
Continuous-flow synthesis of fluorine-containing fine chemicals with integrated benchtop NMR analysis†
Thomas H.
Rehm
 *a,
Christian
Hofmann
a,
Dorothee
Reinhard
a,
Hans-Joachim
Kost
a,
Patrick
Löb
a,
Matthias
Besold
a,
Knut
Welzel
a,
Jan
Barten
b,
Andrey
Didenko
b,
Dmitri V.
Sevenard
b,
Bruce
Lix
c,
Andrew R.
Hillson
c and
Susanne D.
Riegel
c
*a,
Christian
Hofmann
a,
Dorothee
Reinhard
a,
Hans-Joachim
Kost
a,
Patrick
Löb
a,
Matthias
Besold
a,
Knut
Welzel
a,
Jan
Barten
b,
Andrey
Didenko
b,
Dmitri V.
Sevenard
b,
Bruce
Lix
c,
Andrew R.
Hillson
c and
Susanne D.
Riegel
c
aFraunhofer ICT-IMM, Carl-Zeiss-Straße 18-20, 55129 Mainz, Germany. E-mail: thomas.rehm@imm.fraunhofer.de; Tel: +49 6131 990 195
bHansa Fine Chemicals GmbH, BITZ, Fahrenheitstraße 1, 28359 Bremen, Germany
cNanalysis Corp, Bay 4, 4500 5 Street NE, Calgary, Alberta T2E 7C3, Canada
First published on 24th March 2017
Abstract
A compact lab plant was designed for the continuous-flow synthesis of fluorine-containing compounds and was combined with an NMR analysis platform based on a benchtop NMR spectrometer. The approach of a unified synthesis and analysis strategy for fine chemicals was applied to three different reactions, all employing fluorine as a chemical probe for online-19F NMR analysis. A high temperature synthesis for the deprotection of a CF2H group was done as well as Ruppert–Prakash reactions for the perfluoroalkylation of benzaldehyde as a model substrate. The C–H arylation of furan with a trifluoromethylated aryldiazonium salt was performed as an example of a photochemically catalyzed reaction. All three reaction classes challenge the synthesis and analysis setup differently according to sample preparation (premagnetization of bubble-free sample) and spectrometer sensitivity (signal to noise ratio, spectral resolution, scan number, substrate concentration and flow rate), but nonetheless prove the successful application of the continuous-flow synthesis of fluorinated fine chemicals with integrated online NMR analysis.
Introduction
Continuous-flow synthesis has become a widely explored alternative to conventional batch synthesis.1–4 Innovations in flow technology, including the development of microstructured hardware, have driven the rapid development of continuous-flow applications in both academia and industry.5–12 In addition to the development of novel process windows that can carry out reactions at the borderline of physical parameters (e.g. high temperature and high pressure),13 the intensive mixing and the increased phase contact engineered in the microstructured flow reactors has been shown to drastically increase the reaction rate and, in many cases, has also been shown to improve product selectivity.14,15Additionally, flow technology has enabled rapid and easy process development through systematic optimization of key parameters, including system pressure and reactor temperature, substrate concentration and flow rate of the reaction solution.16,17 Inline and online sensor technology based on FT-IR, Raman or UV/vis spectroscopy or GC and HPLC analysis has been well established for reaction control.18–25 Interestingly, despite being one of the most powerful analytical methodologies available, NMR spectroscopy has not yet become a major player in process analytical technology (PAT).26–30 The exclusion of NMR spectrometers from a PAT portfolio is largely owing to the three main limitations of a traditional superconducting magnet: (1) the physical size, (2) the vulnerability of this instrumentation to the physical and chemical hazards of an industrial environment, and (3) the high cost, not only in the initial purchasing expense, but also in the recurring operating costs (i.e. maintenance and cryogen fills) required to maintain a superconducting magnet. With the advent of compact, so-called ‘benchtop’ NMR spectrometers, which use permanent rare earth magnets to generate a homogenous magnetic field, the application of NMR technology as a PAT sensor has now become conceivable.31–37 A detailed comparison between a costly high-resolution NMR spectrometer and a price-conscious mid-resolution benchtop NMR spectrometer reveals an excellent price-performance ratio for the latter.38 Recent publications by Cronin et al.,39 Maiwald et al.38 and Ley et al.40 highlight several examples of online 1H and/or 1H/19F NMR analysis via benchtop spectrometers and illustrate the rapid development of research within this field. Kinetic studies and self-optimization of reaction parameters show the potential and versatility of benchtop NMR for continuous-flow synthesis.
Herein we describe the development of a continuous-flow synthesis lab plant combining the advantages of flow technology with benchtop NMR analysis for the synthesis of fluorinated compounds. Fluorine (19F) is an ideal NMR handle due to its advantageous nuclear magnetic properties. Fluorine-19 is a 100% naturally abundant, high-frequency, spin ½, high receptivity nucleus. Consequently, 19F NMR experiments are comparable to proton NMR measurements.41,42 Unlike 1H NMR experiments, however, the acquired 19F spectrum will be devoid of strong background solvent signals that may interfere with the desired analyte resonances. The safe and reproducible synthesis of fluorinated compounds in a compact lab plant with integrated 19F NMR analysis is an attractive playground for pharmaceutical43 and agrochemical44,45 industries.
