
Experimental and numerical insights into the formation of zirconia nanoparticles: a population balance model for the nonaqueous synthesis†
Pierre
Stolzenburg
and
Georg
Garnweitner
 *
*
Institute for Particle Technology and Laboratory for Emerging Nanometrology, Technische Universität Braunschweig, Volkmaroder Str. 5, 38104 Braunschweig, Germany. E-mail: g.garnweitner@tu-bs.de
First published on 10th February 2017
Abstract
The nonaqueous synthesis of zirconia nanoparticles was investigated to elucidate the complex interplay between process parameters and final nanoparticle properties. We determined the chemical reaction kinetics and derived a model equation for the reaction constant to describe the course of the synthesis as a function of temperature and precursor concentration. While investigating the nucleation kinetics, we were able to show via small-angle X-ray scattering that particles nucleate in roundish shapes in a size regime of 2 nm. The current understanding of the tetragonal-to-monoclinic phase transformation was extended by a novel model that relates the probability of particles undergoing the phase transition to the particle size. The derived model functions result in a comprehensive population balance equation framework that can simulate the entire particle formation process to predict final nanoparticle properties as well as their evolvement during the synthesis. Supported by the simulation results, we unravel why monoclinic particles are present in the final product in smaller sizes than tetragonal particles which at first seems to be in conflict with thermodynamics.
1. Introduction
Over the last two decades, the nonaqueous sol–gel-method has become a prosperous synthesis route for metal oxide nanoparticles.1–5 A variety of nonaqueous pathways have been proposed that lead to binary, ternary and doped nanoparticles such as TiO2,6 Fe3O4,7 Nb2O5,8 WO3,9 ZnO,10 AZO,11 BaTiO3,12 or Ga2O3:Eu3+.13 The advantage over aqueous sol–gel routes is the good control over the particle formation process through the synthesis parameters14 which can be exploited to precisely adjust nanoparticle properties, leading to highly crystalline and well-defined nanoparticles. Although the fundamental mechanisms of particle formation were investigated with great effort during recent years,15–18 to the best of our knowledge, no simulation approaches of the nonaqueous synthesis have been presented so far. In this study, we focus on the synthesis of zirconia nanoparticles and model the underlying mechanisms of particle formation, which leads to a comprehensive process model based on population balance equations (PBE) that can simulate the entire course of the synthesis and predict final particle properties.The method of population balance equations is widely used to simulate crystallization and precipitation processes for particles from the micron19 to the submicron and nanometer20,21 size range. Heine et al. simulated the flame spray synthesis of zirconia nanoparticles via PBE22 whereas Perala et al. used PBE to model nucleation during a synthesis of transition-metal nanoparticles.23 Segets et al. simulated the formation of ZnO quantum dots, including oriented growth and ripening mechanisms, during a synthesis in ethanol.24 A further PBE approach to simulate the disintegration of surface stabilized sol–gel-derived titania nanoparticles was published by Gokhale,25 but did not comprise the particle formation during synthesis.
In order to set up a population balance framework for the nonaqueous synthesis, it is essential to investigate the kinetics of the chemical reaction and particle formation, which we obtained by performing a thorough process study. In our previous work,26 we reported the formation of fractal zirconia nanoparticles and investigated the progress of the synthesis over time, as illustrated in Fig. 1 by the decrease of the soluble zirconium concentration [Zr]i. The zirconium concentration in solution was derived by separating the solid and liquid (supernatant) phases of a sample by centrifugation at high g, subsequent heating of 5 mL supernatant up to 1000 °C and consecutive gravimetric analysis of the resulting ZrO2. Particle formation starts after a certain induction time and is governed by the preceding chemical reactions which directly lead to the tetragonal polymorph of zirconia. Over the course of the synthesis, the primary particles grow to larger sizes and exhibit a phase transformation to the monoclinic form which is accompanied by a characteristic change in morphology from quasi-spherical into dendritic shapes. We have shown that the growth can be altered via the process conditions by varying temperature and initial precursor concentrations and thereby used to adjust final nanoparticle properties. The kinetics of particle formation are in good accordance with the chemical reaction kinetics and follow a similar trend as previously observed in the nonaqueous synthesis of TiO2 nanoparticles.27,28 Thereby, we resolve in this study the complex interplay between process conditions and molecular kinetics as well as their influence on nucleation and the tetragonal-to-monoclinic phase transition to derive model equations that describe the course of the synthesis in the population balance framework.
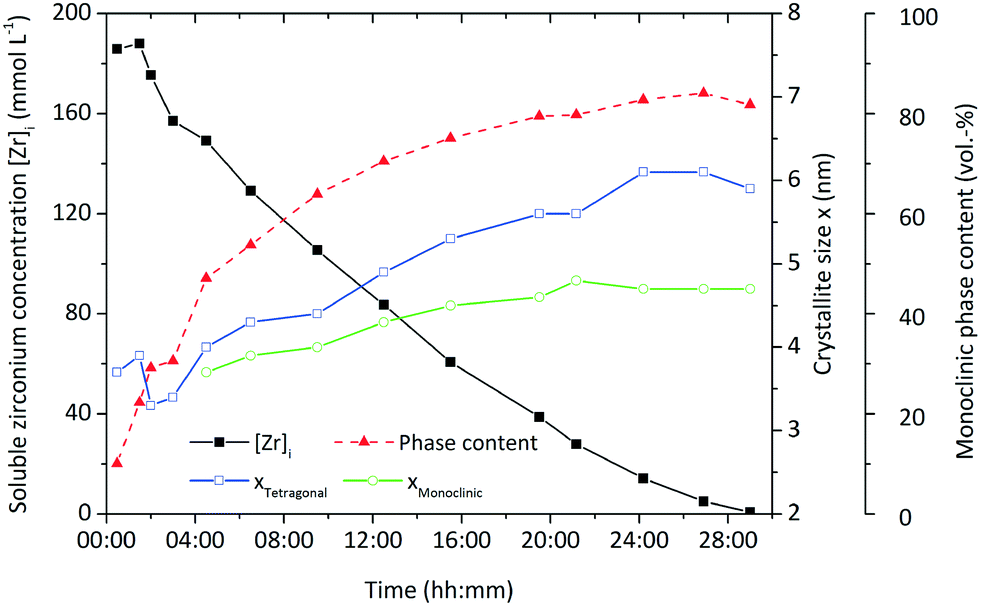 | ||
| Fig. 1 Crystallite growth, progress of the chemical reaction represented by the development of zirconium ion concentration [Zr]i and evolution of the monoclinic phase content to illustrate the tetragonal-to-monoclinic phase transformation. | ||
On a molecular level, the zirconia synthesis comprises several reaction steps starting with the precursor zirconium(IV) propoxide (Zr(OnPr)4) which first reacts with the solvent benzyl alcohol (BnOH) via a ligand exchange to an intermediate product.29–31 Further on, the intermediate product reacts in a subsequent ether condensation reaction to a monomer species that when exceeding a certain concentration nucleates to ZrO2 nanoparticles. Intermediate species triggering nucleation have been shown to exist during the aqueous synthesis of zirconia nanoparticles32,33 and were lately identified during the nonaqueous synthesis for titania nanoparticles.34 Hence, one can assume that intermediate species acting as nucleation monomers play a crucial role during this particular synthesis and are modeled in the simulation as the soluble zirconia monomer species which transforms into the solid phase.
2. Materials and methods
2.1 Experimental
The syntheses were performed under solvothermal conditions in a 1.5 L and a 250 mL batch reactor system as illustrated in Fig. 2. Temperature and pressure were constantly monitored, and the reaction mixtures were continuously stirred in both reactor systems. A typical reaction was started by pouring the precursor zirconium n-propoxide (70 wt-% in 1-propanol, Sigma Aldrich) and the solvent benzyl alcohol (97 wt-%, Merck) into the respective reactor system according to initial precursor concentrations [Zr]0 of 180, 240 and 360 mmol L−1. The systems were then heated to process temperature which was realized in a range of 220 °C to 270 °C. A standard synthesis comprised a reaction at 250 °C with a precursor concentration of 180 mmol L−1. | ||
| Fig. 2 a. 1.5 L reactor system; b. 250 mL reactor system with (laser) transmission measurement. | ||
We conducted larger scale syntheses in a 1.5 L reactor (poly-clave type 3/1, Büchi Glas Uster) (Fig. 2a) to follow the kinetics of particle growth and the chemical reaction by analyzing the solid and liquid phases over the course of the synthesis by taking samples of the reaction mixture. The system was heated through a heating circuit using an external thermostat (Huber Tango HT). After sampling, the liquid (supernatant) and solid phases of the reaction mixture were separated by centrifugation. We determined the kinetics by following the zirconium ion concentration in solution [Zr]i, which we obtained by heating 5 mL supernatant up to 1000 °C and consecutive gravimetric analysis of the resulting ZrO2, over the course of synthesis. The solid phase was analyzed after multiple washing steps with ethanol and vacuum drying with respect to the present crystallite size and phase composition by powder X-ray diffraction (XRD, Cu Kα radiation; Empyrean Cu LEF HR goniometer; Empyrean series 2, PANalytical, PIXcel-3D detector; Si wafer; 20–90°, step size 0.05°) and Rietveld refinement using the tetragonal (ICSD code: 98-006-6789) and monoclinic (ICSD code: 98-008-0045) reference patterns.
