
Expedient Diels–Alder cycloadditions with ortho-quinodimethanes in a high temperature/pressure flow reactor†
Jennifer
Tsoung
,
Ying
Wang
 * and
Stevan W.
Djuric
* and
Stevan W.
Djuric
Discovery Chemistry and Technologies, Abbvie Inc., 1 North Waukegan Road, North Chicago, Illinois 60064, USA. E-mail: wang.ying@abbvie.com
First published on 23rd May 2017
Abstract
We describe herein Diels–Alder cycloadditions enabled by the efficient ring-opening of benzocyclobutenes and benzothiophene-2,2-dioxides using a high temperature/pressure flow reactor. The resultant ortho-quinodimethanes were generated and subsequently reacted with a wide range of dienophiles to provide complex heterocyclic ring systems. The use of flow technology allowed these reactions to be completed within minutes using conventional, readily removed solvents.
Drug discovery has oftentimes been described as a “race” in which several firms pursue similar chemical structures or mechanisms of action before any drug in the class obtains regulatory marketing approval.1 Given this observation, it has become imperative that platform technologies be introduced in drug discovery to accelerate the overall synthetic process while maintaining quality. In this context, we have recently initiated a high temperature/pressure (high T/P) flow technology program focused on exploring fast and reliable small molecule synthesis to support medicinal chemistry programs.2 For example, we recently disclosed the development of an automated Phoenix™ flow reactor, which is a commercially available instrument capable of achieving up to 450 °C and 200 bar of pressure. Our in-house modifications included addition of a fully automated two-arm Tecan Miniprep to handle autosampling and fraction collecting functions (Fig. 1), which allowed us to rapidly optimize and synthesize several libraries of fused pyrimidinone and quinolone derivatives via the Gould–Jacobs reaction.2c
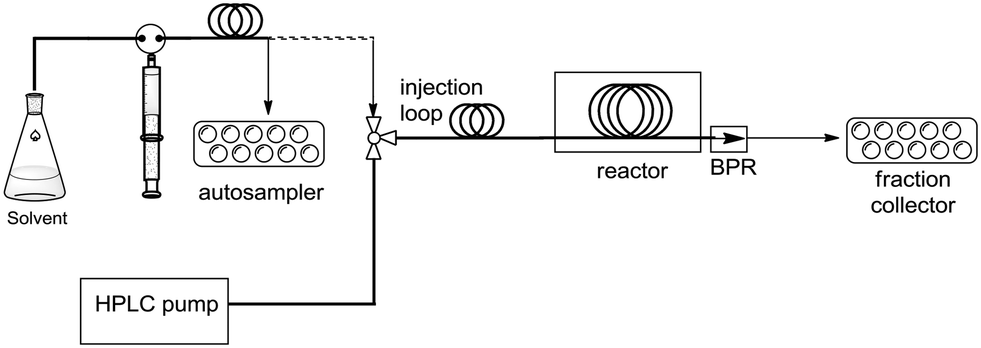 | ||
| Fig. 1 Schematic view of the automated Phoenix™ flow reactor platform. | ||
In this communication, we describe the application of this automated high T/P flow reactor towards the synthesis of benzoisoindolines, benzoisoquinolines and other Rule-of-3 compliant heterocyclic fragments. These stereochemically dense structures are well-known ligands for biological targets such as 5-HT receptors.3 One of the most direct methods to access these compounds is through Diels–Alder (DA) cycloaddition of ortho-quinodimethanes (o-QDMs) with various dienophiles (Scheme 1).4 These highly reactive dienes are most commonly accessed through the thermal electrocyclic ring-opening of benzocyclobutenes. Due to the aromaticity of these systems, this reaction occurs typically with flash vacuum pyrolysis or with prolonged heating in high-boiling solvents, thus greatly limiting its application in drug discovery programs. By adapting this methodology to a high T/P flow reactor where superheated conventional solvents can be used, and reaction times can be dramatically shortened, we can now rapidly evaluate chemical space around these privileged scaffolds.
 | ||
| Scheme 1 Inter- and intra-molecular Diels–Alder cycloadditions of o-QDMs. | ||
We chose to open our investigations with the intermolecular Diels–Alder reaction between commercially available 1-benzocyclobutenecarbonitrile 1a and imine dienophiles that would yield valuable tetrahydroisoquinoline products.5 Initial efforts showed ring-opening of 1a and subsequent trapping with dihydroisoquinoline 2a occurred in just 30 seconds at 300 °C and 120 bar in the Phoenix flow reactor.6 Complete conversion to cycloadduct 3a was observed, with an isolated yield of 85% (Table 1). In comparison, the conventional batch reaction in refluxing 1,2-dichlorobenzene (180 °C) required 6 hours for full conversion.7 Furthermore, microwave heating in THF for 1 hour at 180 °C resulted in only 7% conversion.