Results and discussion
Lab plant design
The concept for the lab plant was to integrate a multi-purpose, continuous-flow synthesis apparatus with an online NMR sensory apparatus, including both a liquid control system and an NMR spectrometer, within the constraint of a standard fume hood (140 cm in width, 90 cm in height) (Fig. 1). The lab plant was designed to be compatible with a variety of reactions by using integrated cartridge systems for immobilized reagents, gas–liquid splitting and incorporating both temperature and pressure management into the flow network. A detailed flow chart of the lab plant can be found in the ESI.† The core element of the synthesis apparatus is the microstructured tubular reactor with a heating cartridge and integrated temperature sensor (Tube Heat Transfer Micro Device XL; THTMD).46 The THTMD has an internal volume of 6 mL and allows rapid heat transfer from the integrated heating cartridge over the microstructured inner wall of the tube reactor to the reaction mixture (Fig. 2, left; for details, see the ESI†). The electrical control unit of the THTMD receives feedback from the temperature sensor for accurate temperature control and stability inside the microreactor. The THTMD can heat to a maximum temperature of T = 300 °C and sustain a maximum pressure of p = 120 bar. The substrate solution is fed to the THTMD using a HPLC pump. | ||
| Fig. 1 Multi-purpose lab plant with integrated NMR analysis using a benchtop NMR spectrometer. | ||
 | ||
| Fig. 2 Assembled THTMD with Swagelok connectors for a reaction stream and temperature sensor (left); capillary photoreactor with integrated LED light source (right). © Fraunhofer ICT-IMM. | ||
Stainless-steel cartridges of various capacities can be integrated in series either before or after the tubular reactor. This flexibility allows the reaction mixture to come in contact with the immobilized catalysts or quenching reagents as is required. Additionally, a gas–liquid splitter was integrated after the reactor. This device becomes necessary either when using gaseous reagents or if gas is evolved throughout the course of a reaction to ensure that the reaction mixture is a bubble-free, homogenous solution prior to entering the NMR flow cell. We adapted the evaporation device designed by Ley et al. for our purpose of phase separation.47 Usually the evaporator applies a strong nitrogen gas stream with a flow rate of approx. 10 L min−1 at elevated temperature to diffuse and strip off the liquid phase of a reaction stream to a condenser. In our case, we found it possible to achieve sufficient gas–liquid splitting even at lower nitrogen gas stream flow rates (1–2 L min−1) at room temperature. Reactive or evolving gases were stripped off, diluted with nitrogen gas and expelled through the exhaust air of the fume hood.
Besides the permanently installed THTMD flow reactor we also used a capillary photoreactor which was connected to the plant in line with the HPLC pump and the gas–liquid splitter (Fig. 2, right; for details, see the ESI†). The capillary photoreactor is based on the reactor design of Booker-Milburn48–51 and consists of a fluorinated ethylene propylene (FEP) capillary with a 14 mL inner volume (ID = 0.8 mm, OD = 1.6 mm) wrapped around a borosilicate glass cylinder. As light source light emitting diodes (LEDs) of different wavelengths (royal blue, λmax = 455 nm; green, λmax = 518 nm; red, λmax = 630 nm) are placed on a 3D-laser sintered hexagonal heat exchanger unit inside the capillary photoreactor (for details, see the ESI†). The electrical power of each LED array ranged from 6 W for red light to 10 W for green and blue light.
Liquid control system
In order to amalgamate the synthesis and analysis components, we employed a liquid control system (LCS) to bridge the platforms (for details, see the lab plant flow chart in the ESI†). The LCS module features a set of four magnetic valves that can be electronically controlled by a LabView-based program (see below). With this software, the experimenter can decide whether the reaction solution is routed continuously through the NMR flow cell for a ‘real-time’ measurement or as a static sample volume in the flow cell for a stopped-flow experiment. In the latter case, the magnetic valves are switched to by-pass mode for directing the incoming reaction solution from the lab plant to the collecting vessel after filling the flow cell. Additionally, a second HPLC pump is directly connected to the magnetic valves of the LCS. This allows the flow cell to be purged with pure solvent either between experiments or during a reaction. This is useful to clean the flow cell and simplify maintenance shimming routines without requiring flow cell exchange.NMR analysis platform
The NMR analysis platform is based on a Nanalysis NMReady-60e 1.4 T benchtop spectrometer tuned to 1H (58.63 MHz) and 19F (55.17 MHz). Unlike conventional NMR experiments, with the flow experiments we used proteo-solvents instead of deuterated ones, and locked and shimmed on the solvent 1H NMR signal and observed changes in 19F NMR spectra. Prior to the development of our lab plant concept, the NMReady was compatible with only standard glass NMR tubes with a diameter of 5 mm. To convert the NMReady into an online analyser, we designed a flow cell based on a hybrid open glass tube with an approximate sample volume of 0.49 mL inside the magnet connected with polyether ether ketone (PEEK) (Fig. 3, top). The flow cell is connected to the LCS with standard chromatography connectors and is filled from bottom to top. The head of the flow cell is fixed to the top plate of the spectrometer for stable operation (Fig. 3, bottom). | ||
| Fig. 3 Bottom-to-top flow cell assembled on holder for safe storage (top left) and close-up view of the glass tube of the flow cell (top right); installed flow cell with head attached to the top plate of the NMR spectrometer. © Fraunhofer ICT-IMM. | ||
After installation of the flow cell we investigated the impact of flow rate and substrate concentration on the peak shape, spectral resolution and signal to noise ratio (SNR). A 0.6 mol L−1 stock solution of 4-(trifluoromethyl)benzyl alcohol in proteo-DMSO was prepared and diluted to 0.3 and 0.15 mol L−1. This concentration series was pumped through the flow cell at various flow rates (0.5, 2 and 4 mL min−1) and a 4, 8, 16 and 64 scan 19F NMR spectrum was acquired. Expectedly, the higher molar concentration and scan number afforded spectra with better SNR (Table 1).52 In an application-oriented sense, a low scan number is desirable to reduce measurement time. Correspondingly, a high substrate concentration is desirable to reduce solvent consumption and improve SNR, but this will be limited by solubility.
| Flow rate fl [mL min−1] | ||||
|---|---|---|---|---|
| c [mol L−1] | Scan # | 0.5 | 2 | 4 |
| 0.6 | 4 | 29.6 | 15.3 | 13.0 |
| 8 | 41.7 | 21.9 | 15.7 | |
| 16 | 60.4 | 31.5 | 22.2 | |
| 64 | 133.6 | 67.3 | 45.0 | |
| 0.3 | 4 | 18.7 | 12.4 | 8.0 |
| 8 | 25.7 | 15.9 | 11.0 | |
| 16 | 36.8 | 21.6 | 14.8 | |
| 64 | 78.1 | 40.4 | 25.1 | |
| 0.15 | 4 | 9.6 | 5.5 | 4.9 |
| 8 | 14.3 | 7.7 | 5.9 | |
| 16 | 20.2 | 10.9 | 6.8 | |
| 64 | 46.6 | 21.5 | 16.3 | |
The acquisition parameters were chosen with a short recycle delay to average quickly (approx. 2 s per scan) and reduce total scan time. With a necessary SNR > 20 for suitable signal integration, 8 scans (approx. 16 s measuring time) were sufficient for concentrated samples at a low flow rate. Increasing the flow rate effectively reduces the perceived SNR as there is incomplete pre-magnetization of the nuclei prior to pulsing them. For example, at 4 mL min−1 16 scans (ca. 32 s) are necessary for a SNR > 20.
