
Synthesis of polymeric nano-objects of various morphologies based on block copolymer self-assembly using microporous membranes†
Sri
Agustina
ad,
Masayoshi
Tokuda
b,
Hideto
Minami
 b,
Cyrille
Boyer
*ac and
Per B.
Zetterlund
*a
b,
Cyrille
Boyer
*ac and
Per B.
Zetterlund
*a
aCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au; p.zetterlund@unsw.edu.au
bGraduate School of Engineering, Kobe University, Rokko, Nada, Kobe 657-8501, Japan
cAustralian Centre for NanoMedicine, School of Chemical Engineering, UNSW Australia, Sydney, NSW, Australia
dDepartment of Chemical Engineering, University of Sultan Ageng Tirtayasa (UNTIRTA), Banten, Indonesia
First published on 18th May 2017
Abstract
Membrane emulsification entails the use of two immiscible liquids for preparation of oil-in-water or water-in-oil emulsions of well-defined droplet size. In this work, we have developed a novel approach for preparation of polymeric nano-objects of various morphologies based on self-assembly of diblock copolymers in tandem with microporous membrane technology (Shirasu porous glass (SPG) membranes). Diblock copolymers poly(oligoethylene glycol acrylate)-b-poly(styrene) (POEGA-b-PSt) comprising different block lengths were prepared by RAFT polymerization. Self-assembly using SPG membrane technology was subsequently conducted by first dissolving the polymer in tetrahydrofuran, followed by continuous passage of this copolymer solution through the membrane pores into the aqueous phase. The results indicate that it is possible to conveniently tune the nano-object morphology via the process parameters of pressure and pore size for a given formulation. The technique also holds promise with regards to high throughput, in particular in the case of vesicles.
Molecular self-assembly into nano-objects of various shapes is a widespread occurrence in nature, for example as illustrated by the variety of shapes formed by viruses. Synthetic amphiphilic diblock copolymers are also able to self-assemble into a range of different nano-sized structures such as spherical micelles, rods and vesicles, providing an attractive route to functional nano-objects with potential applications in nanotechnology.
Pioneered by Eisenberg and coworkers,1–5 self-assembly by diblock copolymers is typically achieved by the gradual decrease in quality of a common solvent by addition of a selective solvent. In more recent years, an alternative synthetic route has gained popularity, namely polymerization-induced self-assembly (PISA).6–13 According to the PISA process, conducted as a dispersion or emulsion polymerization, the solvophobic block of the block copolymer is prepared in situ, resulting in self-assembly in the polymerization mixture. Overall, one of the challenges is to develop efficient methods of morphology control, i.e. methods that enable one to prepare a given morphology in pure form. For example, it is often difficult to access pure rod morphology due to the narrow parameter widow during which such structures form.7,9–11
Microfluidic technologies have been employed for preparation of polymeric nanoparticles based on self-assembly, often with a view to incorporation of drugs within the nanoparticles.14–20 Microfluidic methods offer control over the mixing process (solvent/non-solvent), which in turn enables better control over particle size and size distribution, as well as potential for high throughput. Alternatively, Prud'homme and co-workers have developed a nanoprecipitation process,21–23 whereby rapid self-assembly/precipitation of both block copolymer and drug to be encapsulated results in improved encapsulation efficiency. Preparation of polymeric nano-objects in the form of cylindrical micelles based on diblock copolymer self-assembly has also been reported by use of a nanoscale pore membrane to extrude spherical micelles and transform them into cylindrical micelles.24,25
In this communication, we describe a novel approach to prepare polymeric nano-objects of various morphologies based on self-assembly of diblock copolymers in tandem with microporous membrane technology (Shirasu porous glass (SPG) membranes). Membrane emulsification, which involves generation of a liquid dispersed phase by passage through the pores of a microporous membrane directly into the continuous phase, entails the use of two immiscible liquids for preparation of oil-in-water or water-in-oil emulsions of well-defined droplet size. In this work, this approach has been adapted to self-assembly of amphiphilic diblock copolymers by passage of a copolymer solution through the microporous membrane into a poor solvent for one of the blocks of the copolymer (Scheme 1). Importantly, the two liquids in the present work are miscible, i.e. an emulsion is not generated. Two different poly(oligoethylene glycol acrylate)-b-poly(styrene) (POEGA-b-PSt) copolymers were prepared by RAFT polymerization (ESI†); POEGA17-b-PSt160 (Đ = 1.17) and POEGA17-b-PSt290 (Đ = 1.60). Self-assembly using SPG membrane technology was subsequently conducted by first dissolving the polymer in tetrahydrofuran (THF; good solvent; 11 mg polymer ml−1), followed by continuous passage of this copolymer solution through the membrane pores into an aqueous phase to yield a final polymer concentration of 1 mg polymer mL−1 in the THF/water mixture. As the THF solution was dispensed into the aqueous phase, the solution gradually turned turbid, consistent with the formation of polymeric nano-objects via self-assembly of the diblock copolymer.