|
|
||
|---|---|---|
| a Reported as isolated yields. Cis/trans ratios reported in parantheses and determined by 1H NMR spectroscopic analysis of the crude reaction mixture. | ||
 3a, 85% (1.3
3a, 85% (1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
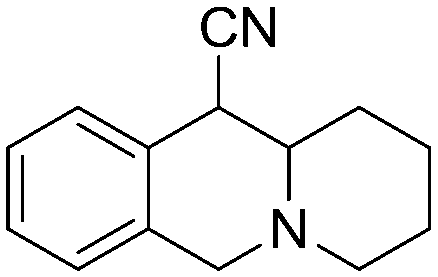 3b, 41% (1.9:1)
3b, 41% (1.9:1) |
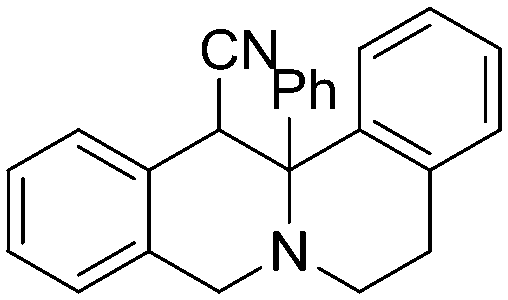 3c, 69% (3:1)
3c, 69% (3:1) |

Exclusive regiospecificity was observed in all cases, and the endo/exo ratio is similar to what is observed in refluxing 1,2-dichlorobenzene.8 Evaluation of various common solvents (CH3CN, THF, toluene) and additives (H2O, pTsOH) showed little effect on product yield or distribution, thus THF was chosen due to its low boiling point and its moderate polarity. Using the optimized reaction conditions, hetero-DA reaction of 1a with a variety of cyclic imines afforded the corresponding pyridoisoquinoline products (Table 1).9 Cyclic imines without activating arenes are also reactive, albeit with a lower yield (3b). Ketimines are also viable dienophiles, giving the highly substituted polycyclic compound 3c with 3:1 diastereoselectivity.
Intramolecular Diels–Alder (IMDA) cycloadditions of o-QDMs bearing a pendant dienophile have also been used to great effect in the synthesis of natural products and biologically active compounds.10 Exploiting the entropic advantage of intramolecular reactions allows use of non-activated dienophiles, leading readily to complex fused ring systems with high regio- and stereo-specificity. Our investigations focused on the generation of benzoisoindoline-like products, where the starting materials could be conveniently generated from commercially available 1-benzocyclobutenecarbonitrile 1a and 1-benzocyclobutenecarboxylic acid. In these cases, we found that more dilute conditions (0.05 M vs. 0.2 M) were required to prevent dimerization of starting materials.
Both olefinic and acetylenic systems were tolerated as dienophiles, and IMDA cycloaddition under these reaction conditions furnished exclusively the cis-fused product in good yields (Table 2). For olefinic products, migration of the double bond was observed to cleanly give the more stable product 5b. Siloxy tethers were also effective, giving alkyl silyl ether (5c) and vinyl silyl ether (5d) products which can be further derivatized with well-known protocols such as Tamao oxidations (Scheme 2), cross-couplings or other fluoride-activated reactions.11 Furthermore, hetero-DA reactions using tethered dienophiles such as aldehydes and nitriles are also possible.12 While both isomers of isochromane 5e can be isolated separately, the cis product can also be exclusively obtained upon acid-catalyzed epimerization of the product. IMDA cycloaddition with an isolated nitrile lead to formation of the stable isoquinoline product 5f in good yields, presumably through isomerization and dehydration processes.