A special 250 mL scale reactor (Fig. 2b; additional information can be found in the ESI†) was designed to perform in situ light transmission measurements with a laser-detector system (wavelength 635 nm). Thereby, we studied the nucleation event by determining the onset of particle formation by performing turbidity measurements. Also, samples from the reaction mixture were withdrawn during the nucleation event and analyzed by small-angle X-ray scattering (SAXS) using an SAXSess mc2 system (Anton Paar) with Cu Kα radiation and a CCD detector. The samples were measured for 10 s, and the obtained scattering curves were desmeared and corrected for transmission and instrument background. Finally, transmission electron microscopy was performed on a JEOL JEM-2100F-UHR device to determine the size and shape of the formed nuclei and hence to confirm the validity of the SAXS and XRD results.
2.2 Modeling and simulation
Matlab (MathWorks, R2015a) was used to determine the model parameters by fitting the experimental data to the kinetic model equations. The particle formation and the tetragonal-to-monoclinic phase transition were simulated by solving the integrodifferential form of the population balance for the tetragonal and monoclinic phases with the software Parsival© (Wulkow CIT GmbH, version 7.6a)35 applying the Garlekin h–p-method. Afterwards, the post-processing of the simulation data was carried out in Matlab.3. Results and discussion
As a prerequisite for the PBE model, we first focused on the process itself and investigated the chemical reactions, nucleation and tetragonal-to-monoclinic phase transition over the course of the synthesis. Hence, we clarified and quantified the respective kinetics and determined the size of the formed nuclei. In the following we will present the experimental results which were used to derive the model equations for the simulation of the particle formation process.3.1 Chemical reaction kinetics
The overall reaction kinetics (eqn (1)) were investigated in detail by following the reaction through the soluble zirconium concentration [Zr]i which is defined as the sum concentration of the precursor as well as all intermediates prior to nanoparticle formation. For better comparison, the zirconium concentration was normalized by the initial precursor concentration to [Zr]i/[Zr]0. It can clearly be observed that after a certain induction time, (linear) pseudo-zero-order kinetics (Fig. 3) best describe the overall kinetics of the nonaqueous sol–gel synthesis of zirconia nanoparticles. Hence, we applied the differential method36 to determine the overall reaction order n. The negative derivative of the zirconium concentration was plotted versus the zirconium concentration in a log–log-graph to yield the reaction order from eqn (1), resulting in an empirical order value n of 0.3 (Fig. S. 1†) for a reaction at standard conditions. It has to be taken into account that the yield represents the overall reaction kinetics which, in addition to the ligand exchange and ether condensation, are also influenced by several sequential reactions, side reactions, and the nucleation event. Thus, the proposed reaction order value of 0.3 is the total of all reaction orders and might explain why the yield apparently follows zero-order kinetics (n = 0). | (1) |
 | ||
| Fig. 3 Normalized zirconium concentration over time for experiments investigating different initial precursor concentrations and the seeding effect at 250 °C. | ||
The integral form of the reaction rate equation for a zero-order reaction corresponds to
| [Zr]i = [Zr]0 − krt | (2) |
 | (3) |
Fig. 3 shows the development of the normalized zirconium concentration over time for runs performed at different initial concentration [Zr]0. We fitted the experimental data to eqn (3) and observed that the slope, corresponding to kr/[Zr]0, appears to be constant for all investigated runs, which is due to faster kinetics for higher [Zr]0 (also illustrated by the determined reaction rates kr in Table 1). This seems to be a direct conflict with the assumption of zero-order kinetics where the chemical reaction rate kr should be independent of the initial precursor concentration. The differential method (ESI Fig. S. 3†) reveals an increase in the reaction order n from 0.3 to 0.55 for higher initial concentrations [Zr]0. This indicates that the synthesis might follow first-order kinetics when higher concentrations are used. According to classical nucleation theory,37 the nucleation rate is a function of the zirconium concentration and thus might result in faster overall reaction kinetics when higher initial precursor concentrations are used. Another possible explanation for the zero-order kinetics might be a superimposed surface nucleation event38 that triggers particle formation and is induced by a seeding effect caused by the reactor walls or by impurities within the precursor solution. We have observed similar phenomena for the nonaqueous synthesis of titania nanoparticles where we have shown that residues from previous experiments (even after extensive cleaning procedures of the reactor) adhere to the reactor wall and cause a seeding effect which accelerates the particle formation.39 As apparently, the chemical reaction and nucleation kinetics are directly coupled to each other, we investigated the impact of heterogeneous nucleation kinetics by performing seeding experiments and added a freshly prepared ZrO2 product suspension, containing one mole percent of zirconia nanoparticles with respect to the initial precursor amount, synthesized under standard process conditions, to the original reaction mixture before heating. As a result, the zirconium concentration decreases more rapidly as shown in the steeper decrease of the corresponding curve in Fig. 3.
Inevitably, the reaction rate is determined by heterogeneous nucleation and by the impact of the initial zirconium concentration on the nucleation rate, which will be discussed in more detail in section 3.2. For syntheses carried out at lower temperatures (Fig. 4), the impact of the zirconium concentration becomes more significant in a lower concentration range for endured reaction times, which leads to an apparent change towards pseudo-first-order kinetics. Furthermore, the normalized zirconium concentration for all runs performed at 270 °C did not decrease to zero but remained constant at 0.05, which might be an effect of enhanced zirconium solubility or a stabilizing effect which led to nanoparticles that were not separated by centrifugation and thereby incorporated into the [Zr]i determined by the gravimetric analysis. The temperature impact on the reaction rates kr (Table 1) follows the law of Arrhenius (eqn (4)) and the activation energy EA was determined to 117.98 kJ mol−1 (ESI Fig. S. 4†), which is in the same range as for the nonaqueous TiO2 synthesis.27
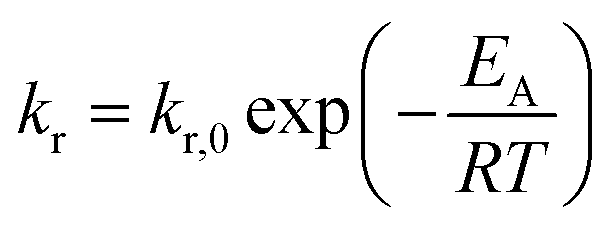 | (4) |
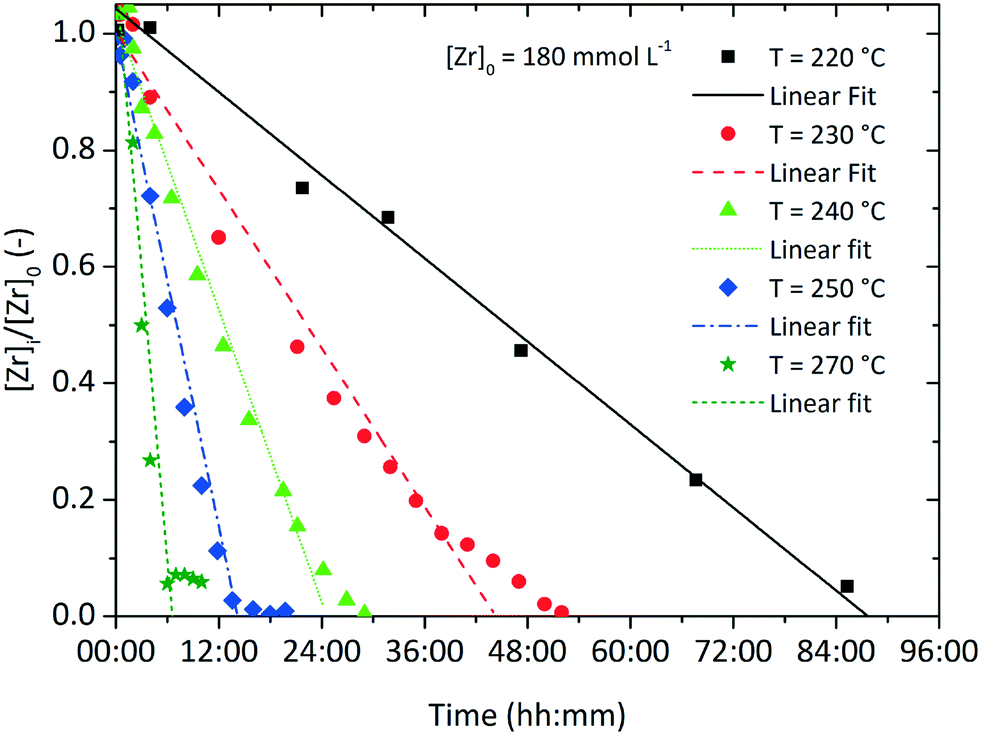 | ||
| Fig. 4 Normalized zirconium concentration over time for various process temperatures with an initial precursor concentration of [Zr]0 = 180 mmol L−1. | ||
The influence of the precursor concentration [Zr]0 on the reaction rate can be described by a hyperbolic function in the form of eqn (5). Hyperbolic functions are widely applied to describe the influence of the concentration of a reagent species on the reaction, e.g. in Michaelis–Menten kinetics to model the reaction rate in enzymatic reactions or the Langmuir isotherm expression for heterogeneous catalytic reactions. In combination with eqn (4), we derived a kinetic model (eqn (6)) for the reaction rate kr as a function of temperature and initial precursor concentration as the process conditions.
