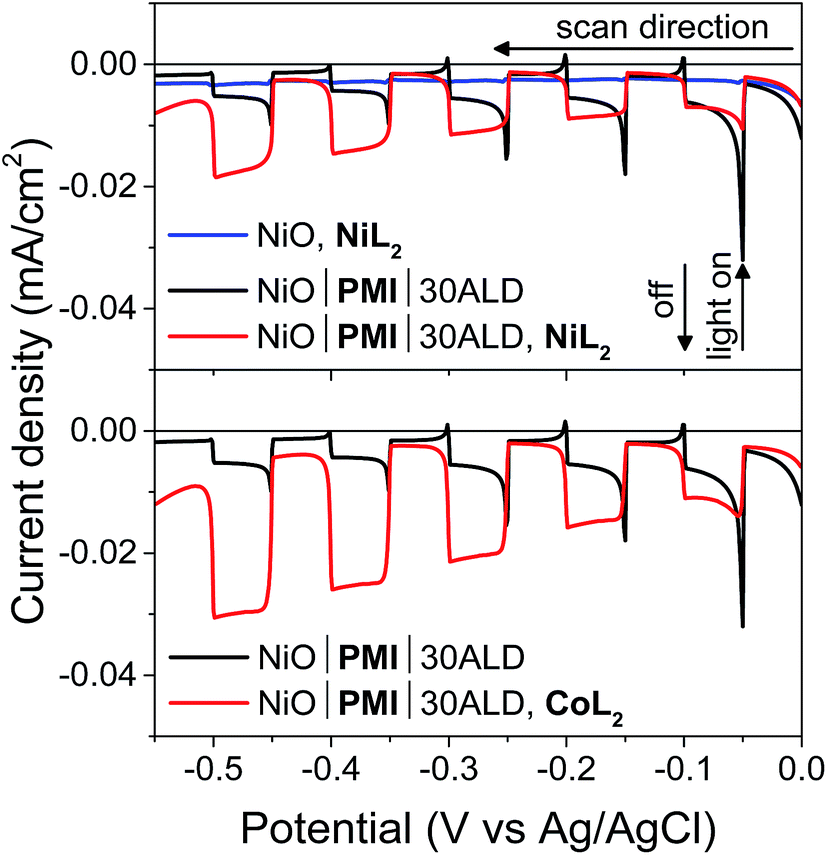 | ||
| Fig. 4 Linear sweep voltammetry measurements with 10 s light on/off cycles on a NiO working electrode with 0.5 mM NiL2 (blue) and on a single NiO|PMI|30ALD working electrode without catalyst (black) and with 0.5 mM NiL2 or 0.5 mM CoL2 (red) (conditions: 0.1 M H2SO4 and 0.1 M Na2SO4 in 1:1 H2O:MeCN, 5 mV s−1). | ||
Films with 0–20 cycles of Al2O3 ALD and either catalyst also display characteristics of proton reduction for experiments run on duplicate batches of films (Fig. S13–S15†). The shapes of the photocurrent responses for films exposed to either catalyst display no sharp capacitive peak when the light is turned on and higher intensity at more negative bias as observed for the films with 30 cycles of ALD. Some of the films, however, displayed a decrease in absolute photocurrent density in the presence of catalyst relative to those without. The experiments without and with catalyst were performed consecutively on the same films for ease of comparison. Dye degradation and/or desorption at strongly negative potentials,70 such as those used in the LSV experiments, result in less photocurrent on the second sweep for most films even without catalyst addition (data not shown). In contrast, films that have only been exposed to a −0.40 vs. Ag/AgCl bias, even with catalyst, do not display the photocurrent loss because the PMI molecules are much more stable at these mild potentials (Fig. 6B, S15 and S16†). Gratifyingly, photocurrent is stable over a 10 minute experiment for NiO|PMI|0–30ALD films with either catalyst at this potential (Fig. S19†). Alternatively, degradation at more negative potentials decreases as we increase the number of cycles of Al2O3. The color of the 30ALD films is preserved throughout the LSV experiment, unlike films with fewer ALD layers, and the photocurrent density on the second sweep is higher relative to the first sweep with increasing ALD layers (Fig. S13†). Thus, we postulate that the shapes of the photocurrent traces suggest successful hydrogen evolution occurs despite a lack of enhanced photocurrent in the presence of catalyst in some instances, and the loss of photocurrent due to degradation can be avoided on our experimental timescale by applying mild potentials and by applying sufficient Al2O3 coverage by ALD. We note that Meyer et al. have also observed a decrease in absolute photocurrent for their TiO2|dye|ALD|catalyst|ALD samples but were able to assign a change in photocurrent signal shape to an enhanced rate of oxidative catalysis.53
Photoelectrochemistry: analysis of rates
In light of the preceding discussion, we propose that it is most relevant to compare photocurrent response shapes to understand the differences in charge transfer dynamics between samples. These shapes are quantified using the ratio of photocurrent density at the end of a 10 s illumination period during the LSV experiment to that at the beginning (I−0.10 V/I−0.05 V or Ilight off/Ilight on, Fig. 5). Small ratios for the films without catalyst reflect the strong capacitive current spike, whereas larger current ratios for catalyst-containing