Software interface
For convenient operation of the integrated lab plant, we designed a software interface based on a LabView program (for details, see the ESI†) that facilitates control over both the synthesis and analysis components of the lab plant design. The front end of the interface provides control over the LCS, allowing the user to specify flow rates and measuring mode (i.e. continuous measurement or stopped-flow measurement) and set subsequent flow cell purging (number of purge cycles and flow rate). The connection between LabView and the NMR spectrometer utilizes an application programmatic interface (API) that enables the control of the observed nuclei (1H or 19F), the lock nuclei (deutero- or proteo-), number of scans, recycle delay, spectral centre and spectral width. The obtained spectral data are automatically transferred from the spectrometer to LabView, where they are displayed on the interface front end to allow automated data processing and/or quick interpretation of the results.Chemistry and online NMR analysis
Three reactions were chosen to explore the versatility and range of our unified synthesis and analysis platform: (1) Krapcho decarboxylation, (2) Ruppert–Prakash reaction, and (3) dye-sensitized visible light C–H arylation.The Krapcho decarboxylation was chosen to explore the temperature and pressure range manageable within the lab plant (Scheme 1).53 This reaction proceeds stepwise via a) mid-temperature ester cleavage forming gaseous chloroethane and the corresponding lithium carboxylate and b) high-temperature decarboxylation followed by protonation to afford the desired product, 2. Both reaction steps require a polar solvent mixture (DMSO and water) to solubilize the LiCl and stabilize the anionic intermediates. These conditions are challenging for the lab plant, as they require managing temperature gradients, pressure irregularities and difficult gas–liquid separation of chloroethane and CO2 from the reaction solution for a bubble-free, homogenous sample feed to the NMR flow cell.
 | ||
| Scheme 1 Krapcho decarboxylation for the formation of CF2H groups. | ||
The pyridine derivative 1 was dissolved in a DMSO–water mixture (97/3 v/v; 0.4 M) and pumped into the reactor at a flow rate of fl = 1 mL min−1. The temperature of the THTMD was set to 70 °C, and the nitrogen gas flow rate of the gas–liquid splitter was set to fg = 2 L min−1. Under these mild conditions, the first 19F NMR spectrum acquired had only one singlet resonance (δ = −108.3 ppm) indicative of the CF2 group in the starting material (red colour code; spectrum A in Fig. 4). With a reduced flow rate of fl = 0.5 mL min−1 and an increased reactor temperature of 116 °C, a second singlet resonance appeared at δ = −107.5 ppm. This was attributed to the mid-temperature lithium carboxylate intermediate (violet colour code; spectrum B in Fig. 4). With an increasing reaction temperature from 150 °C to finally 193 °C, the doublet expected for the product can be clearly observed δ = −123.9 ppm with a coupling constant of 2JH–F = 53 Hz (green colour code, spectra C–F in Fig. 4).54
 | ||
| Fig. 4 19F NMR spectra (A–F) of the reaction solution from the lab plant (fl = 0.5 mL min−1, 32 scans). Signal colour code: starting material in red, intermediate compound in violet, and product in green. | ||
Unfortunately, full conversion to the desired product was not observed under these reaction conditions since both starting material and intermediate compound were not completely consumed.
In summary, a total volume of approx. 80 mL was processed under various conditions. The crude product was collected and then subjected to an aqueous workup followed by purification via flash chromatography. The 1H NMR spectrum of purified 2 shows the anticipated triplet signal for the CF2H proton at δ = 7.17 ppm with a coupling constant of 2JH–F = 53 Hz to the CF2 group (Fig. 5). A high-resolution 1H NMR spectrum of purified 2 can be found in the ESI.†
 | ||
| Fig. 5 1H NMR spectrum of purified 2 in DMSO-d6 (offline measurement in a standard NMR tube; residual signals are observed for water at δ = 3.3 ppm and DMSO at δ = 2.50 ppm).55 | ||
The Ruppert–Prakash perfluoroalkylation was chosen to explore the optimization of reaction conditions using a series of reagent derivatives.56–58 This reaction transfers a perfluoroalkyl group to a carbonyl-containing substrate (e.g. aldehyde) via a TMS-protected perfluoroalkyl reagent. Typically a nucleophilic fluoride initiator, such as a quaternary ammonium fluoride salt or CsF, is added under water-free conditions to facilitate the reaction. In the case of continuous-flow synthesis immobilized fluoride sources can be used. There are commercially available spherical polymer-bound ammonium salt beads (e.g. polystyrene-based PS-NEt3F).59 For the first approach we filled approx. 400 mg of these spherical beads (F− loading: 3 mmol g−1) into the small cartridge located inline immediately before the reactor. A solution of benzaldehyde in THF (0.17 M) was mixed with 1.2 eq. of TMS-CF3 (0.2 M) and pumped through the fixed catalyst bed contained inside the small cartridge into the reactor. The resultant reaction mixture was then directly passed from the reactor into the NMR flow cell and analysed via online 19F NMR. Although the expected doublet of the addition product was observed (δ = −78.5 ppm, 3JH–F = 6.1 Hz), it disappeared only a few minutes after the first online measurement was acquired, while the signal for the starting material appeared again as a singlet signal at δ = −66.7 ppm. This result suggested rapid consumption and complete leaching of the fluoride source from the cartridge. In order to circumvent this problem we changed our synthetic strategy to a catalyst-free protocol. Iwanami and Oriyama suggest that performing the reaction in DMSO in the presence of molecular sieves as a dehydrating agent (mesh 8–10, 3 Å pore) will allow the solvent to activate TMS-CF3 (Scheme 2).60 We therefore charged the small cartridge with 0.57 g and the large cartridge (being installed after the reactor) with 5.43 g of molecular sieves.