 | (5) |
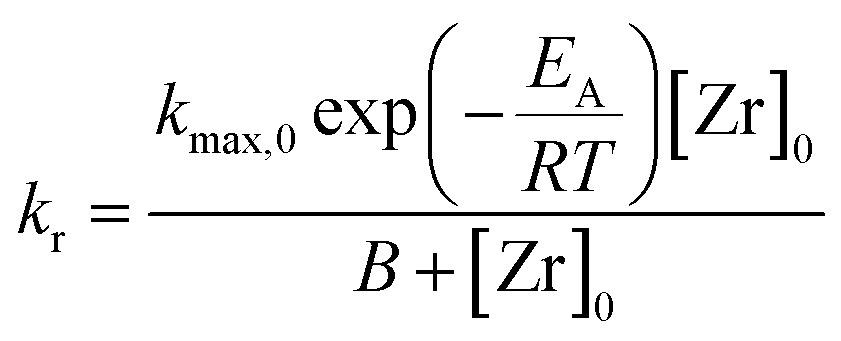 | (6) |
The data in Table 1 was used to determine the factors kmax,0 to 1.554 × 1014 mmol L−1 h−1 and B to 3771.15 mmol L−1. Here, kmax,0 is the maximum frequency (also denoted as preexponential factor) at the given precursor concentration and B is the theoretical initial precursor concentration at which kr would be half of kmax,0. Fig. 5 shows the reaction rate as a function of synthesis temperature and initial precursor concentration which was plotted from the model function (eqn (6)) and the corresponding experimental data. The graph illustrates a steep increase of the reaction constant towards reaction temperatures of 270 °C which becomes more pronounced at higher precursor concentrations. All in all, the derived model agrees well with the experimental data however deviations (seem to) become distinctive at 270 °C which might be due to the fast reaction kinetics. These fast kinetics lead to a characteristic change to smaller crystal sizes, very low monoclinic phase contents and different morphologies of the resulting nanoparticles.26Eqn (6) is used to model the influence of the process conditions on the overall reaction and more importantly, is used to connect the process conditions to the overall population balance model of particle formation.
 | ||
| Fig. 5 Reaction constant kr for the investigated process conditions determined from experimental data and the model function. | ||
The chemical reaction is assumed to yield an intermediate monomer species [ZrO2]m which is the precursor for the particle formation reaction and is believed to be in accordance with classical nucleation theory (CNT). The existence of such monomer species – which are distinct from the used precursor – has been shown for the aqueous synthesis of ZrO2 nanoparticles by Ambrosi et al.40 and Hu et al.33 and hence, we assume a similar monomer species in our model as the driving force of nucleation. Furthermore, we assume that no other zirconia species are formed and with respect to the simulation model, the rate at which monomers are formed can be described by eqn (7) with the reaction constant kr and order n.
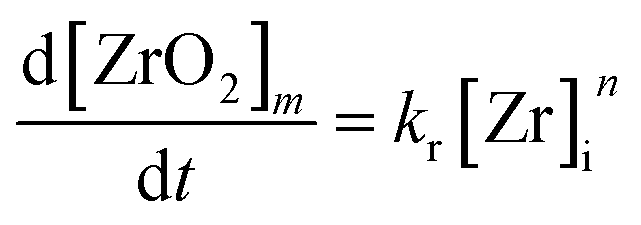 | (7) |
3.2 Nucleation
The observed induction time might be explained by a two-step formation mechanism as proposed by Watzky et al.,41 whereby the slow nucleation step is followed by the autocatalytic growth of the resulting particles.23 Although the slow nucleation step proposed in the Finke–Watzky model would explain the induction time observed in our experiments, the linear kinetics (i.e. a constant reaction rate) oppose this mechanism, as according to Watzky, continuously formed nuclei accelerate the reaction rate and lead to an exponential trend over the course of synthesis.To elucidate the nucleation phenomena in more detail, we performed in situ light transmission measurements during the synthesis in the 250 mL reactor system. The light transmission technique was realized by a pair of borosilicate glass windows situated on opposite sides of the self-designed reactor to align the laser source and photodiode detector along the optical axis. This allowed the light sensor to be continuously illuminated and fully saturated when the optical pathway within the reaction mixture was free of particles. The induction time was determined as the period between the time when the system reaches the reaction temperature and the time when the detector first records a decrease in intensity, which is caused by the scattering effect of particles passing the laser beam. For a synthesis temperature of 250 °C, an induction time of about 32 min was found. Subsequently, the reaction mixture increased in turbidity (Fig. 6), containing an increasing amount of solid material. As primary nanoparticles, especially at low concentrations, lead to much lower scattering intensities than agglomerates and therefore generate no detectable signal, small-angle X-ray scattering was applied to investigate the nucleation phenomena during the induction time.
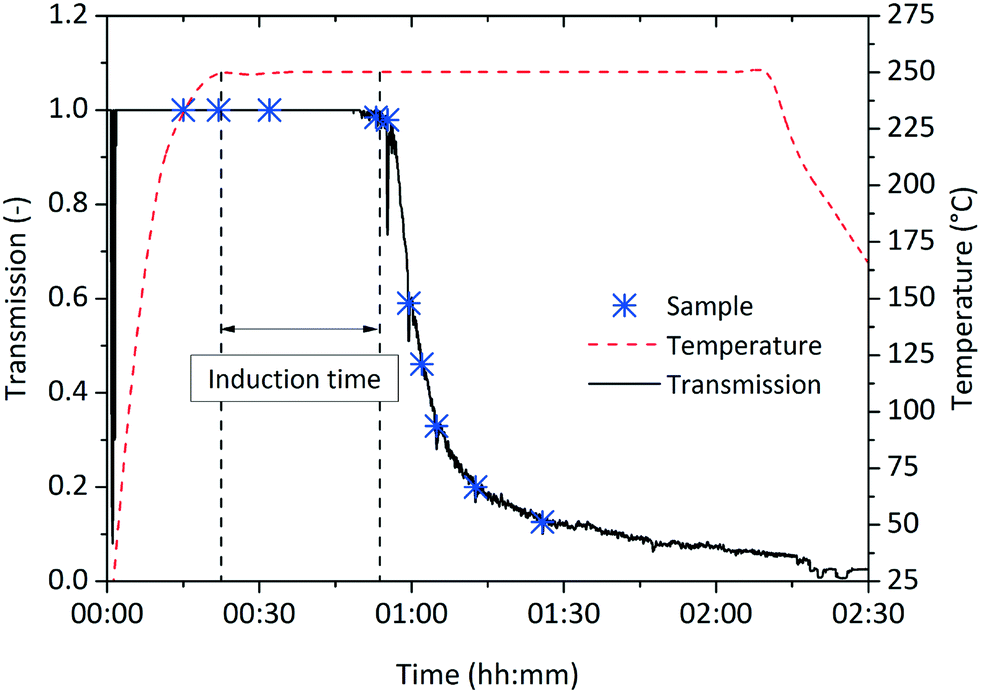 | ||
| Fig. 6 Induction time measurements during a synthesis at standard conditions in the 250 mL reactor with an induction time determined to 32 minutes. Samples were withdrawn at marked times. | ||
Samples of the reaction mixture were withdrawn during the synthesis and analyzed via SAXS. The scattering curves were desmeared and corrected for background. Samples withdrawn before and during the heating procedure showed no scattering signals (data not shown) whereas the scattering curves (Fig. 7a) of four samples directly withdrawn when reaching synthesis temperature (reaction time of 22 minutes), at the beginning (51 minutes), during (1 hour 4 minutes) and after the intensity decay (1 hour 25 minutes) showed scattering signals that can be related to newly formed particles. In general, the scattering intensity strongly increases over time due to higher particle concentrations as particles are constantly formed during the synthesis. Interestingly, the scattering curve of the sample taken after 1 hour 25 minutes shows a slight shoulder at q ≈ 1.2 nm−1 which can be interpreted as particle agglomeration. Gossard et al.42 have observed that similar signals appear at smaller q values for an aqueous synthesis of zirconia nanoparticles which is assigned to inter-particle distances in defined agglomerates. As we have found that evolving nuclei during a nonaqueous TiO2 synthesis6 instantly agglomerate, we assume a similar behavior during the zirconia synthesis.