samples reflect less decreasing or even increasing photocurrent densities during illumination. The current ratios approaching or greater than unity with catalyst indicate that rapid dye regeneration is sustained over time as the catalyst species regenerating the dye performs catalytic turnovers. Faster electron transfer to NiL2 from the reduced dye (and slower recombination) compared to CoL2 is evidenced by a larger current ratio I−0.10 V/I−0.05 V for the former, likely due to the less negative catalytic onset and lower degree of reversibility of the first catalyst reduction.12 A comparison of ratios of current densities at −0.40 V (Fig. S18†), where there is no difference in photocurrent response shape between the two catalysts, confirms that driving force is sufficiently strong and charge recombination is sufficiently slowed with the −0.40 V applied bias for the rapid second reduction of either catalyst to occur.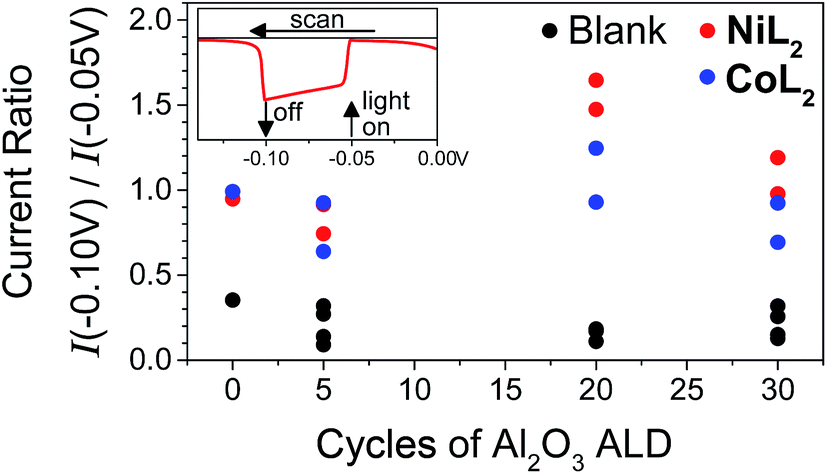 | ||
| Fig. 5 Ratios of current densities at −0.10 V (light off) to those at −0.05 V (light on) during LSV in duplicate and for NiO|PMI|ALD films with 0–30 cycles of Al2O3 ALD (black, “blank”) and again after additional 0.5 mM NiL2 (red) or 0.5 mM CoL2 (blue). The raw data for the two sets of films are reported in Fig. S13 and S15† (conditions: 0.1 M Na2SO4 and 0.1 M H2SO4 in 1:1 H2O:MeCN). | ||
In potentiostatic photocurrent experiments at −0.40 V, photocurrent enhancement alone with either catalyst is a strong indicator of hydrogen evolution (Fig. 6 and S16†). The amplitude of the recorded photocurrent does not increase during the entire 10 s of illumination because a constant bias is applied throughout. In contrast to the LSV experiments discussed above, where the change in the photocurrent response shape is stronger for NiL2 due to its milder catalytic onset potential, in these potentiostatic experiments the photocurrent enhancement with catalyst is more pronounced for CoL2 than for NiL2 (Fig. 6B). The higher photocurrent density for CoL2 compared to NiL2 may result from an easier approach of CoL2 to the NiO|PMI electrode due to its less sterically bulky ligand so that catalyst reduction is faster and thus more competitive with charge recombination. Overall, however, the differences in photocurrent densities between the NiO|PMI electrodes with the two different catalysts are minimal compared to the differences between the catalytic currents of the two catalysts in solution driven by a glassy carbon electrode (Fig. S4†). The current densities for electrocatalysis in that case are much larger than with the photoelectrodes, which suggests that the catalyst turnover is not rate-limiting at the NiO|PMI|ALD electrodes. This finding emphasizes the importance of electrode design and agrees with Ji et al. that the shapes of the photocurrent responses observed for dye-sensitized electrodes are more dependent on the electron transfer dynamics within the electrode and between the dye and the catalyst than on the inherent kinetics of the catalysts.16 Thus, the findings for this system should apply to systems that incorporate other catalysts as well.