 | ||
| Scheme 2 Catalyst-free trifluoromethylation of benzaldehyde as a model reaction with DMSO as an activating solvent. | ||
The optimization steps of the experiment are depicted in the stacked 19F NMR spectra in Fig. 6. The first two spectra represent a fresh (A) and aged solution (B) of TMS-CF3 in DMSO measured in a standard NMR tube. In the case of the fresh solution, the significant signal of the CF3 group can be observed at δ = −66.7 ppm. Two smaller signals are also visible at δ = −79.1 and −80.2 ppm, and these increase in intensity if the sample is stored for one day at room temperature. These signals were attributed to CHF3, formed through the hydrolysis of the silyl reagent (i.e. Me3SiOH).61 This side reaction can happen when the DMSO-activated reagent encounters traces of water. The observed doublet resonance results from the coupling of the fluorine atoms with the hydrogen atom with a coupling constant of 2JH–F = 79.5 Hz.61 The corresponding coupling constant is also observed in 1H NMR spectra for the proton quartet signal at δ = 7.3 ppm (for details, see the ESI†). Regardless, we prepared a fresh reaction mixture with the non-dried DMSO and benzaldehyde (0.17 M with 1.2 eq. TMS-CF3) and used it for a test run at a flow rate of fl = 0.5 mL min−1. Under these catalyst-free conditions the anticipated doublet, attributed to the product, appeared and persisted throughout the complete run time of 120 min. In spectrum C of Fig. 6 it can be observed that the starting material has been completely consumed (δ = −66.7 ppm), whereas the desired product doublet dominates the spectrum. It resonates at δ = −78.5 ppm with a coupling constant of 3JH–F = 6.1 Hz between the fluorine atoms and the α-CH proton of the trimethylsilyl-protected alcohol. We then increased the temperature from 22 °C to 55 °C and finally to 65 °C (spectra D and E in Fig. 6), showing an increasing product signal. This result indicates that the product formation is faster than the hydrolysis of TMS-CF3 at elevated temperatures. Correspondingly, this is evidenced by the decreased intensity of the CHF3 by-product.
 | ||
| Fig. 6 19F NMR spectra of pure TMS-CF3 in DMSO (spectra A and B; offline measurement) and of the reaction solution from the flow plant (fl = 0.5 mL min−1, 64 scans; spectra C–E). Signal colour code: starting material in red, side product (CHF3) in violet, and product in green. | ||
In summary, a total volume of approx. 60 mL was processed under various conditions as described above. The collected volume containing the crude product was then subjected to an aqueous workup and purified with flash chromatography for standard NMR analysis. The 1H NMR spectrum shows the desired quartet signal for the α-CH proton of the trimethylsilyl-protected alcohol at δ = 5.37 ppm with a coupling constant of 3JH–F = 6.8 Hz to the CF3 group (Fig. 7). A high-resolution 1H NMR spectrum of purified 4 can be found in the ESI.†
 | ||
| Fig. 7 1H NMR spectrum of purified 4 in DMSO-d6 (offline measurement in a standard NMR tube; residual signals are observed for water at δ = 3.3 ppm, DMSO at δ = 2.50 ppm and cyclohexane at δ = 1.37 ppm).55,62 | ||
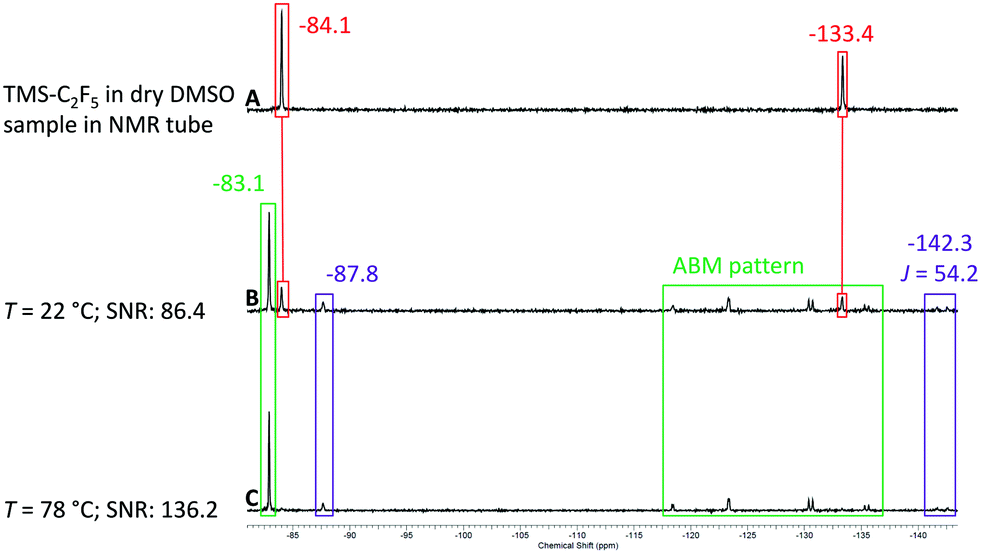
After the successful test of the Ruppert–Prakash reaction with TMS-CF3, we decided to perform the same reaction with TMS-C2F5 (Scheme 3). The pentafluoroethylation of benzaldehyde allows us to evaluate the sensitivity and resolution of the benchtop NMR spectrometer with higher order coupling. In the case of pentafluoroethylation there is an expected ABMX3 pattern resulting from strong coupling between the CF2 group, α-CH proton and the CF3 group. Similarly to the previous conditions with TMS-CF3 we prepared a solution of TMS-C2F5 in non-dried DMSO and analysed the hydrolysis rate of the reagent activated by DMSO. In contrast to its shorter chain CF3-homologue, a significant amount of TMS-C2F5 was already converted to C2HF5 after one hour (violet colour code in spectrum A in Fig. 8). The CF3 group of the starting material shows a singlet signal at δ = −84.1 ppm, and the CF2 shows a singlet signal at δ = −133.4 ppm. The resonances of hydrolyzed pentafluoroethane are located at δ = −87.8 ppm as a singlet signal for the CF3 group and at δ = −142.3 ppm as a doublet signal with a coupling constant of 2JH–F = 51 Hz for the CF2 group of C2HF5.61 As with the methyl derivative, the corresponding coupling constant is also observed in the 1H NMR spectrum as a triplet centred at δ = 6.99 ppm (for details, see the ESI†). Employing the same procedure as that for the methyl derivative, we prepared a solution of benzaldehyde in DMSO (0.17 M) with 1.2 eq. of TMS-C2F5 (0.2 M) and pumped it with a flow rate of fl = 0.5 mL min−1 through the cartridge–reactor–cartridge cascade filled with fresh molecular sieves inside the cartridges.