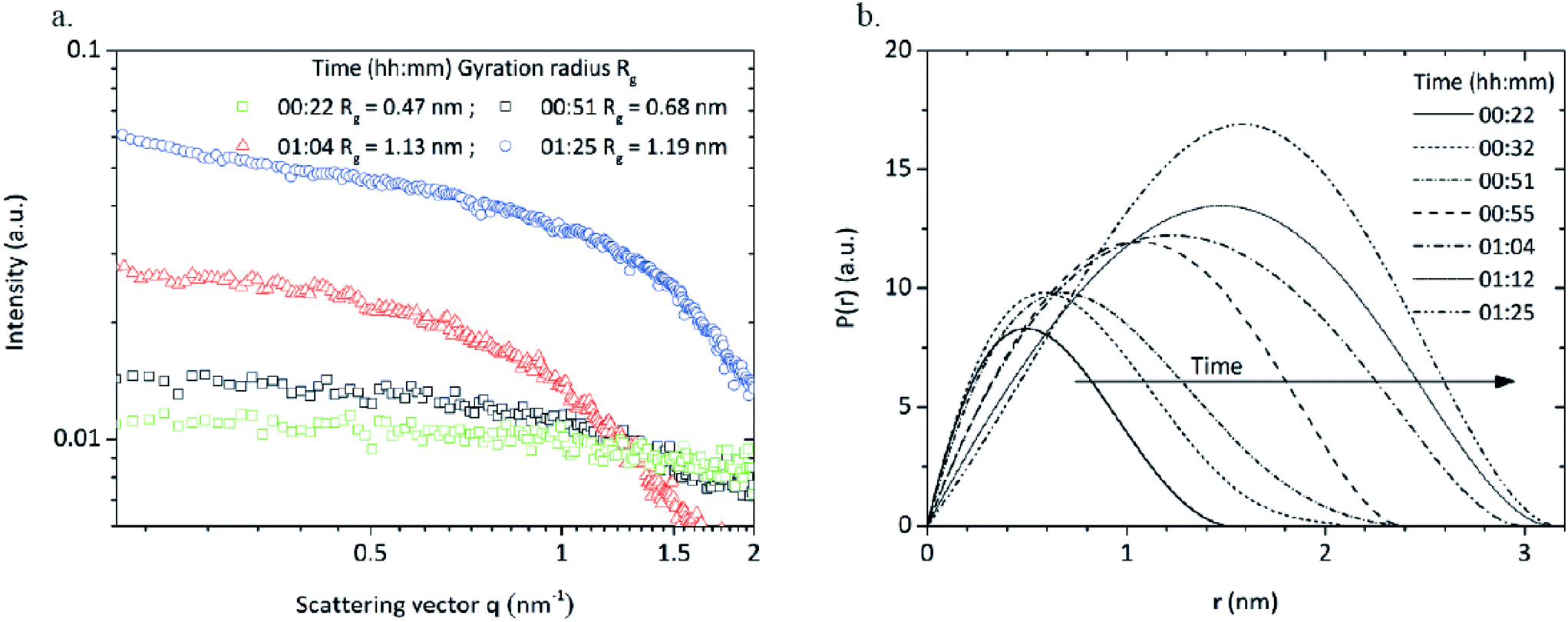 | ||
| Fig. 7 a. Scattering curves and respective radii of gyration of samples taken during induction time (00:22), after induction time (00:51) as well as during (01:04) and after (01:25) the decay of light intensity during a standard synthesis; b. pair distance distribution functions calculated from the scattering curves. | ||
We determined the radius of gyration (RG) from the scattering curves of the withdrawn reaction mixtures via a Guinier fit from a log-intensity-over-q2 plot. The sample withdrawn after 22 minutes (right after reaching the synthesis temperature) shows a gyration radius of 0.47 nm (Fig. 7a) which was surprising as particle formation was expected to start after the previously determined induction time. The scattering intensity was rather low due to a minor particle concentration which explains why the onset of particle formation was not detected during light transmission measurements and the induction time shows a certain delay. Hu et al.33 investigated an aqueous zirconia nanoparticle synthesis via SAXS and observed cluster species with a gyration radius of 0.45 nm. Singhal et al.32 showed in a very similar synthesis the presence of octameric cluster species Zr8(OH)20(H2O)24Cl12 with a size of 0.51 nm which upon further growth can be considered as zirconia nanoparticles. We assume that in our system, very similar cluster species are present that play a crucial role in the formation of zirconia nanoparticles. The SAXS results indicate that the detected cluster species grow further and upon reaching a certain size, might be considered as ZrO2 nuclei.
Pair distance distribution functions (PDDF) were calculated for all samples (Fig. 7b) to determine size and morphology of the formed species. The PDDFs correspond almost to a Gaussian curve which indicates that the nuclei are formed with a roundish shape and a diameter growing from 1.5 to 3 nm.
A sample taken shortly after induction time was stabilized according to Grote et al.43 to investigate the size and shape of the nucleated primary particles with transmission electron microscopy. The obtained micrograph (Fig. 8) shows that the particles are formed with a roundish morphology and mean size of 2.3 nm which is in good accordance with the SAXS results. The particle size distribution (histogram inset Fig. 8) was determined by manually measuring around 120 particles using the freeware ImageJ. Moreover, the particle sizes correspond well to the crystallite sizes determined by XRD and Rietveld refinement, which confirms that the particles are highly crystalline, and the crystallite size can be assumed to represent the actual particle size.