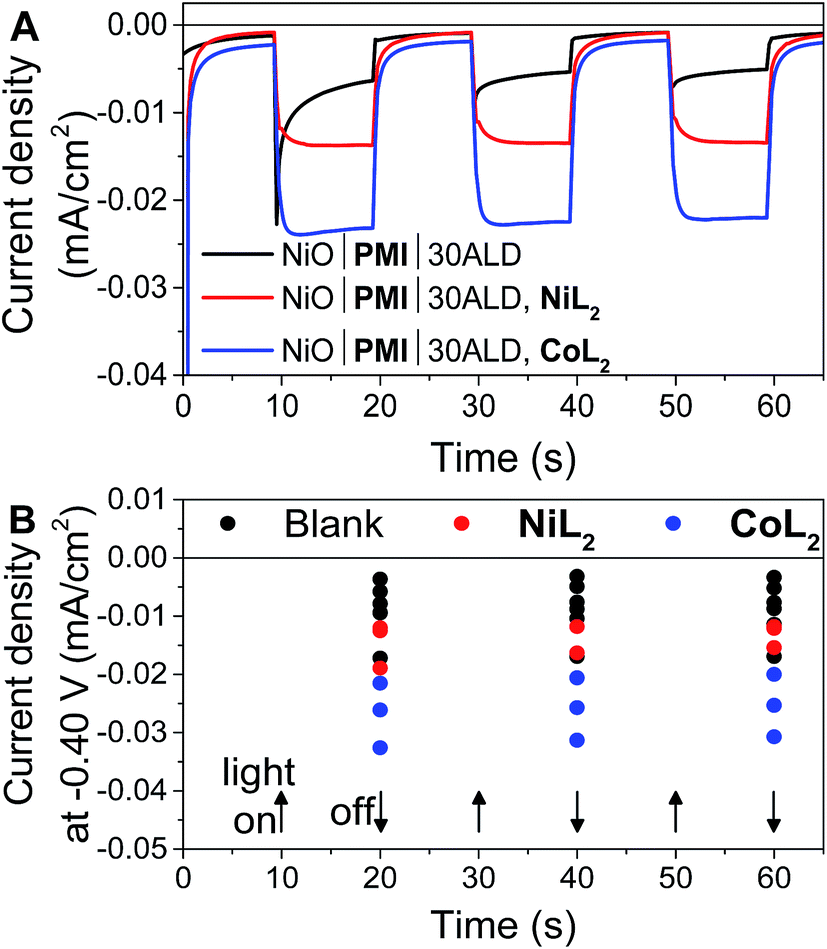 | ||
| Fig. 6 (A) Current density measurements with three 10 s light on/off cycles with −0.40 V applied bias on a single NiO|PMI|30ALD working electrode without catalyst (black, “blank”) and again after additional 0.5 mM NiL2 (red) or 0.5 mM CoL2 (blue). (B) Results of the same experiment with 5–30 cycles of Al2O3 ALD from Fig. S16.† The number of cycles of ALD is not distinguished for clarity (conditions: 0.1 M H2SO4 and 0.1 M Na2SO4 in 1:1 H2O:MeCN). | ||
The photocurrent traces also corroborate the hypothesis that hole recombination to PMI and to the catalyst before release of a hydrogen molecule are successfully slowed by the Al2O3 (ref. 69) without catastrophic losses in hole injection or catalyst reduction yields. The similar amplitude of photocurrent with the different amounts of ALD demonstrates the similar yields of charge injection and long-lived holes between samples. The effect of ALD on slowing charge transfer between the dye and the catalyst can be observed at the rise of the photocurrent traces over the initial 3 s after the light is turned on. This rise occurs as the rate of charge collection at the electrode (not necessarily the rate of charge injected into NiO) comes into equilibrium with that of charge transfer to the catalyst. With 0ALD or 5ALD and catalyst, photocurrent increases to the maximum much more slowly compared to films with more ALD (Fig. S13 and S15†). This observation is consistent with fast dye regeneration by hole transfer from the catalyst in the absence of ALD. As layers of Al2O3 are added, the initial maximum photocurrent is reached rapidly (followed by the slow rise with increasing potential), which indicates that dye regeneration is likely slowed by the insulating barrier. We note that permanent changes to the Al2O3 could contribute to the observed behavior. However, if charge transfer to the catalyst is slowed by Al2O3, we also expect to observe slowed recombination. An increase is observed in the absolute photocurrent densities at the end of 10 s of illumination with increasing Al2O3 thickness especially in the presence of catalyst (Fig. S17†). The NiO|PMI|30ALD, NiL2 sample in Fig. S17† has low current density and does not match the overall trend, but a comparison with Fig. 4, 6, and S13† suggests that this film is an outlier, potentially due to poor NiO quality. These data suggest that slower recombination and longer-lived charge separated states are achieved by addition of the ALD layer, which favors catalysis. Thus, the overall effect of ALD is to slow undesirable charge recombination events without preventing forward electron transfer.