 | ||
| Scheme 3 Catalyst-free pentafluoroethylation of benzaldehyde with DMSO as activating solvent. | ||
 | ||
| Fig. 8 19F NMR spectra of pure TMS-C2F5 in DMSO (spectrum A; offline measurement) and of the reaction solution from the flow plant (fl = 0.5 mL min−1, 64 scans; spectra B–D). Signal colour code: starting material in red, by-product (HC2F5) in violet, product in green. | ||
To our delight, we observed all relevant signals with sufficient resolution in the 19F NMR spectrum of the reaction mixture. Starting with spectrum B in Fig. 8 the signals of the product (green colour code) can be observed at δ = −83.1 ppm for the CF3 group and as the complex ABMX3 pattern for the CF2 group between −118 and −135 ppm. With increasing temperature from 22 °C to 78 °C the intensity of the product signal increased, and similarly to the trifluoromethyl derivative, the intensity for the C2HF5 by-product decreased, showing that the addition reaction is faster at elevated temperatures (spectra C and D in Fig. 8).
In the upper part of Fig. 9 the ABM pattern of 5 is shown as a magnified section from spectrum D depicted in Fig. 8. The resolution of the pattern clearly proves the capability of the benchtop NMR spectrometer to analyse more complex structures under flow conditions still with a SNR of approx. 15. For the AB part of the pattern a coupling constant of JAB = 271 Hz can be observed. The smaller coupling constants of the AM and the BM parts are JAM = 6 Hz and JBM = 19 Hz, respectively. In the bottom part of Fig. 9 the 1H NMR spectrum of purified 5 is depicted, showing the corresponding coupling constants of JMA = 6 Hz and JMB = 19 Hz. A high-resolution 1H NMR spectrum of purified 5 can be found in the ESI.†
 | ||
| Fig. 9 (Top) Magnified ABM pattern of 5 from spectrum D in Fig. 8 (online analysis). (Bottom) 1H NMR spectrum of purified 5 in DMSO-d6 (offline measurement in a standard NMR tube; residual signals are observed for water at δ = 3.32 ppm, DMSO at δ = 2.50 ppm and cyclohexane at δ = 1.39 ppm).55,63 | ||
The previous examples confirmed that the effect of the dehydrating agent was not sufficient when the molecular sieves were integrated solely in the cartridges of the plant. In order to circumvent a gradual hydrolysis of the DMSO-activated reagent already in the substrate vessel we prepared a fresh solution of benzaldehyde in DMSO (0.34 M) and a fresh solution of TMS-C2F5 in dry DMSO (0.4 M). Both solutions were pumped using separate HPLC pumps at equal flow rates into a static T-mixer (PEEK T-piece) and afterwards through the cartridge–reactor–cartridge cascade filled with fresh molecular sieves inside the cartridges. This setup allows the continuous preparation of a substrate solution with a 0.2 M concentration of the fluorinated reagent. In spectrum A of Fig. 10 only unreacted TMS-C2F5 is detected; the hydrolysis by-product was not observed under these conditions. We then started the experimental run with the lab plant at a temperature of T = 22 °C and a flow rate of 0.5 mL min−1 (0.25 mL min−1 for each HPLC pump). In spectrum B of Fig. 10 the higher order spin system of the desired alkylation product could be observed. In addition, the starting material was also still available with a negligible amount of HC2F5 as the hydrolysis by-product. By increasing the temperature to 78 °C we were able to convert all TMS-C2F5 to the alkylated product (spectrum C in Fig. 10). Only trivial amounts of HC2F5 were detected under these conditions.
 | ||
| Fig. 10 19F NMR spectra of pure TMS-C2F5 in dry DMSO (spectrum A; offline measurement) and of the reaction solution from the flow plant (fl = 0.5 mL min−1, 64 scans; spectra B and C). Signal colour code: starting material in red, by-product (HC2F5) in violet, and product in green. | ||
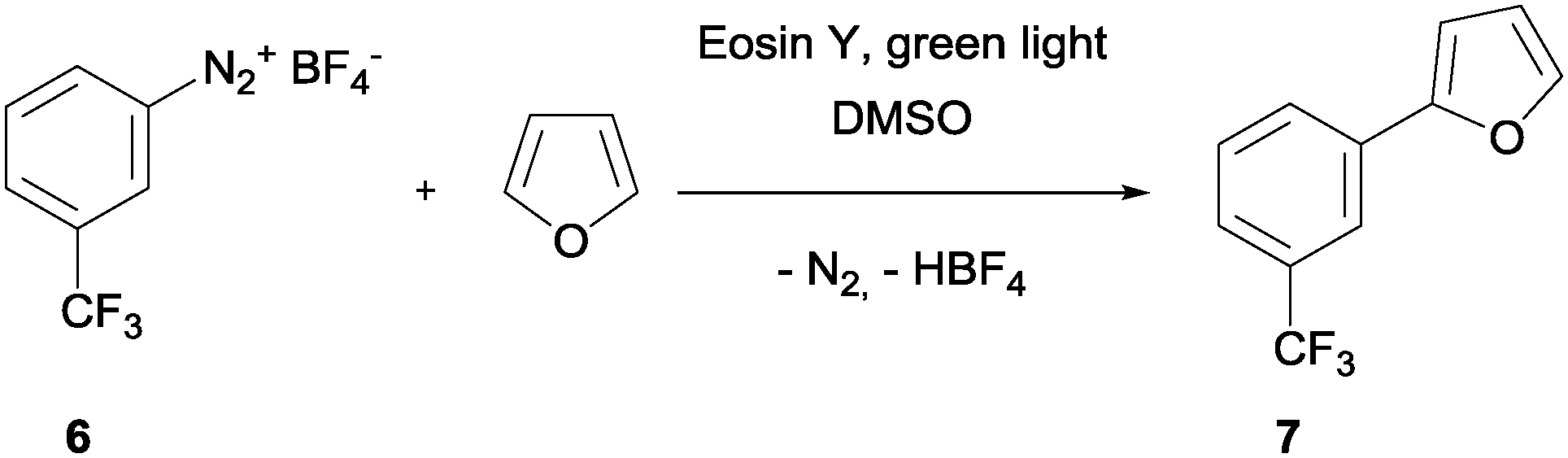
Finally, we used the capillary photoreactor to carry out the dye-sensitized C–H arylation of furan with a trifluoromethylated aryldiazonium salt (Scheme 4).64 Aryldiazonium salt 6 was dissolved in DMSO (0.4 M) whereas the sensitizer Eosin Y was dissolved in a DMSO–furan mixture (85.5/14.5 v/v; 1 mol%). The two starting material solutions were pumped separately with identical flow rates into a static micromixer (PEEK T-piece) that was installed in front of the capillary photoreactor. Under these conditions the final concentration of the diazonium salt remained 0.2 M. The reaction solution inside the capillary was illuminated with green light for a perfect match with the main absorption band of the sensitizer in the visible light region (λabs = 525 nm). Analogously to the Krapcho decarboxylation, we also used the gas–liquid splitter for separating the liquid reaction solution from the evolving nitrogen gas. For this reaction the gas flow rate was set to fg = 1 L min−1.
