
Combining batch and continuous flow setups in the end-to-end synthesis of naturally occurring curcuminoids†
Christian C.
Carmona-Vargas
a,
Leandro
de C. Alves
b,
Timothy J.
Brocksom
a and
Kleber T.
de Oliveira
 *a
*a
aDepartamento de Química, Universidade Federal de São Carlos, 13565-905, São Carlos, SP, Brazil. E-mail: kleber.oliveira@ufscar.br; Web: www.lqbo.ufscar.br
bAccert Chemistry and Biotechnology, Rua Alfredo Lopes 1717 Sala 7E, 13560-460, São Carlos, SP, Brazil
First published on 15th February 2017
Abstract
A successful end-to-end continuous flow synthesis of pure curcumin (1) and two other natural derivatives present in turmeric is described. Batch and continuous flow conditions were combined in order to develop the end-to-end process. Three operations were telescoped yielding the three different natural curcuminoids in a safe, gram-scale and reproducible manner.
Introduction
Curcumin (1), bis-demethoxycurcumin (2) and demethoxycurcumin (3) are the three main curcuminoids present in turmeric, the golden powder used as colouring in foods obtained from the dried rhizomes of the plant Curcuma longa, originating from India.1The medicinal properties exhibited by turmeric have been attributed to the presence of the curcuminoids 1–3 which have gained increasing attention,2 due to their significant biological activities such as antioxidant,3 antibacterial,4 anti-cancer,5 and antifungal.6 Furthermore, curcumin (1) has demonstrated a number of applications in photonic therapies due to its excellent photodynamic activity as a sensitizer, covering oral treatments,7 elimination of dengue mosquito larvae,8 and decontamination of foodstuffs.9
In 1964, Pabon10 developed a two-step synthesis of 1 in which a boron–acetylacetone complex was formed by reacting acetylacetone with boron anhydride/(n-BuO)3B in ethyl acetate, followed by an aldol condensation reaction between vanillin and the enolate formed by the addition of n-butylamine, giving 1 in 73% yield.
Chemical syntheses have usually been performed batch-wise in standardized glassware or reactors, but with some limitations. However, new enabling techniques for organic synthesis are improving the manner in which reactions can be carried out, with continuous flow processes being an important tool.11,12
Continuous flow systems have attracted particular attention, due to the possibility of combining chemical synthesis and chemical engineering, improving the efficiency of processes from laboratory to larger scales.13,14 Controlled mixing,15 fast heat transfer,16 control of residence time,17 handling of hazardous reagents,18–20 process automation,21,22 and easy scale-up/scale-out23 are some of the major advantages of this enabling technology. Numerous single-step and multi-step transformations under continuous flow conditions24,25 have been reported for the synthesis of active pharmaceutical ingredients (APIs),26,27 as well as natural products,28 sugars,29 peptides,30 cross-coupling reactions,31 biocatalysed processes,32 and other transformations.33–35
Despite all the aforementioned advantages of continuous flow systems, so far it is not possible (or desirable) to abandon batch conditions completely and carry out a reaction sequence totally under continuous flow conditions. Recently, Ley's group28 has shown that chemical synthesis should not necessarily be limited to batch or flow conditions. If combining the two is better, why not use them both?36–39 Therefore, choosing the best of both synthetic approaches40 can offer a straightforward means of meeting process safety41 and preserving reaction efficiency42 without the need for special or sophisticated equipment. Most importantly, new alternatives for synthesis, creativity and broader operating conditions may become possible by integrating these two techniques.43