Photodriven hydrogen generation
In order to monitor the performance of the films under conditions where hydrogen was expected to form, the electrodes were held at −0.40 V under illumination for 2 h, after which evolved hydrogen was detected by gas chromatography (Fig. S20 and S21†). The applied bias, rather than detracting from the viability of the electrode design, mimics the effect of incorporating the photocathode into a tandem device with a water oxidation photoanode based on TiO2, where the conduction band of TiO2 is at approximately this potential.13,15 A small but detectable amount of hydrogen was produced by films exposed to light but not to catalyst, in accordance with a report by Tong et al. that PMI-based chromophores alone can drive hydrogen evolution.37 No hydrogen was detected for films with catalyst present in the absence of light. NiO|PMI|30ALD films in the presence of both catalyst and light, however, formed hydrogen with faradaic efficiencies of 98 ± 4% and 60 ± 10% for NiL2 and CoL2, respectively (Table S2†). A lower hydrogen yield was observed for CoL2 compared to NiL2, despite the larger photocurrents recorded for CoL2 with shorter irradiation times (Fig. 6), which is likely due to the known instability of CoL2 to hydrolysis71 and to the overall higher activity of NiL2 under these conditions (Fig. S4†). Finally, films without ALD in the presence of NiL2 produced less hydrogen compared to those with ALD and exhibited a lower faradaic efficiency of 80 ± 10%. These films displayed a nearly complete loss of detectable PMI by the end of the two hour experiment, in contrast to the Al2O3-protected films on which PMI remains (Fig. S22†). This comparison reflects the improvements in stability achieved through incorporation of the Al2O3 layer into the electrode design.Some high-performing electrodes resulted from variability during the preparation of the underlying NiO. One such champion NiO|PMI|30ALD film with NiL2 produced about four times the hydrogen with the same faradaic efficiency compared to a typical film (Fig. S20–S21 and Table S2†). From the fsTA results, it is evident that charge recombination between the hole in NiO and the electron on PMI occurs very rapidly and in high yield, as is common for dye-sensitized NiO devices.12–17 The power conversion efficiency of our high-performing electrode is as a result still very low (<1%). Notably, this champion electrode did produce 1.82 ± 0.02 μmol hydrogen, which corresponds to a turnover number of about 1.5 with respect to catalyst and demonstrates that the hydrogen evolution observed here is indeed catalytic. An improved preparation of NiO, such as a recently reported treatment to passivate defect sites72 or a single cycle of ALD before dye loading,16 would be expected to raise the amplitude of the photocurrent response and overall performance of a device by increasing hole collection.