|
|
||
|---|---|---|
| a Reported as isolated yields. Cis/trans ratios are reported in parantheses and determined by 1H NMR spectroscopic analysis of the crude reaction mixture. | ||
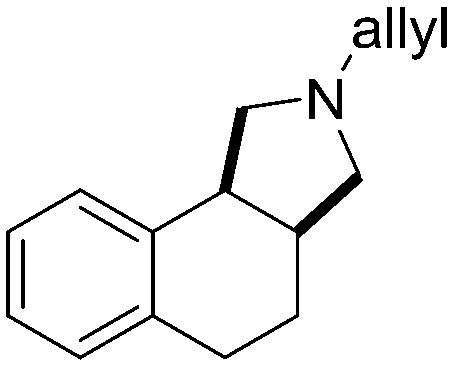 5a, 64%
5a, 64% |
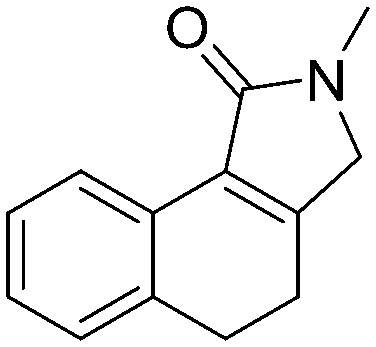 5b, 79%
5b, 79% |
 5c, 75%
5c, 75% |
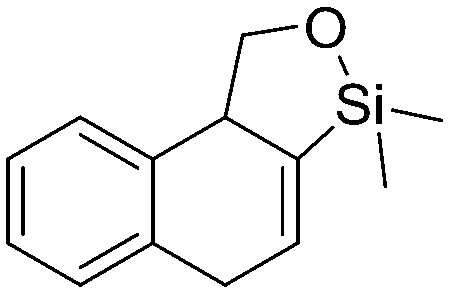 5d, 21%
5d, 21% |
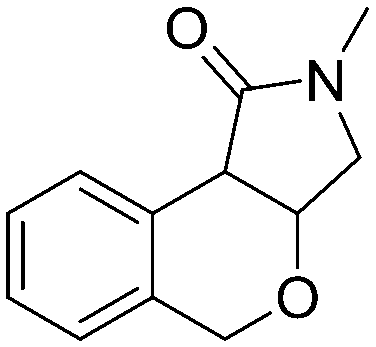 5e, 51% (1.7:1)
5e, 51% (1.7:1) |
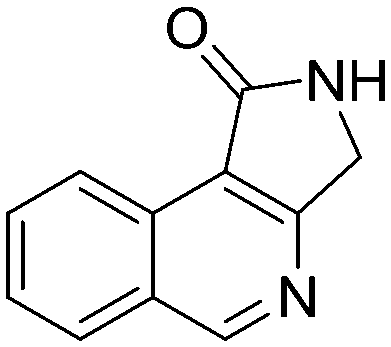 5f, 58%
5f, 58% |

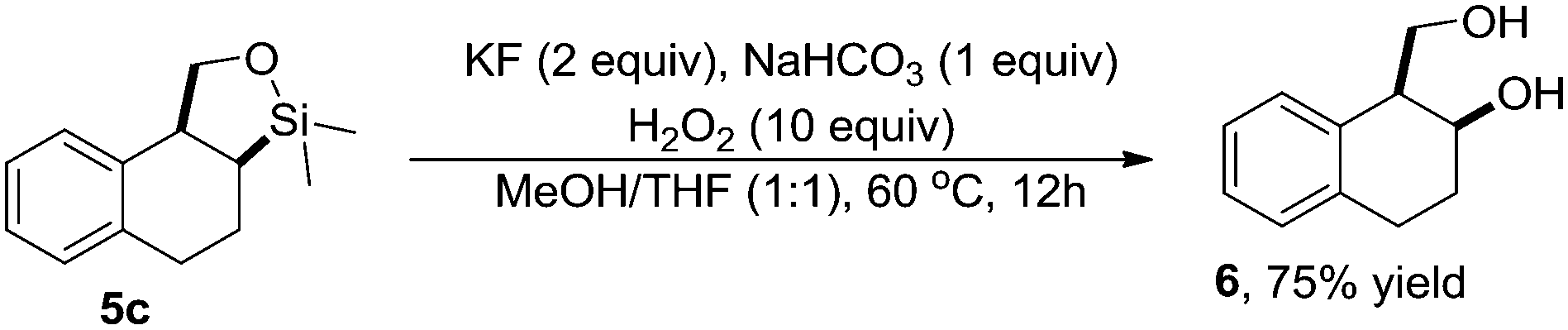 | ||
| Scheme 2 Derivatization of silyl ether 5c. | ||
Other challenging dienophiles such as 1,2-disubstituted alkenes and furans can also participate in DA reactions with o-QDMs (Scheme 3). The 1,2-disubstituted alkene substrate 4g gave rise to a mixture of two separable diastereomeric products (5g). In both instances of 4h and 4i, the furan served as the 2π component in the [4 + 2] cycloaddition, which is quite rare for unactivated furans.13 The matched case (4h) fully converted to the expected tetracyclic fused ring-system 5h along with the rearranged product 5h’. Addition of acid to the reaction mixture prior to injection in the Phoenix gave full conversion to 5h’. Incomplete conversion was observed for the mismatched case (4i), giving recovered starting material and cycloadduct 5i. Of note, both reactions were highly site-selective and diastereoselective, thus setting three contiguous stereocentres in a single synthetic operation.
 | ||
| Scheme 3 IMDA reactions with di- and tri-substituted olefins. | ||
Finally, we were also interested in the use of dihydrobenzothiophene-2,2-dioxides 7 in the high T/P flow reactor, which generates the reactive o-QDM intermediate via thermal cheletropic extrusion of SO2.14 These sulfone substrates have the advantage of being readily available and easily functionalized.