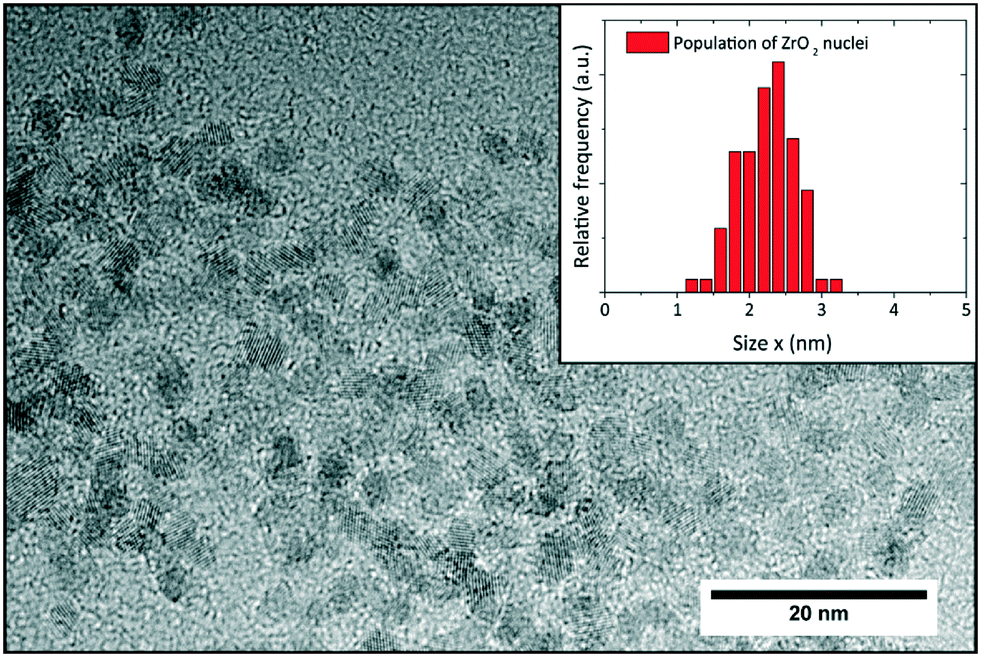 | ||
| Fig. 8 TEM analysis of sample taken from a standard synthesis shortly after induction time; the inset shows the respective particle size distribution with a mean size of 2.3 nm. | ||
3.3 Tetragonal-to-monoclinic phase transformation
The tetragonal-to-monoclinic phase transformation of zirconia nanoparticles has been widely discussed in literature in the pioneering works of Garvie44,45 and Ward.46 We have postulated that the martensitic phase transformation might be governed by kinetics26 and we propose a model that quantifies the probability of this transformation.Garvie states that the polymorphic transformation is triggered by the crystallite size effect,44 where a nanocrystal is thermodynamically more stable in the monoclinic phase above a critical crystallite size xc of 10 nm:47
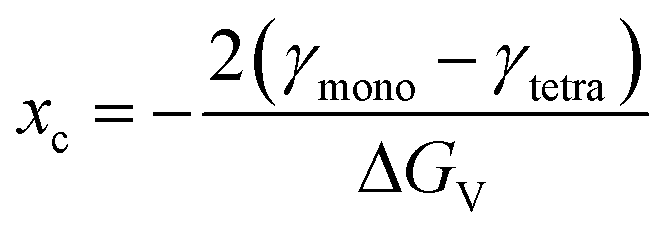 | (8) |
The critical crystallite size is therefore a function of the volume-specific free energy ΔGV of the zirconia bulk phase and the difference of the surface free energies γ of the respective phases. If the transformation were solely driven by thermodynamics, every crystallite that grows above a certain size would immediately transform into the monoclinic phase. Although recent works confirm a stable tetragonal phase for a crystallite size of 7 nm,48 multiple authors have reported coexisting tetragonal and monoclinic crystals in a size range of 4–67 nm31,49,50 which cannot be adequately explained by the thermodynamic model of Garvie. However, this observation can be explained by the embryo theory,46 postulating that a tetragonal crystallite cannot overcome the kinetic energy barrier to transform into the monoclinic phase by way of homogenous nucleation, even though it is thermodynamically more stable in the monoclinic state. Still, the kinetic barrier can be lowered and thus overcome if so-called embryos are present and serve as heterogeneous nucleation sites. An embryo could be an internal structural defect, such as a dislocation loop or stress at the interface caused by a Hertzian contact, but most importantly, occurs with a probability proportional to the crystal size.46 Once an embryo occurs, the crystallite transforms, beginning from the nucleation site, into the monoclinic form as illustrated in the inset of Fig. 9.
 | ||
| Fig. 9 Size-dependent probability P of crystallites undergoing the martensitic transformation from the tetragonal to the monoclinic phase; the inset shows a schematic illustration of this transformation induced by an embryo occurring at the crystallite surface. | ||
At quasi-equilibrium conditions, one can assume that the average number of embryos N found in a tetragonal crystal with the size xt can be described by a volume-specific embryo number density a0 (number of embryos per volume unit, nm−3) according to eqn (9), whereby kV represents the volume-specific shape factor.
| N = a0kVxt3 | (9) |
We assume that the probability P of finding m embryos in a single crystallite can be described with a Poisson distribution as follows:
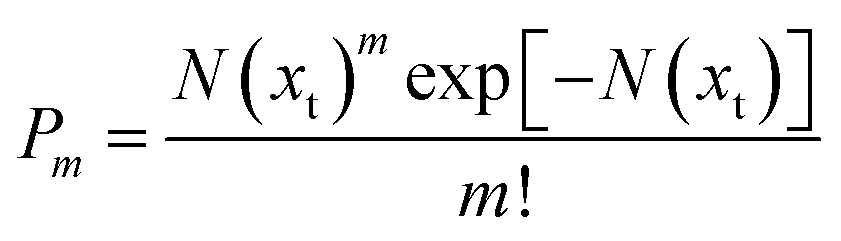 | (10) |
Hence, the probability of finding no embryos (m = 0) accounts to
| P0(xt) = exp[−N(xt)] | (11) |
| P0(xt) = 1 − exp[−N(xt)] = 1 − exp[−a0kVxt3] | (12) |
We calculated the number of particles that transformed into the monoclinic phase from the respective mean crystallite sizes and phase volume fractions which were obtained by XRD analysis of a sample withdrawn at the end of the synthesis. At this point, we observed that the primary crystallite growth has ended and that the monoclinic phase content has stabilized at a constant value, which justifies the assumption of quasi-equilibrium conditions. Then, the probability P of tetragonal crystallites transferring into the monoclinic phase at a certain size xt corresponds to the ratio between the number of monoclinic and the total number of crystals which is proportional to the phase content ratio of the monoclinic to the tetragonal phase. We plotted the probability P over the tetragonal crystallite size xt (Fig. 9) and fitted eqn (12) to our obtained data (a tabulated summary of the respective XRD data and calculated crystal numbers can be found in the ESI, Table S. 1†). Thereby, the specific embryo density a0 was determined to 1.13 embryos per cubic nanometer (nm−3) with a sphere-specific shape factor kV.
3.4 Simulation
We applied population balance equations (PBE) to simulate the formation of both crystal fractions using the commercially available software Parsival©. A set of integral-partial differential and nonlinear differential equations were numerically solved by the Garlekin h–p-method to yield the evolution of the crystallite number density of the tetragonal phase n(t, xt) and the monoclinic phase v(t, xm) with xt, xm ∈![[Doublestruck R]](https://www.rsc.org/images/entities/i_char_e175.gif) >0 for the respective crystallite sizes and times t ∈ >0. To formulate the population balance equation model, a closed batch system at isothermal conditions, with no nanocrystals entering or leaving via the system boundaries, is assumed. The system is assumed to be ideally mixed, which allows us to neglect any concentration- or heat gradients in the control volume and hence justifies a homogenous crystallite number density distribution within the system. Thus, the crystallite number densities can be described by one-dimensional population balance equations that are solely functions of the crystallite size coordinate x and independent from space coordinates. Parsival© derives the mass balances from the defined reaction system, e.g. reaction volume, operating conditions or initial concentrations of the species, and solves the equations in parallel to solving the PBE.
>0 for the respective crystallite sizes and times t ∈ >0. To formulate the population balance equation model, a closed batch system at isothermal conditions, with no nanocrystals entering or leaving via the system boundaries, is assumed. The system is assumed to be ideally mixed, which allows us to neglect any concentration- or heat gradients in the control volume and hence justifies a homogenous crystallite number density distribution within the system. Thus, the crystallite number densities can be described by one-dimensional population balance equations that are solely functions of the crystallite size coordinate x and independent from space coordinates. Parsival© derives the mass balances from the defined reaction system, e.g. reaction volume, operating conditions or initial concentrations of the species, and solves the equations in parallel to solving the PBE.
A set of ordinary differential equations (ODE) in the form of eqn (1) for the respective species was used to represent the ligand exchange as well as the ether condensation reactions. The ligand exchange reaction commences already at room temperature and is assumed to be completed before the reaction system reaches the synthesis temperature.2 Moreover, the ligand exchange is faster than the subsequent condensation reaction which is the kinetic bottleneck (kLigand ≫ kr). Therefore, the condensation reaction can be represented by an overall reaction using the reaction rate constant kr from the introduced model (eqn (6)). Hence, the condensation reaction yields the intermediate species that serve as the nucleation monomers and is modeled via an ordinary differential equation corresponding to eqn (7). The reaction order n was set to zero, as the chemical reactions follow pseudo-zero-order kinetics in good approximation. One has to note that the reaction rate is calculated individually for each time step and thus changes in temperature, e.g. during the heating period, are considered. The calculated monomer concentration is only theoretical and represents the assumed cluster species. Unfortunately, zirconia is practically insoluble in the solvent benzyl alcohol and the solubility concentration is below the detection limit of commonly used analytical methods which prevented a validation of theoretical with experimental concentration data. Again, the monomer concentration couples mass and population balances and serves as the driving force of the particle formation process.