Herein, we present an approach for the end-to-end synthesis of curcumin (1), which, according to the market research company Grand View Research Inc., shows a global market with a growing demand for the pharmaceutical industry.44 Additionally, their naturally occurring analogues bis-demethoxycurcumin (2) and demethoxycurcumin (3) are synthesized using the same integrated protocols. A fully telescoped approach under continuous flow conditions has been developed and compared to the combined flow–batch and all batch systems.10,45
Results and discussion
Operational planning
The end-to-end synthesis of curcumin (1) was initially planned and divided into three unit operations as shown in Fig. 1. Each operation was designed to be executed independently and aimed for telescoping. The first operation (in red) is the synthesis of the boron complex 6 starting from acetylacetone (4) and B2O3/B(O-n-Bu)3. In the second operation (in blue), complex 6 was reacted with vanillin (5) through four aldol condensations to yield the complex 7. In the third operation (in green), boron decomplexation was executed by using HCl (1.0 mol L−1), integrating simple phase-separation and curcuminoid production. The main challenges were found during the upstream processes which involved operation 1 (slurries) and 2, as well as the integration of both. Operation 2 is the most critical since it involves a one-pot four-step enolate formation, addition and elimination sequence, the reason that it was chosen as the first process to be optimized. In general, we describe herein two new strategies to furnish a protocol for the synthesis of curcuminoids 1–3. The first successful protocol involves both batch and flow conditions in a combined tactic. The second protocol uses only end-to-end flow conditions, and we have demonstrated the advantages of the combined tactic.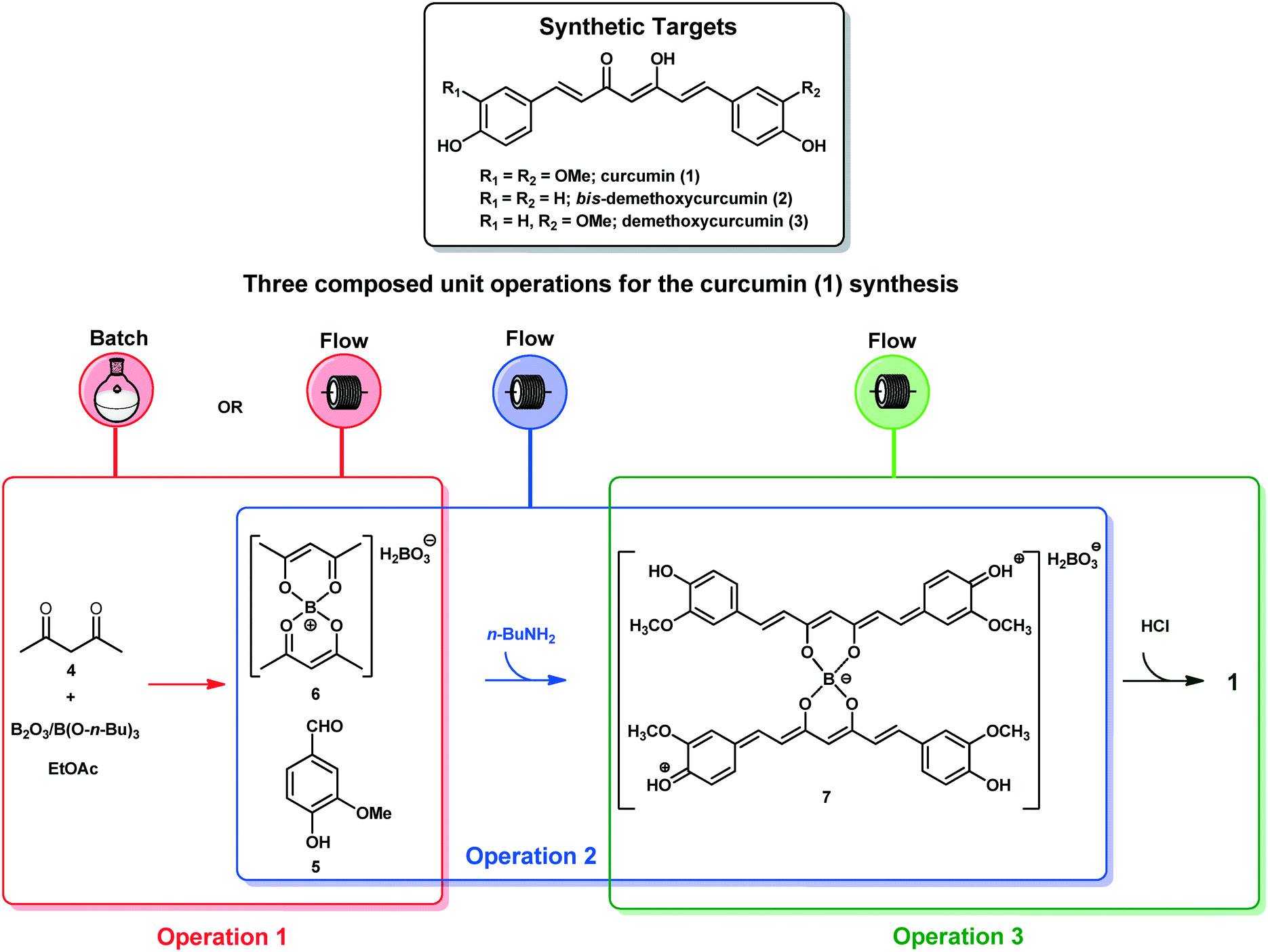 | ||
| Fig. 1 Operational planning for end-to-end curcumin (1) synthesis. | ||
Trial-and-error optimization of operation 2
In order to optimize operation 2, we decided to start by using a batch approach for performing operation 1, since it involves an initial slurry formation (Fig. 1). Thus, a mixture of acetylacetone (4) (10 mmol) and B2O3 (5.0 mmol)/B(O-n-Bu)3 (40 mmol) in EtOAc (4 mL) was heated under batch conditions at 70 °C for 30 min, and a homogeneous solution of complex 6 was obtained. Vanillin (5) (35.2 mmol) in EtOAc (24 mL) was added to the complex 6 solution, thus achieving the first operation in batch (Scheme 1 – in red). Then, operation 2 was optimized under continuous flow conditions using a heated tubular reactor (40 mL) and a double channel Syrris® syringe pump (Scheme 1). The reagents were pumped (Pump 1: n-butylamine (BuNH2), 2.0–8.0 mmol, in EtOAc; Pump 2: complex 6 + vanillin (5) in EtOAc) into a mixing zone (T-piece) and then to the tubular reactor heated at different temperatures (Table 1). Initially, we established a 0.2 mL min−1 flow rate for both Pump 1 and Pump 2 in order to achieve adequate dilution of these reactants, and a residence time of 100 min. The boron decomplexation was performed in batch by using HCl (1.0 mol L−1) at 50 °C (Scheme 1), and pure curcumin (1) was obtained after extraction and crystallization from ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:1).