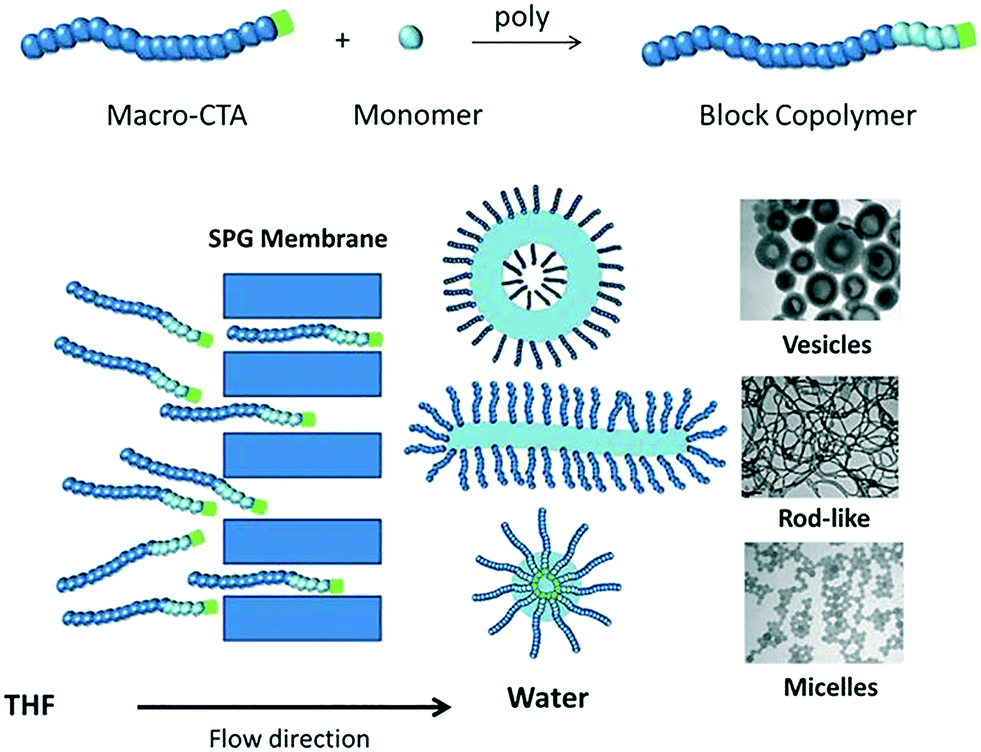 | ||
| Scheme 1 Schematic illustration of self-assembly of amphiphilic diblock copolymers by passage of a copolymer solution through a microporous membrane. | ||
When using membrane emulsification to generate emulsions, the pore size is a crucial parameter as it dictates the obtained droplet size.26–31 The greater the pore size, the lower is the minimum pressure required to obtain passage of the solution through the membrane (the critical pressure).26
Experiments were first conducted for both block copolymers using different membrane pore sizes (0.2–5.2 μm) at the respective critical pressures (Table 1) while keeping the block copolymer concentration in THF, the pressure, and the water volume constant. As evidenced by the TEM images in Fig. 1, a variety of polymeric nano-objects formed. Particle size distributions (DLS; Fig. 1) showed a shift towards larger sizes with increasing membrane pore size (DLS theory is based on spherical objects and as such these data are semi-quantitative). For the block copolymer with the shorter PSt block (POEGA17-b-PSt160), the smallest pore size of 0.2 μm yielded spherical particles (Fig. 1A).