The PBE for the tetragonal crystallite fraction is formulated in eqn (13) that describes the change in number density over time ∂n(xt)/∂t. The right-hand side of the equation comprises a nucleation term BNuc([ZrO2]m, xt) and a growth term G([ZrO2]m)∂n(xt)/∂t as well as a term PPhase([ZrO2]m, xt) for the number of crystallites that transform into the monoclinic phase. As the latter term represents the death term of the tetragonal PBE, a term using the same factors, except for the algebraic sign, serves as the birth term in the PBE of the monoclinic fraction (eqn (14)). In fact, our model postulates that the nucleation and growth of particles occurs only in the tetragonal phase and that the number of monoclinic particles ∂v(xm)/∂t in the monoclinic PBE (eqn (14)) is only changed by the term that accounts the number of particles that transform from the tetragonal to the monoclinic phase.
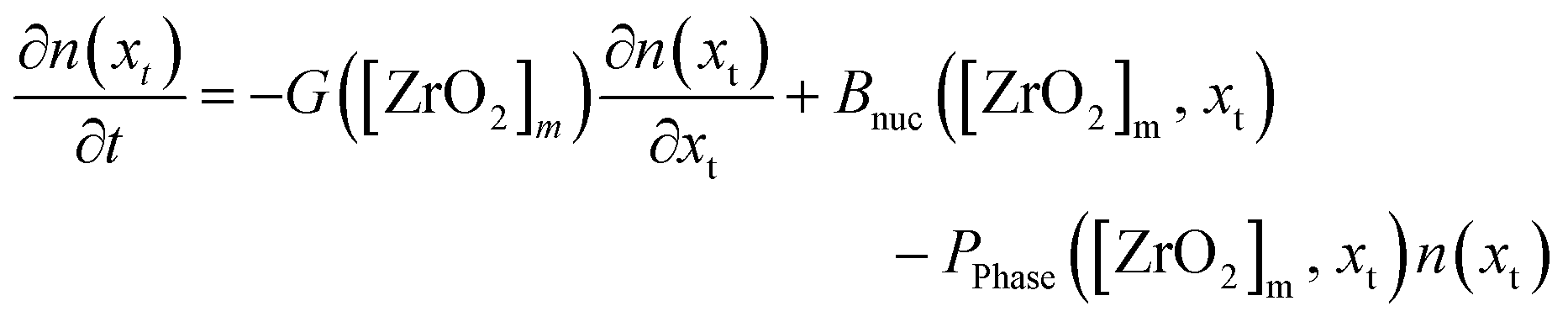 | (13) |
 | (14) |
The nucleation term is a power law function (eqn (15)) that yields the temporal number of nuclei. Here, knuc is the nucleation rate constant and ϕ(xt) a normal distribution function with a mean μ of 2 nm and a variance σ of 0.2 nm, which was set according to the obtained SAXS results (Fig. 7) assuming monodisperse nanoparticles. We found that the simulation results best fit the experimental data with a factor b of 2.2. The driving force of this term is the zirconia monomer concentration that is directly determined by the chemical reaction kinetics. The growth rate seems to be independent of the crystallize size and therefore only driven by the chemical reaction. Therefore we used a power law approach (eqn (16)), similar to the nucleation term, to model the growth kinetics, whereby we found that an exponent g set to 1 best reproduces our experimental observations.
| Bnuc = knuc[ZrO2]mbϕ(xt) | (15) |
| G = kg[ZrO2]mg | (16) |
We model the tetragonal-to-monoclinic phase transition as follows:
| PPhase = kphaseP(xt)[ZrO2]m | (17) |
Fig. 10 shows the simulation results of the size development for the tetragonal and monoclinic crystal fractions over the course of a standard synthesis carried out at 250 °C with an initial precursor concentration of 180 mmol L−1. The simulation results are in good accordance with the experimental data, although after 16 hours the growth ceases and the crystal sizes remain constant. This is due to the character of the linear overall reaction kinetics which provides a discontinuous drop of the monomer concentration d[ZrO2]m/dt to zero after complete yield (Fig. 11) and does not steadily converge to zero. The onset of particle formation is delayed due to the heating procedure, which was simulated via a heat ramp from ambient to synthesis temperature within 30 minutes. Since the model for the reaction constant kr (eqn (6)) is temperature dependent, the condensation reaction starts with an apparent time offset. The kink in the tetragonal crystallite evolution is due to the pronounced phase transition rate (eqn (17)) at larger crystallite sizes which causes an enhanced amount of particles to cross the phase transition barrier as simultaneously solely smaller tetragonal particles are nucleated. The tetragonal crystallite size appears to increase more rapidly after 10 h owing to term G([ZrO2]m) in eqn (13) that forces growth upon the remaining amount of tetragonal crystallites.
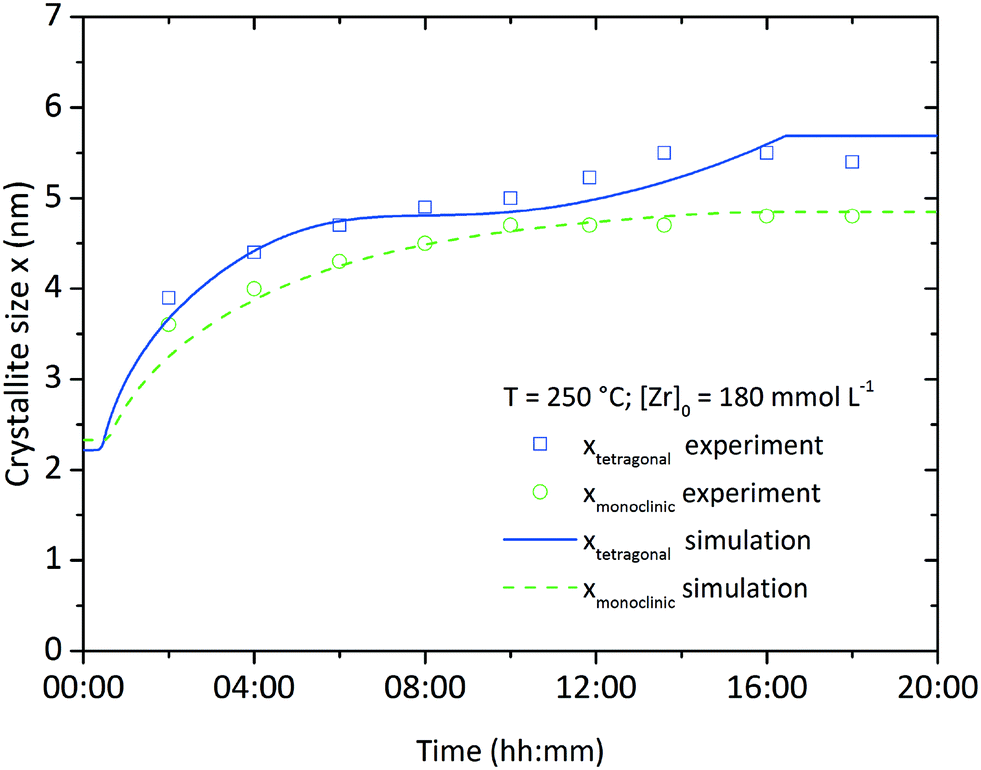 | ||
| Fig. 10 Simulation and experimental results for the growth of the tetragonal and monoclinic crystallite fractions over time at standard conditions. | ||
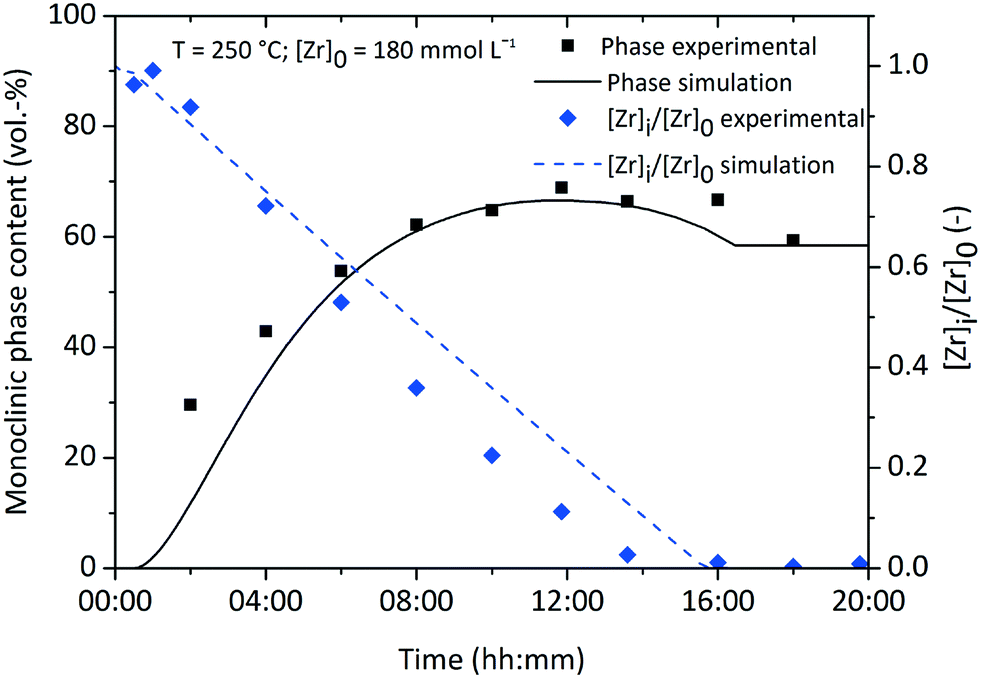 | ||
| Fig. 11 Normalized zirconium concentration and monoclinic volume fraction over the course of a standard synthesis. | ||
Throughout all experiments, we observed that monoclinic crystallites are smaller than tetragonal crystallites. This seems to be a direct conflict with the observations of Ward and Garvie stating that crystallites transform from the tetragonal to the monoclinic phase when exceeding a certain size. Hence it is unlikely to find smaller monoclinic than tetragonal crystallites. To deal with this problem, we propose that the chemical reaction leads solely to the growth of tetragonal crystallites which stop to grow once transformed into the monoclinic phase, i.e. there is no monoclinic particle growth via the incorporation of monomers or driven by the chemical reaction itself. Our experimental data (Fig. 10) shows that the apparent growth trends of both fractions are very similar, with the only major difference being an offset in crystallite size or an apparent time lag in the growth of the monoclinic fraction, which can be interpreted as being caused by a single growth process (of the tetragonal crystallites) with subsequent conversion of crystallites to the monoclinic phase. Moreover, Kuo et al.52 investigated the growth kinetics of tetragonal and monoclinic ZrO2 crystallites (albeit for high-temperature growth processes) and found that the growth constant of the monoclinic fraction is not only lower, but also features a substantially higher activation energy. This might reduce or even prevent monoclinic growth, especially at mild temperature conditions. We support this proposition by our simulations, as we included no growth term into the monoclinic population balance equation and are thereby able to reproduce the change in monoclinic crystal size observed experimentally.
Still, one has to consider the development of the monoclinic volume fraction (Fig. 11) which appears to pass a maximum and decline afterwards until reaching a constant value. This relates to the interplay between the phase change, nucleation and growth terms which all contain [ZrO2]m as a driving force. The monoclinic PBE provides only the birth term PPhase([ZrO2]m, xt) which sharply decreases towards the end of the simulation, whereas the tetragonal PBE still gains growing crystallites through the birth and growth terms. Nevertheless, the simulation seems to be in good accordance with our obtained experimental data by using one set of parameters in a single run which yielded the evolution of the chemical reaction kinetics, crystallite growth, and the tetragonal-to-monoclinic phase transformation.