 | ||
| Scheme 4 Dye-sensitized arylation of furan with an aryldiazonium salt. | ||
We started with a combined flow rate of fl = 0.5 mL min−1 corresponding to a flow rate of 0.25 mL min−1 for each starting material solution. The progress of the reaction can be observed directly in the capillary of the reactor; the slightly pink reaction solution changes to yellow immediately upon green light illumination. After travelling approximately 10 centimetres through the capillary, the liquid phase is interrupted by gas slugs that result upon the expulsion of molecular nitrogen from the diazonium salt after reaction with the excited sensitizer. A picture of this process can be found in the ESI.† As depicted in Fig. 11, spectrum A, the starting material 6 appears at δ = −66.6 ppm, whereas spectrum B shows only conversion to the product singlet resonance at δ = −66.1 ppm, suggesting complete conversion under these conditions. As internal reference we used the chemical shift of the tetrafluoroborate counterion of the diazonium salt with a signal at δ = −152.6 ppm. With complete conversion at fl = 0.5 mL min−1, we looked to increasing the combined flow rate to fl = 0.75 mL min−1 and finally to fl = 1.0 mL min−1. Spectra C and D in Fig. 11 clearly confirm that still no starting material is left at these flow rates. Unfortunately, it was not possible to increase the flow rate to higher values since the maximum pressure of the capillary photoreactor was reached at p = 20 bar supplied by the nitrogen gas evolving during the reaction. At the highest flow rate we then switched off the light source implemented inside the capillary reactor (spectrum E in Fig. 11). Without the light source, there was negligible conversion to the desired product. However, turning the green light on immediately initiates the reaction (spectrum F in Fig. 11). The small residual signals for the starting material and product result in preventing the last two spectra E and F from back-mixing due to too short exchange times of the reaction solution in the gas–liquid splitter.
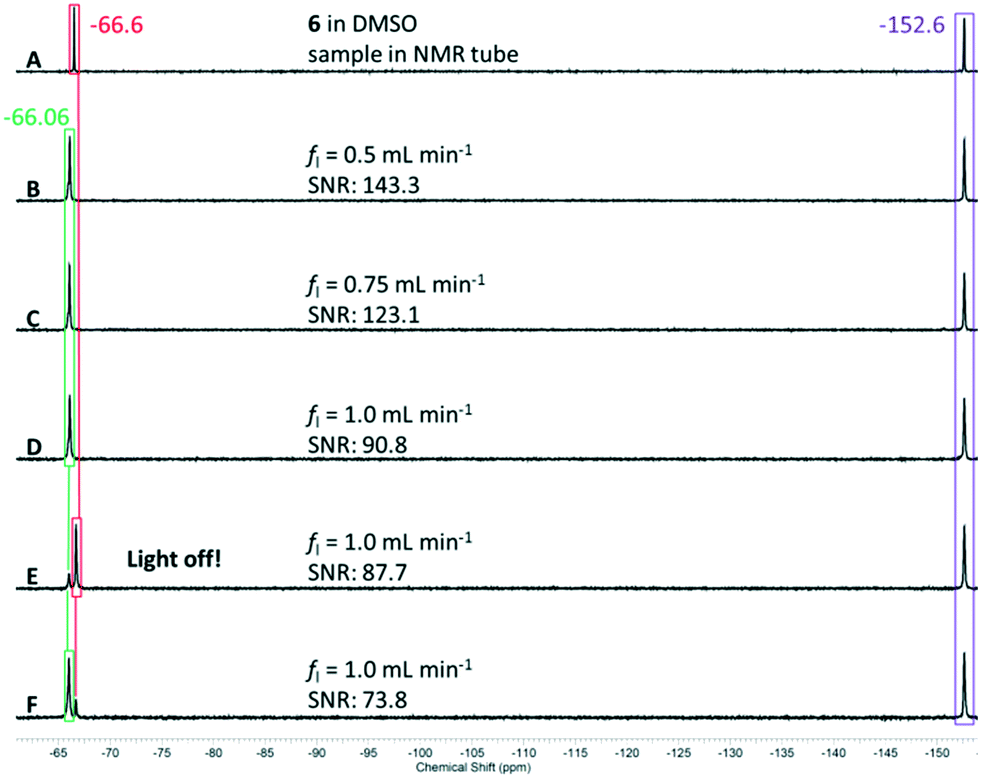 | ||
| Fig. 11 19F NMR spectra (A–F) of the reaction solution from the lab plant (T = 22 °C, 64 scans). Signal colour code: starting material in red, reference in violet, and product in green. | ||
In conclusion, a total volume of approx. 90 mL was processed for this reaction. The collected crude product was purified by flash chromatography and analysed with 1H NMR spectroscopy, showing the relevant signals for the desired compound (Fig. 12).64 A high-resolution 1H NMR spectrum of purified 7 can be found in the ESI.†
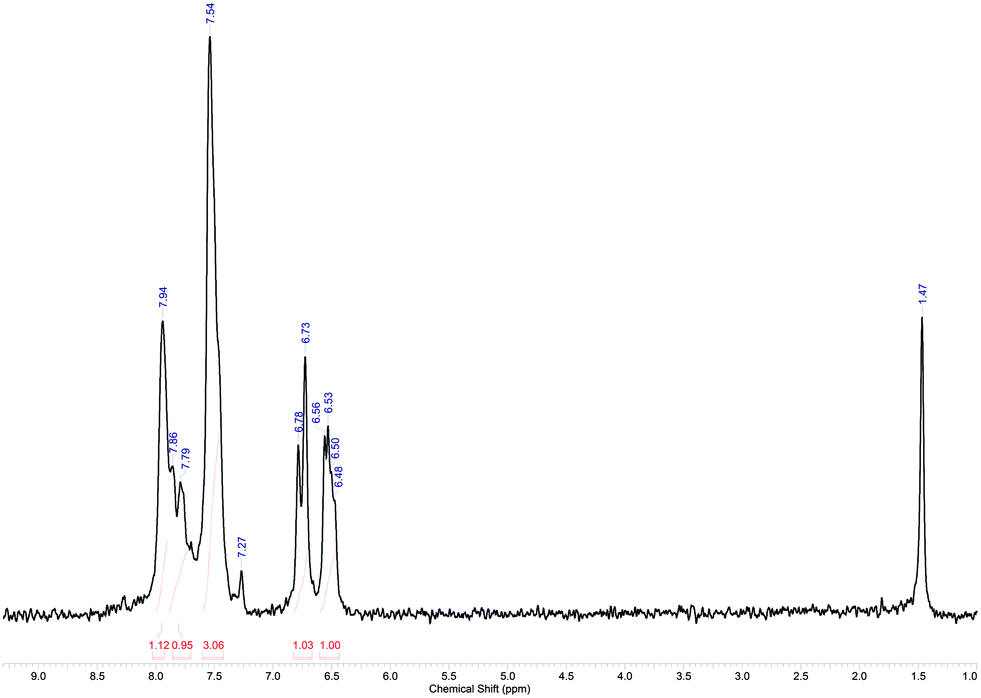 | ||
| Fig. 12 1H NMR spectrum of purified 7 in CDCl3 (offline measurement in a standard NMR tube; residual signals are observed for CHCl3 at δ = 7.27 ppm and water at δ = 1.47 ppm).55,65 | ||
Conclusions