| Pore size (μm) | Pressure (kPa) | Time (min) | Morphology | |
|---|---|---|---|---|
| POEGA17-b-PSt160 | POEGA17-b-PSt290 | |||
| 5.2 | 1 | 3 | Rod-like, vesicles | Vesicles |
| 0.2 | 70 | 3 | Rod-like, vesicles | Vesicles |
| 2.1 | 5 | 5 | Branched tubular vesicles | Vesicles |
| 0.2 | 50 | 7 | Rod-like, vesicles | Branched tubular vesicles |
| 0.8 | 8 | 13 | Rod-like | Vesicles |
| 0.2 | 30 | 13 | Rod-like | Vesicles |
| 0.3 | 12 | 25 | Rod-like | Rod-like, vesicles |
| 0.2 | 15 | 30 | Spherical micelles | Rod-like |
 | ||
| Fig. 1 TEM micrographs and dynamic light scattering size distributions (number-based) of nanoparticles prepared using SPG membranes with different pore sizes as indicated: (A & C) POEGA17-b-PSt160; (B & D) POEGA17-b-PSt290. | ||
Increasing the pore size to 0.3, 0.8, 2.1 and finally 5.2 μm was accompanied by the morphology changing into rod-like structures followed by bicontinous bilayers and vesicles consistent with DLS data indicating formation of gradually larger structures. In case of the block copolymer with the longer PSt block (POEGA17-b-PSt290), rods formed already for the lowest pore size of 0.2 μm, and the morphology gradually developed towards vesicles with increasing pore size. Thus, for both block copolymers, the morphology shifted in the direction spheres–rods–vesicles with increasing pore size. In general, it is well-known that the morphology of self-assembled low molecular weight surfactants as well as amphiphilic block copolymers change from spheres–rods–vesicles with an increase in the packing parameter (p),32 for example as caused by an increase in the degree of polymerization of the insoluble block. As such, the differences (especially for the lowest pore size) between the two block copolymers can be readily rationalized, i.e. for the same conditions the morphology is shifted towards vesicles for POEGA17-b-PSt290 (Fig. 1Avs. B).
The effect of membrane pressure on morphology was subsequently investigated. According to Darcy's law, an increase in the membrane pressure will lead to an increase in the flux of copolymer passing through the pores.28 Using the 0.2 μm membrane for POEGA17-b-PSt160 (Fig. 2A), spherical micelles were obtained at 15 kPa. Increasing the pressure resulted in the morphologies transforming to mainly rods (30 kPa) with a gradual presence of vesicles (50 and 70 kPa). In the case of