We found that these masked o-QDMs were also highly reactive in the IMDA reaction with both olefins and acetylenes, though longer residence times were required (Table 3). Use of longer tethers (6- vs. 5-membered ring formation) resulted in a mixture of cis/trans products, which can be readily separated.15 Entropic assistance with a substituted amide tether resulted in high yields of the desired product (8a), favouring the trans product. With unsubstituted amide tethers, lower yields and selectivities were observed with a significant amount of byproducts likely resulting from a [1,5]-hydride shift from the reactive intermediate.16 Furthermore, ortho-quinonemethane imine (o-QMI) intermediates could also be generated from 2,1-benzoisothiazoline 2,2-dioxides under the same reaction conditions, undergoing IMDA cycloaddition to yield the corresponding pyrroloquinoline derivatives (8c–d). Comparatively, the batch reaction of 7d in refluxing 1,2,4-trichlorobenzene (215 °C) showed full consumption of the starting material in 2 hours, but the product 8d was not recoverable due to co-elution with the solvent.17,18
|
|
|||
|---|---|---|---|
| a Reported as isolated yields. Cis/trans ratios are reported in parantheses and determined by 1H NMR spectroscopic analysis of the crude reaction mixture. | |||
 8a, 93%, (2.9:1)
8a, 93%, (2.9:1) |
 8b, 69% (1:1)
8b, 69% (1:1) |
 8c, 55%
8c, 55% |
 8d, 14%
8d, 14% |

Conclusions
As part of our ongoing efforts to introduce modern chemical technologies to our drug discovery programs, we have disclosed our findings on the application of a high T/P flow reactor to the electrocyclic ring opening of benzocyclobutenes, benzothiophene-2,2-dioxides and benzoisothiazoline-2,2-dioxides, and subsequent Diels–Alder cycloaddition of the resulting highly reactive o-QDM intermediates with a range of dienophiles to give a broad scope of complex heterocyclic products.19 The use of flow technology allowed reaction times to be brought down to less than 4 minutes, and for conventional solvents to be superheated in a safe and controlled fashion, thus replacing high-boiling, challenging-to-use solvents. As the synthetic power of this fundamental transformation can hardly be overstated, we envisage this rapid and efficient method to have great implications in medicinal chemistry.Acknowledgements
We thank the staff in the AbbVie structural chemistry group for NMR support and the staff in the Abbvie analytical and purification sciences group for purification support. We also thank IMSERC Mass Spectrometry at Northwestern University for assistance with HRMS. All authors are employees or former employees of AbbVie. AbbVie contributed to the study design, research, interpretation of data, writing, reviewing and approving the manuscript. The financial support for this research was provided by AbbVie.Notes and references
- J. A. DiMasi and L. B. Faden, Nat. Rev. Drug Discovery, 2011, 10, 23–27 CrossRef CAS PubMed.
- (a) A. R. Bogdan, M. Charaschanya, A. Worthy, Y. Wang and S. W. Djuric, Org. Lett., 2016, 18, 1732–1735 CrossRef CAS PubMed; (b) M. Charschanya, A. R. Bogdan, Y. Wang and S. W. Djuric, Tetrahedron Lett., 2016, 57, 1035–1039 CrossRef; (c) J. Tsoung, A. R. Bogdan, S. Kantor, Y. Wang, M. Charaschanya and S. W. Djuric, J. Org. Chem., 2017, 82, 1073–1084 CrossRef CAS PubMed.
- (a) L. A. Walter and W. K. Chang, J. Med. Chem., 1975, 18, 206–208 CrossRef CAS PubMed; (b) F. Z. Basha, J. F. DeBernardis and R. J. Altenbach, US Pat., US5248677(A), 1993 Search PubMed; (c) N. M. Barnes and T. Sharp, Neuropharmacology, 1999, 38, 1083–1152 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. L. Charlton and M. M. Alauddin, Tetrahedron, 1987, 43, 2873–2889 CrossRef CAS; (b) J. L. Segura and N. Martin, Chem. Rev., 1999, 99, 3199–3246 CrossRef CAS PubMed; (c) G. Mehta and S. Kotha, Tetrahedron, 2001, 57, 625–804 CrossRef CAS; (d) A. K. Sadana, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539–1602 CrossRef CAS PubMed.