This work presents the successful application of a unified synthesis and analysis platform for fluorine-containing compounds. A multi-purpose synthesis lab plant was designed for a broad application range of reaction classes with different reaction conditions and challenges. We illustrated the preparation of CF2H, CF3 functional groups and a carbon–carbon bond forming reaction in flow via a high-temperature Krapcho decarboxylation, the Ruppert–Prakash and a photochemically catalyzed C–H arylation reaction with a trifluoromethylated aryldiazonium salt, respectively. In all cases, fluorine was used as an NMR probe to monitor the reaction in continuous-flow. In the case of the pentafluoroethylation of benzaldehyde our online-NMR platform proved its efficacy even in analysing complex, higher-order NMR spectra. The benchtop NMR spectrometer was demonstrated to be a suitable partner for the analysis of continuously produced fine fluorinated chemical compounds. The combination of a powerful flow technology with sensitive and quantitative NMR analysis hardware opens the door for many future applications in continuous-flow process development.Acknowledgements
The work leading to these results was funded in large part by Zentrales Innovationsprogramm Mittelstand (ZIM, BMWI) and the German-Canadian Centre for Innovation and Research (GCCIR).Notes and references
- D. Webb and T. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- J. Némethné-Sóvágó and M. Benke, Mater. Sci. Eng., A, 2014, 39, 89–101 Search PubMed.
- T. H. Rehm, Chem. Eng. Technol., 2015, 16, 66–80 Search PubMed.
- C. Mallia and I. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS.
- T. Wirth, Microreactors in organic synthesis and catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- A. Kirschning, W. Solodenko and K. Mennecke, Chem. – Eur. J., 2006, 12, 5972–5990 CrossRef CAS PubMed.
- N. Kockmann, M. Gottsponer, B. Zimmermann and D. Roberge, Chem. – Eur. J., 2008, 14, 7470–7477 CrossRef CAS PubMed.
- N. Kockmann and D. Roberge, Chem. Eng. Technol., 2009, 32, 1682–1694 CrossRef CAS.
- D. Roberge, B. Zimmermann, F. Rainone, M. Gottsponer, M. Eyholzer and N. Kockmann, Org. Process Res. Dev., 2008, 12, 905–910 CrossRef CAS.
- T. H. Rehm, S. Gros, P. Löb and A. Renken, React. Chem. Eng., 2016, 1, 636–648 CAS.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- M. Baumann and I. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- V. Hessel, D. Kralisch and N. Kockmann, Novel Process Windows: Innovative Gates to Intensified and Sustainable Chemical Processes, Wiley-VCH, Weinheim, 2015 Search PubMed.
- W. Ehrfeld, V. Hessel and H. Löwe, Microreactors, Wiley-VCH, Weinheim, 2000 Search PubMed.
- V. Hessel, A. Renken, J. Schouten and J.-I. Yoshida, Micro Process Engineering, Wiley-VCH, Weinheim, 2009 Search PubMed.
- N. Kockmann, M. Gottsponer and D. Roberge, Chem. Eng. J., 2011, 167, 718–726 CrossRef CAS.
- X. Zhang, S. Stefanick and F. Villani, Org. Process Res. Dev., 2004, 8, 455–460 CrossRef CAS.
- D. Browne, S. Wright, B. Deadman, S. Dunnage, I. Baxendale, R. Turner and S. Ley, Rapid Commun. Mass Spectrom., 2012, 26, 1999–2010 CrossRef CAS PubMed.
- D. Fabry, E. Sugiono and M. Rueping, React. Chem. Eng., 2016, 1, 129–133 CAS.
- D. Fabry, E. Sugiono and M. Rueping, Isr. J. Chem., 2014, 54, 341–350 CrossRef CAS.