POEGA17-b-PSt290 (Fig. 2B), rods and vesicles were present already at the lowest pressure of 15 kPa, with branched tubular structures and mainly vesicles dominating at higher pressures. The differences observed between the two different block copolymers can be rationalized as for Fig. 2. The DLS data (Fig. 2) show a shift to larger structures with increasing pressure for both polymers.
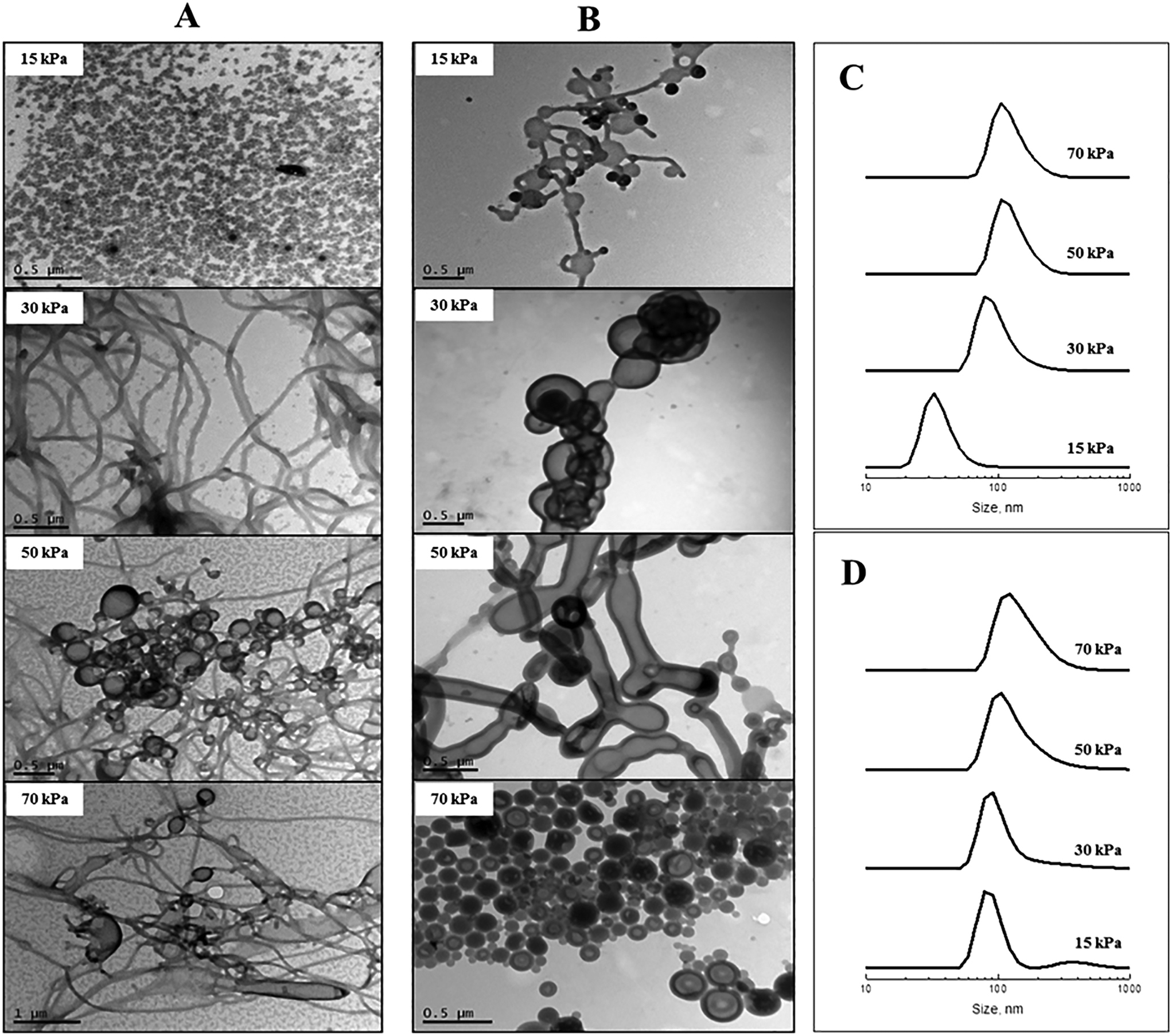 | ||
| Fig. 2 TEM micrographs and dynamic light scattering size distributions (number-based) of nanoparticles prepared using SPG membranes with different membrane pressures as indicated: (A & C) POEGA17-b-PSt160; (B & D) POEGA17-b-PSt290. | ||
The above results demonstrate that the nano-object morphology can be tuned by altering the process parameters of membrane pore size and pressure without changing the nature of the diblock copolymers nor experimental parameters such as block copolymer concentration. Importantly, the process itself is very rapid, in sharp contrast with other typically time-consuming approaches employed to achieve self-assembly via e.g. dialysis or slow addition of non-solvent utilized in the Eisenberg approach.5 The higher the pressure and the larger the membrane pore size, the more rapid is the process. The experiments of Fig. 1 and 2 concerning POEGA17-b-PSt160 have been tabulated (Table 1) in order of increasing time taken for completion of the process (i.e. passing 5 ml of polymer solution through the membrane). For the larger pore sizes, the process is extraordinarily rapid – with a pore size of 5.2 μm the process is complete in 3 min. Further examination of the data in Table 1 reveals that very rapid completion can be achieved in case of formation of vesicles and also rods, whereas the experiments generating spherical micelles are more time consuming (30 min in the longest case). These data also indicate that the morphologies correlate with the time taken to complete the process. In other words, vesicle-rich systems can be obtained using a large pore size (5.2 μm/1 kPa) or by employing a smaller pore size with higher pressure (0.2 μm/70 kPa), both processes taking similar lengths of time of 3 min (first two entries in Table 1). This observation may provide important insight into the mechanism underlying the formation of nano-objects of different morphology using this novel experimental approach.