- T. A. Kshirsagar and P. S. Portoghese, J. Org. Chem., 1998, 63, 1706–1708 CrossRef CAS.
- See ESI† for further information on optimization studies.
- For similar reactions, see: T. Kametani, H. Yukawa, Y. Suzuki and T. Honda, J. Chem. Soc., Perkin Trans. 1, 1985, 2151–2154 RSC.
- C. W. Jefford, G. Bernardinelli, Y. Wang, D. C. Spellmeyer, A. Buda and K. N. Houk, J. Am. Chem. Soc., 1992, 114, 1157–1165 CrossRef CAS.
- For reports on imino-DA with o-QDMs, see: (a) W. Oppolzer, Angew. Chem., Int. Ed. Engl., 1972, 11, 1031–1032 CrossRef CAS; (b) T. Kametani, T. Takahashi, K. Ogasawara and K. Fukumoto, Tetrahedron, 1974, 30, 1047–1051 CrossRef CAS; (c) W. F. Berkowitz and T. V. John, J. Org. Chem., 1984, 49, 5269–5271 CrossRef CAS; (d) D. Craig, M. J. Robson and S. J. Shaw, Synlett, 1998, 1381–1383 CrossRef CAS; (e) M. F. Hentemann, J. G. Allen and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 1937–1940 CrossRef CAS.
- For a review, see: W. Oppolzer, Synthesis, 1978, 11, 793–802 CrossRef.
- For a review on silicon-tethered reactions, see: M. Bols, Chem. Rev., 1995, 95, 1253–1277 CrossRef CAS.
- For reports on intramolecular oxo-DA with o-QDMs, see: (a) R. L. Funk and K. P. C. Vollhardt, J. Am. Chem. Soc., 1976, 98, 6755–6757 CrossRef CAS; (b) R. L. Funk and K. P. C. Vollhardt, J. Am. Chem. Soc., 1980, 102, 5245–5253 CrossRef CAS.
- For examples of furans serving as dienophiles, see: (a) C.-H. Chen, P. D. Rao and C.-C. Liao, J. Am. Chem. Soc., 1998, 120, 13254–13255 CrossRef CAS; (b) Y. Matsua, Y. Sasaki, M. Nagaoka, H. Kakuda, N. Toyooka, N. Imanishi, H. Ochiai and H. Nemoto, J. Org. Chem., 2004, 69, 7989–7993 CrossRef PubMed , and references contained therein; (c) S. E. Steinhardt and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 7546–7547 CrossRef CAS PubMed.
- M. P. Cava and A. A. Deana, J. Am. Chem. Soc., 1959, 81, 4266–4268 CrossRef CAS.
- W. Oppolzer, Tetrahedron Lett., 1974, 12, 1001–1004 CrossRef.
- T. Kametani, M. Tsubuki, Y. Shiratori, Y. Kato, H. Nemoto, M. Ihara and K. Fukumoto, J. Org. Chem., 1977, 42, 2672–2676 CrossRef CAS.
- (a) R. D. Bowen, D. E. Davies, C. W. G. Fishwick, T. O. Glasbey, S. J. Noyce and R. C. Storr, Tetrahedron Lett., 1982, 23, 4501–4504 CrossRef CAS; (b) K. Wojciechowski, Tetrahedron, 1993, 49, 7277–7286 CrossRef CAS.
- Low yield of 8d can be attributed to isomerization of the reactive intermediate followed by [1,5]-hydride shift.
- For other recent examples of high temperature Diels–Alder cycloadditions in flow, see: (a) T. Razzaq and C. O. Kappe, Chem. – Asian J., 2010, 5, 1274–1289 CAS; (b) S. Abele, S. Hock, G. Schmidt, J.-A. Funel and R. Marti, Org. Process Res. Dev., 2012, 16, 1114–1120 CrossRef CAS; (c) R. Martin, F. Morawitz, C. Kuratli, A. M. Alker and A. I. Alanine, Eur. J. Org. Chem., 2012, 47–52 CrossRef CAS; (d) J. Lehmann, T. Alzieu, R. E. Martin and R. Britton, Org. Lett., 2013, 15, 3550–3553 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization study details, experimental procedures, characterization data, copies of 1H, 13C and 2D NMR spectra. See DOI: 10.1039/c7re00058h |
| This journal is © The Royal Society of Chemistry 2017 |