- C. Carter, H. Lange, S. Ley, I. Baxendale, B. Wittcamp, J. Goode and N. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS.
- A. O’Brien, O. Luca, P. Baran and D. Blackmond, React. Chem. Eng., 2016, 1, 90–95 Search PubMed.
- N. Holmes, G. Akien, R. Savage, C. Stanetty, I. Baxendale, J. Blacker, B. Taylor, R. Woodward, R. Meadows and R. Bourne, React. Chem. Eng., 2016, 1, 96–100 CAS.
- A. Perro, G. Lebourdon, S. Henry, S. Lecomte, L. Servant and S. Marre, React. Chem. Eng., 2016, 1, 577–594 CAS.
- F. Benito-Lopez, M. Verboom, M. Kakuta, J. Gerdeniers, R. Egberink, E. Oosterbroek, A. van der Berg and D. Reinhoudt, Chem. Commun., 2005, 2857–2859 RSC.
- L. Colnago, F. Andrade, A. Souza, R. Azeredo, A. Lima, L. Cerioni, T. Osán and D. Pusiol, Chem. Eng. Technol., 2014, 37, 191–203 CrossRef CAS.
- M. Maiwald, H. Fischer, Y.-K. Kim, K. Albert and H. Hasse, J. Magn. Reson., 2004, 166, 135–146 CrossRef CAS PubMed.
- M. Bernstein, M. Števinović and C. Sleigh, Magn. Reson. Chem., 2007, 45, 564–571 CrossRef CAS PubMed.
- T. Eifert and M. Liauw, React. Chem. Eng., 2016, 1, 521–532 Search PubMed.
- D. Folay, E. Bez, A. Codina, K. Colson, M. Fey, R. Krull, D. Piroli, M. Zell and B. Marquez, Anal. Chem., 2014, 86, 12008–12013 CrossRef PubMed.
- M. Elipe, Nucl. Magn. Reson., 2016, 45, 190–216 Search PubMed.
- M. Elipe and R. Milburn, Magn. Reson. Chem., 2016, 54, 437–443 CrossRef PubMed.
- M. Goldbach, E. Danieli, J. Perlo, B. Kaptein, V. Litvinov, B. Blümich, F. Casanova and A. Duchteau, Tetrahedron Lett., 2016, 57, 122–125 CrossRef CAS.
- B. Blümich, TrAC, Trends Anal. Chem., 2016, 83A, 2–11 CrossRef.
- K. Singh and B. Blümich, TrAC, Trends Anal. Chem., 2016, 83A, 12–26 CrossRef.
- K. Meyer, S. Kern, N. Zientek, G. Guthausen and M. Maiwald, TrAC, Trends Anal. Chem., 2016, 83A, 39–52 CrossRef.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS PubMed.
- N. Zientek, C. Laurain, K. Meyer, M. Kraume, G. Guthausen and M. Maiwald, J. Magn. Reson., 2014, 249, 53–62 CrossRef CAS PubMed.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2015, 6, 1258–1264 RSC.
- B. Ahmed-Omer, E. Sliwinski, J. Cerruti and S. Ley, Org. Process Res. Dev., 2016, 20, 1603–1614 CrossRef CAS.
- H. Günther, NMR Spectroscopy - Basic Principles, Concepts and Applications in Chemistry, Wiley-VCH, Weinheim, 2013 Search PubMed.
- NMR Analysis in pharmaceutical analysis, ed. U. Holzgrabe, I. Wawer and B. Diehl, Elsevier, Oxford, 2008 Search PubMed.
- J. Wang, M. Sánchez-Roselló, J. Aceña, C. del Pozo, A. Sorochinsky, S. Fustero, V. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- T. Fujiwara and D. O’Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- P. Jeschke, ChemBioChem, 2004, 4, 570–589 CrossRef PubMed.
- U. Krtschil, V. Hessel, H.-J. Kost and D. Reinhard, Chem. Eng. Technol., 2013, 36, 1010–1016 CrossRef CAS.
- B. Deadman, C. Battilocchio, E. Sliwinski and S. Ley, Green Chem., 2013, 15, 2050–2055 RSC.
- B. Hook, W. Dohle, P. Hirst, M. Pickworth, M. Berry and K. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- J. Knowles, L. Elliott and K. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- E. Blackham, J. Knowles, J. Burgess and K. Booker-Milburn, Chem. Sci., 2016, 7, 2302–2307 RSC.
- K. Maskill, J. Knowles, L. Elliott, R. Alder and K. Booker-Milburn, Angew. Chem., Int. Ed., 2013, 52, 1499–1502 CrossRef CAS PubMed.
- Signal to noise ratios were determined with the ACD/NMR Processor software (academic edition) from ACD/Labs.
- P. Krapcho, J. Weimaster, J. Eldridge, E. Jahngen, A. Lovey and W. Stephens, J. Org. Chem., 1978, 43, 138–147 CrossRef.
- S. Prakash, S. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. Swabeck and G. Olah, Angew. Chem., 2012, 124, 12256–12260 CrossRef.
- H. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- I. Ruppert, K. Schlich and W. Volbach, Tetrahedron Lett., 1984, 25, 2195–2198 CrossRef CAS.
- G. Prakash, R. Krishnamurti and G. Olah, J. Am. Chem. Soc., 1989, 111, 393–395 CrossRef CAS.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed.
- M. Baumann, I. Baxendale, L. Martin and S. Ley, Tetrahedron, 2009, 65, 6611–6625 CrossRef CAS.
- K. Iwanami and T. Oriyama, Synlett, 2006, 1, 112–114 Search PubMed.
- A. Foris, Magn. Reson. Chem., 2004, 42, 534–555 CrossRef CAS PubMed.
- J. Klein, S. Rommel and B. Plietker, Organometallics, 2014, 33, 5802–5810 CrossRef CAS.
- S. Prakash, Y. Wang, R. Mogi, J. Hu, T. Mathew and G. Olah, Org. Lett., 2010, 12, 2932–2935 CrossRef PubMed.
- D. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed.
- S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Lab plant flow chart and details of the microreactors, 19F NMR spectra of flow cell evaluation and product solutions, experimental details. See DOI: 10.1039/c7re00023e |
| This journal is © The Royal Society of Chemistry 2017 |