In order to further understand the results described above, it is instructive to also conduct traditional self-assembly experiments based on (i) dropwise addition of water to polymer/THF, and (ii) dropwise addition of polymer/THF to water.33 According to (i), different total amounts of water were added dropwise to solutions of POEGA17-b-PSt290 in THF (0.9 mg ml−1) at a fixed rate. The times taken for the addition to be complete ranged from 26–70 min for 15–40 wt% total final water content. The TEM images in Fig. 3 show that the particle size increased with increasing water content – lower water content led to formation of spherical particles, whereas unilamellar and multilamellar vesicles as well as possibly large compound micelles (LCMs) formed at higher water contents. These results are consistent with previous work for similar systems.5,33,34 It has been rationalized that an increase in water content leads to an increase in the aggregation number (Nagg), which in turn causes an increase in the degree of stretching of PSt chains of the core. This eventually leads to a change in morphology towards structures with less curvature to reduce the stretching penalty of PSt chains.5
 | ||
| Fig. 3 TEM micrograph of nanoparticles prepared using method (i) (dropwise addition of water to polymer/THF) with POEGA17-b-PSt290 (final wt% of water as indicated). | ||
Experiments were subsequently conducted according to method (ii), whereby a fixed volume of POEGA17-b-PSt290 dissolved in THF (1 ml; 11 mg ml−1) was added dropwise to a fixed amount of water (10 ml) at different addition rates. The total time of addition was 12–100 min. The conditions in terms of overall ratio of THF to water, polymer concentration in THF, as well as stirring rate in the container containing water were the same as for the corresponding membrane experiment. This experiment mimics the membrane approach in that the only difference is the way that the polymer solution is added, i.e. dropwise vs. through membrane pores. At low addition rates, rod-like structures were observed, whereas increasing the addition rate led to formation of mixtures of rod-like, lamellae and vesicular structures (Fig. 4). This is in qualitative agreement with the membrane experiments – higher addition rates (via increase in pore size or increase in pressure) resulted in morphology transitions in the direction spherical micelles–rods–vesicles (progressively lower core–corona curvature). To further mimic the membrane process, in particular with respect to the very high addition rates achieved with high pressure or large pore size, method (ii) was conducted with total addition times of only 50 and 100 s as well as with instantaneous addition (all THF solution added at once) using POEGA17-b-PSt290. In all three cases, only spherical micelles were observed (Fig. S9†). Furthermore, the case of instantaneous addition resulted in large scale precipitation in the form of a white solid.
 | ||
| Fig. 4 TEM micrographs of nanoparticles prepared using method (ii) (dropwise addition of polymer/THF to water) with POEGA17-b-PSt290 (addition rates as indicated). | ||
Given that self-assembly using the membrane approach in many cases (Fig. 1 and 2) yields different morphologies for the same recipe (same block copolymer, block copolymer concentration, volumes of water/THF etc.), it follows that some of these experiments must involve kinetically trapped structures, i.e. kinetic control as opposed to equilibrium having been reached.35 The same applies to method (ii) described above.
The membrane-based approach enables preparation of higher order structures such as rods and vesicles in a much shorter time frame than the traditional techniques of methods (i) and (ii). The membrane approach afforded predominantly vesicles within 3 min using the 5.2 μm membrane or high pressure of 70 kPa (0.2 μm membrane) for POEGA17-b-PSt290. By comparison, methods (i) and (ii) did not generate vesicles under any conditions attempted. Importantly, using a THF/polymer addition rate (method (ii)) similar to that for the fastest membrane experiments (3 min) only generated spherical micelles, with significant precipitation when using instantaneous addition.
As discussed by Langer and coworkers,14,15 the mixing time is a characteristic feature influencing the size and size distribution distributions when self-assembly of amphiphilic diblock copolymers occurs as a result of a change in the solvent quality experienced by the copolymers using microfluidic technologies. However, to the best of our knowledge, such approaches have to date not been employed to control the nano-object shape. In case of rapid microfluidic mixing, the final solvent composition is reached before the self-assembly occurs. Conversely, under conditions of slow mixing, self-assembly begins at a solvent composition differing from the final solvent composition.14 Such circumstances would affect the self-assembly process and the nature of the resulting nano-objects. The mechanistic details of self-assembly in the case of our membrane approach remain to be elucidated. However, one can speculate that both pore size and transmembrane pressure influence the solvent composition experienced by the polymer as self-assembly occurs. Presumably, larger pore size/pressure leads to a higher local concentration of THF (good solvent) during the actual self-assembly as polymer emerges from the pores. This would make it more likely that equilibrium structures can be reached under such conditions, as opposed to the initially formed structures (typically spheres5) becoming kinetically trapped. This also appears consistent with our experimental observations, i.e. larger pore size/pressure leads to a morphology shift in the direction spheres–rods–vesicles. Moreover, higher pore size or higher transmembrane pressure may also lead to higher polymer concentration in the vicinity of the pore where self-assembly occurs – higher polymer concentration is known to shift the morphology in the direction spheres–rods–vesicles,5 consistent with what is observed. One may also speculate that shear-induced aggregation leads to formation of larger structures, as proposed previously in connection with microfluidic systems.18 Importantly, the rapid and tunable mixing afforded by the membrane approach enables effective tuning of nanoparticle morphology via membrane pore size and transmembrane pressure without altering other experimental parameters.