We plotted the simulated number weighted crystal size distributions (CSD) for both fractions over the course of a standard synthesis (Fig. 12). The tetragonal crystallite fraction exhibits a nucleation burst up to a number density of 7.5 × 1030 m−3 in a narrow size distribution with crystallites growing up to 4 nm within the first two hours. Afterwards, the tetragonal CSD decreases in number density and grows as well as broadens in size with longer reaction times. On the contrary, the monoclinic phase experiences no nucleation burst and grows continuously over the entire course of the synthesis with a broader size distribution.
 | ||
| Fig. 12 Development of the crystallite size distribution for the tetragonal (a.) and the monoclinic polymorph (b.) as simulated for a standard synthesis. | ||
The population balance model was used to simulate the particle formation at other process conditions to check its validity for a broader scope of parameters. The simulation was validated in the temperature range from 220 to 270 °C and initial precursor concentrations from 180 to 360 mmol L−1 (additional results are shown in ESI, Fig. S. 5†). Here, we compare the model with experimental data of syntheses carried out at 270 °C and 180 mmol L−1 (Fig. 13), as our former studies26 revealed that the final nanoparticle characteristics change drastically at 270 °C. Notably, the population balance model is still valid for these conditions, but Fig. 13 shows a minor difference between the experimental and simulated crystal size of the tetragonal phase which can be related to the faster reaction kinetics leading to higher monomer concentrations. This leads to pronounced nucleation but less growth of the tetragonal crystals since the nucleation term (eqn (15)) comprises [ZrO2]mb with a value of b determined to 2.2 for these conditions. The development of the normalized zirconium concentration corresponds well to experimental data (Fig. 13b), which confirms that the reaction constant model (eqn (6)) is valid. The experimental monoclinic phase content shows a different behavior and remains rather constant with a slight decrease in the beginning, which is due to impurities from previous experiments. However, the simulation shows only a minor increase in the monoclinic phase content which adjusts to values in the range of the experimental data.
 | ||
| Fig. 13 Validation of experimental and simulation results for a synthesis carried out at 270 °C with an initial precursor concentration of 180 mmol L−1; a. shows the development of the respective crystallite size for both fractions; b. shows the evolution of the normalized zirconium concentration and monoclinic phase content with some deviation between the experimental and simulation data. | ||
4. Conclusion
We advanced our process study on the nonaqueous synthesis of zirconia nanoparticles and thoroughly investigated the kinetics of the chemical reactions that lead to particle formation. The reaction order was quantified to 0.3 which solidifies the assumption that the chemical reaction can be best described by quasi-zero-order kinetics. We derived a model for the reaction constant kr, as a function of the temperature and initial precursor concentration [Zr]0 as process conditions that can be used to describe the evolution of the soluble zirconium concentration [Zr]i over time.The nucleation event was investigated with a specially designed reactor system which allowed the determination of the onset of particle formation and the preceding induction time via in situ light (vis) transmission measurements. Precise sampling during the induction time and subsequent SAXS analysis indicated that the particles nucleate with a roundish shape in a size regime of 2 nm which was confirmed by TEM analysis. Surprisingly, we found via SAXS that early stage particle formation seems to start when the reaction mixture reaches the final temperature, i.e. before the actual induction time. Thereby, we detected species with a gyration radius of 0.47 nm which we believe are the intermediate clusters or zirconia monomers that serve as nucleation precursor.
The tetragonal-to-monoclinic transformation of zirconia nanoparticles was modeled by relating the probability of crystallites undergoing the phase transition to the crystal size. Hereby, we use the Poisson distribution function to predict the statistical occurrence of tetragonal-to-monoclinic nucleation sites in a tetragonal crystal continuum with a size xt that triggers the martensitic transformation of the entire particle.
These findings are comprised in a comprehensive population balance model that simulates the entire crystal formation process. In this population balance framework, nucleation and growth were modeled with empirical equations that are driven by the concentration of zirconia monomers [ZrO2]m. Thus, crystal formation is closely linked to the chemical reaction which highly depends on the process conditions. We applied the tetragonal-to-monoclinic phase transition model and showed that the simulation results can be validated with experimental data, achieving good agreement. Supported by our simulation results, we propose that only tetragonal crystallites grow through the incorporation of monomers into the crystal structure resulting in lower mean crystallite sizes of the monoclinic polymorph. All in all, we quantified important kinetic and thermodynamic parameters and derived model equations for the nonaqueous synthesis. For the first time, a simulation approach for the nonaqueous sol–gel method is presented that can predict the course of the synthesis and final nanoparticle properties for a broad range of process parameters.