Conclusion
We have designed and fabricated a NiO|PMI|Al2O3 ALD photocathode architecture for photodriven hydrogen evolution that incorporates only earth-abundant elements. The PMI chromophores inject holes into NiO in <1 ps in high yield, and ALD of Al2O3 serves to disaggregate PMI and disfavor excimer formation as monitored by steady-state and transient optical spectroscopies. Photoelectrochemical experiments demonstrate successful photodriven reduction of and subsequent hydrogen evolution by two different molecular catalysts in solution. The Al2O3 layers stabilize PMI against desorption and degradation and slow undesirable charge recombination events so that higher photocurrent is achieved. The stabilizing effect further affords photodriven hydrogen evolution at high faradaic efficiencies. These results are promising for further exploration of ALD-embedded organic chromophores in photocathodes for photodriven hydrogen evolution. The findings that ALD of Al2O3 slows charge recombination and stabilizes surface-bound chromophores are expected to apply to other semiconductors that have better inherent charge recombination dynamics and to other catalysts, chromophores, and solution conditions, as long as those solution conditions are selected to allow for stability of the exposed components. Experiments are underway to attach catalysts to the electrode surface in order to improve the rate of catalyst reduction compared to recombination and better probe electron transfer dynamics between the adsorbed dye and the catalyst.Acknowledgements
This work was supported by the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under award number DE-SC0001059. Profilometry and ellipsometry measurements were performed in the Keck-II facility of the NUANCE Center at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. UV-Vis absorption measurements with the integrating sphere were performed in the Keck Biophysics Facility at Northwestern University, which is supported in part by grant NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.References
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- J. R. McKone, H. B. Gray and N. S. Lewis, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- H. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- C. E. Castillo, M. Gennari, T. Stoll, J. Fortage, A. Deronzier, M. N. Collomb, M. Sandroni, F. Legalite, E. Blart, Y. Pellegrin, C. Delacote, M. Boujtita, F. Odobel, P. Rannou and S. Sadki, J. Phys. Chem. C, 2015, 119, 5806–5818 CAS.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farre, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarstrom, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472–16475 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- C. Gimbert-Suriñach, J. Albero, T. Stoll, J. Fortage, M.-N. Collomb, A. Deronzier, E. Palomares and A. Llobet, J. Am. Chem. Soc., 2014, 136, 7655–7661 CrossRef PubMed.
- Y. Xu, X. Yin, Y. Huang, P. Du and B. Zhang, Chem.–Eur. J., 2015, 21, 4571–4575 CrossRef CAS PubMed.
- P. Meng, M. Wang, Y. Yang, S. Zhang and L. Sun, J. Mater. Chem. A, 2015, 3, 18852–18859 CAS.
- Y. Chen, H. Chen and H. Tian, Chem. Commun., 2015, 51, 11508–11511 RSC.
- A. Krawicz, D. Cedeno and G. F. Moore, Phys. Chem. Chem. Phys., 2014, 16, 15818–15824 RSC.
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef CAS PubMed.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- R. J. Lindquist, B. T. Phelan, A. Reynal, E. A. Margulies, L. E. Shoer, J. R. Durrant and M. R. Wasielewski, J. Mater. Chem. A, 2016, 4, 2880–2893 CAS.
- A. Morandeira, J. Fortage, T. Edvinsson, L. Le Pleux, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. C, 2008, 112, 1721–1728 CAS.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, U. Bach and P. Bauerle, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- L. Le Pleux, A. L. Smeigh, E. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarstrom and F. Odobel, Energy Environ. Sci., 2011, 4, 2075–2084 CAS.
- M. Weidelener, A. Mishra, A. Nattestad, S. Powar, A. J. Mozer, E. Mena-Osteritz, Y.-B. Cheng, U. Bach and P. Baeuerle, J. Mater. Chem., 2012, 22, 7366–7379 RSC.
- J. Warnan, J. Gardner, L. Le Pleux, J. Petersson, Y. Pellegrin, E. Blart, L. Hammarstrom and F. Odobel, J. Phys. Chem. C, 2014, 118, 103–113 CAS.
- Z. Liu, D. Xiong, X. Xu, Q. Arooj, H. Wang, L. Yin, W. Li, H. Wu, Z. Zhao, W. Chen, M. Wang, F. Wang, Y.-B. Cheng and H. He, ACS Appl. Mater. Interfaces, 2014, 6, 3448–3454 CAS.
- L. Tong, A. Iwase, A. Nattestad, U. Bach, M. Weidelener, G. Gotz, A. Mishra, P. Bauerle, R. Amal, G. G. Wallace and A. J. Mozer, Energy Environ. Sci., 2012, 5, 9472–9475 CAS.
- C. Prasittichai and J. T. Hupp, J. Phys. Chem. Lett., 2010, 1, 1611–1615 CrossRef CAS.
- M. J. DeVries, M. J. Pellin and J. T. Hupp, Langmuir, 2010, 26, 9082–9087 CrossRef CAS PubMed.