In conclusion, we have developed a novel method for synthesis of polymeric nano-objects of various non-spherical morphology based on self-assembly of amphiphilic diblock copolymers whereby mixing of the solvent (containing the polymer) and non-solvent phases is achieved using microporous membranes. The morphology obtained can be conveniently tuned via the membrane pore size and the transmembrane pressure. The method appears particularly advantageous for rapid synthesis of high-order morphologies such as vesicles.
Acknowledgements
The authors thank UNSW Mark Wainwright Analytical Center.References
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CAS.
- L. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883 RSC.
- J. T. Sun, C. Y. Hong and C. Y. Pan, Soft Matter, 2012, 8, 7753 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174 CrossRef CAS PubMed.
- S. Dong, W. Zhao, F. P. Lucien, S. Perrier and P. B. Zetterlund, Polym. Chem., 2015, 6, 2249 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985 CrossRef CAS PubMed.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558 CrossRef CAS.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984 CrossRef CAS.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean and W. Kyei-Manu, et al. , Nano Lett., 2008, 8, 2906 CrossRef CAS PubMed.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623 CrossRef CAS PubMed.
- J.-M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi and J. Wu, et al. , ACS Nano, 2014, 8, 6056 CrossRef CAS PubMed.
- A. Jahn, W. N. Vreeland, M. Gaitan and L. E. Locascio, J. Am. Chem. Soc., 2004, 126, 2674 CrossRef CAS PubMed.
- C.-W. Wang, D. Sinton and M. G. Moffitt, J. Am. Chem. Soc., 2011, 133, 18853 CrossRef CAS PubMed.
- Q. B. Xu, M. Hashimoto, T. T. Dang, T. Hoare, D. S. Kohane and G. M. Whitesides, et al., Small, 2009, 5, 1575 CrossRef CAS PubMed.
- G. Schabas, H. Yusuf, M. G. Moffitt and D. Sinton, Langmuir, 2008, 24, 637 CrossRef CAS PubMed.
- C. Zhang, V. J. Pansare, R. K. Prud'Homme and R. D. Priestley, Soft Matter, 2012, 8, 86 RSC.
- K. M. Pustulka, A. R. Wohl, H. S. Lee, A. R. Michel, J. Han and T. R. Hoye, et al. , Mol. Pharmaceutics, 2013, 10, 4367 CrossRef CAS PubMed.
- Z. X. Zhu, Mol. Pharmaceutics, 2014, 11, 776 CrossRef CAS PubMed.
- Q. Chen, H. Zhao, T. Ming, J. Wang and C. Wu, J. Am. Chem. Soc., 2009, 131, 16650 CrossRef CAS PubMed.
- Q. Chen, J. Wang and L. Shao, Macromol. Rapid Commun., 2013, 34, 1850 CrossRef CAS PubMed.
- C. Charcosset, I. Limayem and H. Fessi, J. Chem. Technol. Biotechnol., 2004, 79, 209 CrossRef CAS.
- A. Gijsbertsen-Abrahamse, Membrane emulsification: process principles, Wageningen Universiteit, 2003 Search PubMed.
- S. M. Joscelyne and G. Trägårdh, J. Membr. Sci., 2000, 169, 107 CrossRef CAS.
- J. Lee, D. R. Hwang, S. E. Shim and Y.-M. Rhym, Macromol. Res., 2010, 18, 1142 CrossRef CAS.
- R. Liu, G. Ma, F.-T. Meng and Z.-G. Su, J. Controlled Release, 2005, 103, 31 CrossRef CAS PubMed.
- G. T. Vladisavljević and H. Schubert, J. Membr. Sci., 2003, 225, 15 CrossRef.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 2011 Search PubMed.
- L. F. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239 CrossRef CAS.
- H. W. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, and SEC results (Scheme S1, Fig. S1–S9 and Table S1). See DOI: 10.1039/c7re00032d |
| This journal is © The Royal Society of Chemistry 2017 |