Acknowledgements
This study was financed by the Deutsche Forschungsgemeinschaft (grant GA 1492/3-3). The authors would like to thank Bilal Temel for TEM and XRD measurements. Further, the authors are grateful to Adrian Holste and Maximilian Roehe for the great support during the development of the 250 mL reactor system.References
- M. Niederberger, M. H. Bartl and G. D. Stucky, Chem. Mater., 2002, 14, 4364–4370 CrossRef CAS.
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC.
- I.-M. Grabs, C. Bradtmöller, D. Menzel and G. Garnweitner, Cryst. Growth Des., 2012, 12, 1469–1475 CAS.
- G. Garnweitner, N. Tsedev, H. Dierke and M. Niederberger, Eur. J. Inorg. Chem., 2008, 2008, 890–895 CrossRef.
- N. Pinna, M. Karmaoui and M. G. Willinger, J. Sol-Gel Sci. Technol., 2011, 57, 323–329 CrossRef CAS.
- M. Zimmermann and G. Garnweitner, CrystEngComm, 2012, 14, 8562–8568 RSC.
- I. C. Masthoff, M. Kraken, D. Mauch, D. Menzel, J. A. Munevar, E. Baggio Saitovitch, F. J. Litterst and G. Garnweitner, J. Mater. Sci., 2014, 49, 4705–4714 CrossRef CAS.
- S. Ge, H. Jia, H. Zhao, Z. Zheng and L. Zhang, J. Mater. Chem., 2010, 20, 3052–3058 RSC.
- I. Olliges-Stadler, J. Stötzel, D. Koziej, M. D. Rossell, J.-D. Grunwaldt, M. Nachtegaal, R. Frahm and M. Niederberger, Chem. – Eur. J., 2012, 18, 2305–2312 CrossRef CAS PubMed.
- M. Distaso, D. Segets, R. Wernet, R. K. Taylor and W. Peukert, Nanoscale, 2012, 4, 864–873 RSC.
- S. Zellmer, A. Kockmann, I. Dosch, B. Temel and G. Garnweitner, CrystEngComm, 2015, 17, 6878–6883 RSC.
- T. M. Stawski, S. A. Veldhuis, O. F. Göbel, J. E. Ten Elshof and D. H. A. Blank, J. Am. Ceram. Soc., 2010, 93, 3443–3448 CrossRef CAS.
- R. Lorenzi, A. Paleari, N. V. Golubev, E. S. Ignat'eva, V. N. Sigaev, M. Niederberger and A. Lauria, J. Mater. Chem. C, 2015, 3, 41–45 RSC.
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- I. C. Masthoff, A. Gutsche, H. Nirschl and G. Garnweitner, CrystEngComm, 2015, 17, 2464–2470 RSC.
- N. Kranzlin, M. Staniuk, F. J. Heiligtag, L. Luo, H. Emerich, W. van Beek, M. Niederberger and D. Koziej, Nanoscale, 2014, 6, 14716–14723 RSC.
- H. Jensen, M. Bremholm, R. P. Nielsen, K. D. Joensen, J. S. Pedersen, H. Birkedal, Y.-S. Chen, J. Almer, E. G. Søgaard, S. B. Iversen and B. B. Iversen, Angew. Chem., 2007, 119, 1131–1134 CrossRef.
- G. V. Jensen, M. Bremholm, N. Lock, G. R. Deen, T. R. Jensen, B. B. Iversen, M. Niederberger, J. S. Pedersen and H. Birkedal, Chem. Mater., 2010, 22, 6044–6055 CrossRef.
- B. J. Ridder, A. Majumder and Z. K. Nagy, Ind. Eng. Chem. Res., 2014, 53, 4387–4397 CrossRef CAS.
- R. T. Kügler, S. Doyle and M. Kind, Chem. Eng. Sci., 2015, 133, 140–147 CrossRef.
- H.-C. Schwarzer, F. Schwertfirm, M. Manhart, H.-J. Schmid and W. Peukert, Chem. Eng. Sci., 2006, 61, 167–181 CrossRef CAS.
- M. C. Heine and S. E. Pratsinis, Ind. Eng. Chem. Res., 2005, 44, 6222–6232 CrossRef CAS.
- S. R. K. Perala and S. Kumar, Langmuir, 2014, 30, 12703–12711 CrossRef CAS PubMed.
- D. Segets, M. A. J. Hartig, J. Gradl and W. Peukert, Chem. Eng. Sci., 2012, 70, 4–13 CrossRef CAS.
- Y. P. Gokhale, R. Kumar, J. Kumar, W. Hintz, G. Warnecke and J. Tomas, Chem. Eng. Sci., 2009, 64, 5302–5307 CrossRef CAS.
- P. Stolzenburg, A. Freytag, N. C. Bigall and G. Garnweitner, CrystEngComm, 2016, 18, 8396–8405 RSC.
- M. Zimmermann, B. Temel and G. Garnweitner, Chem. Eng. Process.: Process Intesif., 2013, 74, 83–89 CrossRef CAS.
- G. Garnweitner and C. Grote, Phys. Chem. Chem. Phys., 2009, 11, 3767–3774 RSC.
- G. Garnweitner, L. M. Goldenberg, O. V. Sakhno, M. Antonietti, M. Niederberger and J. Stumpe, Small, 2007, 3, 1626–1632 CrossRef CAS PubMed.
- T. Ninjbadgar, G. Garnweitner, A. Börger, L. M. Goldenberg, O. V. Sakhno and J. Stumpe, Adv. Funct. Mater., 2009, 19, 1819–1825 CrossRef CAS.
- T. A. Cheema and G. Garnweitner, CrystEngComm, 2014, 16, 3366–3375 RSC.
- A. Singhal, L. M. Toth, J. S. Lin and K. Affholter, J. Am. Chem. Soc., 1996, 118, 11529–11534 CrossRef CAS.
- M. Z.-C. Hu, J. T. Zielke, J.-S. Lin and C. H. Byers, J. Mater. Res., 1999, 14, 103–113 CrossRef CAS.
- M. Zimmermann, K. Ibrom, P. G. Jones and G. Garnweitner, ChemNanoMat, 2016, 2, 1073–1076 CrossRef CAS.
- M. Wulkow, A. Gerstlauer and U. Nieken, Chem. Eng. Sci., 2001, 56, 2575–2588 CrossRef CAS.
- A. K. Coker, Modeling of Chemical Kinetics and Reactor Design, Elsevier Science, 2001 Search PubMed.
- J. W. Mullin, Crystallization, Elsevier Science, 2001 Search PubMed.
- A. Mersmann, Crystallization Technology Handbook, Taylor & Francis, 2001 Search PubMed.
- M. Zimmermann, Dissertation, TU Braunschweig, 2014 Search PubMed.
- M. Ambrosi, E. Fratini, P. Canton, S. Dankesreiter and P. Baglioni, J. Mater. Chem., 2012, 22, 23497–23505 RSC.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- A. Gossard, F. Grasland, X. Le Goff, A. Grandjean and G. Toquer, Solid State Sci., 2016, 55, 21–28 CrossRef CAS.
- C. Grote, T. A. Cheema and G. Garnweitner, Langmuir, 2012, 28, 14395–14404 CrossRef CAS PubMed.
- R. C. Garvie, J. Phys. Chem., 1965, 69, 1238–1243 CrossRef CAS.
- R. C. Garvie and M. V. Swain, J. Mater. Sci., 1985, 20, 1193–1200 CrossRef CAS.
- D. A. Ward and E. I. Ko, Chem. Mater., 1993, 5, 956–969 CrossRef CAS.
- R. C. Garvie, J. Phys. Chem., 1978, 82, 218–224 CrossRef CAS.
- H. Kim, M. Kim, C. Bae, E. Kim, S. Lee, J. M. Montero-Moreno, H. S. Jung and H. Shin, RSC Adv., 2015, 5, 80472–80479 RSC.
- S. Shukla and S. Seal, Int. Mater. Rev., 2005, 50, 45–64 CrossRef CAS.
- T. Chraska, A. H. King and C. C. Berndt, Mater. Sci. Eng., A, 2000, 286, 169–178 CrossRef.
- G. Madras, B. J. McCoy and A. Navrotsky, J. Am. Ceram. Soc., 2007, 90, 250–255 CrossRef CAS.
- C.-W. Kuo, K.-C. Lee, F.-L. Yen, Y.-H. Shen, H.-E. Lee, S.-B. Wen, M.-C. Wang and M. M. Stack, J. Alloys Compd., 2014, 592, 288–295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00005g |
| This journal is © The Royal Society of Chemistry 2017 |