- V. O. Williams, N. C. Jeong, C. Prasittichai, O. K. Farha, M. J. Pellin and J. T. Hupp, ACS Nano, 2012, 6, 6185–6196 CrossRef CAS PubMed.
- C. Prasittichai, J. R. Avila, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 16328–16331 CrossRef CAS PubMed.
- M. J. Katz, M. J. D. Vermeer, O. K. Farha, M. J. Pellin and J. T. Hupp, Langmuir, 2013, 29, 806–814 CrossRef CAS PubMed.
- M. J. Katz, M. J. DeVries Vermeer, O. K. Farha, M. J. Pellin and J. T. Hupp, J. Phys. Chem. B, 2015, 119, 7162–7169 CrossRef CAS PubMed.
- K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2013, 25, 3–5 CrossRef CAS.
- H.-J. Son, C. H. Kim, D. W. Kim, N. C. Jeong, C. Prasittichai, L. Luo, J. Wu, O. K. Farha, M. R. Wasielewski and J. T. Hupp, ACS Appl. Mater. Interfaces, 2015, 7, 5150–5159 CAS.
- T. W. Hamann, O. K. Farha and J. T. Hupp, J. Phys. Chem. C, 2008, 112, 19756–19764 CAS.
- H.-J. Son, C. Prasittichai, J. E. Mondloch, L. Luo, J. Wu, D. W. Kim, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 11529–11532 CrossRef CAS PubMed.
- K. Hanson, M. D. Losego, B. Kalanyan, G. N. Parsons and T. J. Meyer, Nano Lett., 2013, 13, 4802–4809 CrossRef CAS PubMed.
- N. C. Jeong, H.-J. Son, C. Prasittichai, C. Y. Lee, R. A. Jensen, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 19820–19827 CrossRef CAS PubMed.
- H.-J. Son, X. Wang, C. Prasittichai, N. C. Jeong, T. Aaltonen, R. G. Gordon and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9537–9540 CrossRef CAS PubMed.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS PubMed.
- L. Alibabaei, B. D. Sherman, M. R. Norris, M. K. Brennaman and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5899–5902 CrossRef CAS PubMed.
- A. M. Lapides, B. D. Sherman, K. Brennaman, C. J. Dares, K. R. Skinner, J. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- P. Connolly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684–2688 CrossRef CAS.
- C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- W. A. Hoffert, J. A. S. Roberts, R. M. Bullock and M. L. Helm, Chem. Commun., 2013, 49, 7767–7769 Search PubMed.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197–5202 CrossRef CAS.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS.
- R. M. Young, S. M. Dyar, J. C. Barnes, M. Juríček, J. F. Stoddart, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 12438–12448 CrossRef CAS PubMed.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS.
- A. L. Smeigh, L. L. Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarstrom, Chem. Commun., 2012, 48, 678–680 RSC.
- M. K. Brennaman, M. R. Norris, M. K. Gish, E. M. Grumstrup, L. Alibabaei, D. L. Ashford, A. M. Lapides, J. M. Papanikolas, J. L. Templeton and T. J. Meyer, J. Phys. Chem. Lett., 2015, 6, 4736–4742 CrossRef CAS PubMed.
- M. T. Vagnini, M. W. Mara, M. R. Harpham, J. Huang, M. L. Shelby, L. X. Chen and M. R. Wasielewski, Chem. Sci., 2013, 4, 3863–3873 RSC.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- A. Weller, J. Phys. Chem., 1982, 133, 93–98 CAS.
- D. W. Kim, S. C. Riha, E. J. DeMarco, A. B. F. Martinson, O. K. Farha and J. T. Hupp, ACS Nano, 2014, 8, 12199–12207 CrossRef CAS PubMed.
- B. I. Lemon and J. T. Hupp, J. Phys. Chem. B, 1999, 103, 3797–3799 CrossRef CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- C. J. Flynn, S. M. McCullough, E. Oh, L. Li, C. C. Mercado, B. H. Farnum, W. Li, C. L. Donley, W. You, A. J. Nozik, J. R. McBride, T. J. Meyer, Y. Kanai and J. F. Cahoon, ACS Appl. Mater. Interfaces, 2016, 8, 4754–4761 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; additional electrochemical and photoelectrochemical characterization, UV-Vis spectra, and fsTA results; quantification of evolved hydrogen; and DFT-computed ground state structure of PMI diester. See DOI: 10.1039/c6sc02477g |
| This journal is © The Royal Society of Chemistry 2017 |