:water (1:1).
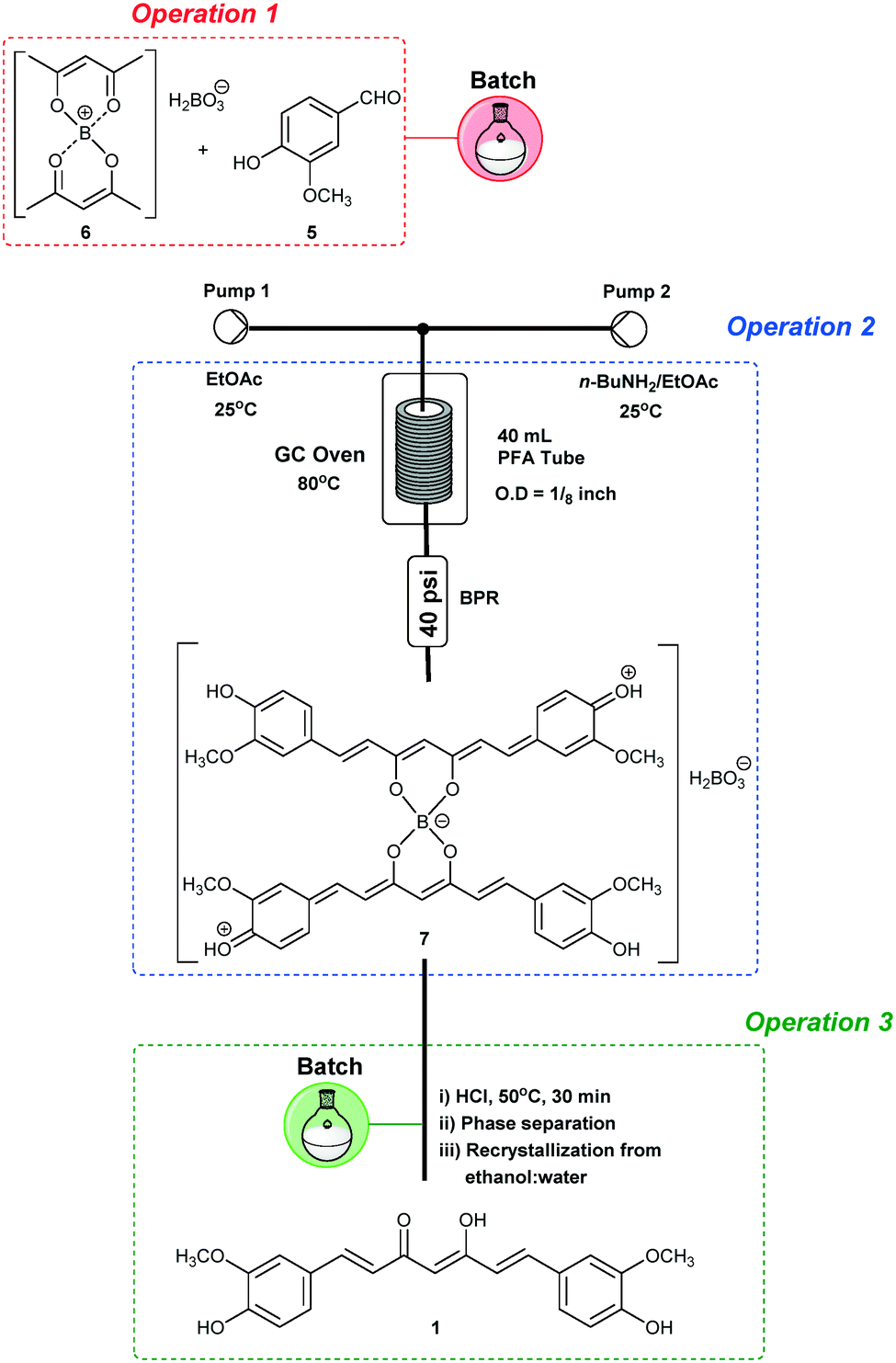 | ||
| Scheme 1 Optimizations combining batch and continuous flow conditions for the synthesis of curcumin (1). | ||
| Entrya,b | n-BuNH2 (mol L−1) | T (°C) | Curcumin yield (%) | STY (g per day) |
|---|---|---|---|---|
| a Residence time = 100 min. b Vanillin (5) conc. of 0.50 mol L−1 (4 equiv.). | ||||
| 1 | 0.05 (0.4 equiv.) | 80 | 50 | 13.2 |
| 2 | 0.05 (0.4 equiv.) | 100 | 45 | 11.9 |
| 3 | 0.05 (0.4 equiv.) | 120 | 40 | 10.6 |
| 4 | 0.075 (0.6 equiv.) | 80 | 59 | 15.6 |
| 5 | 0.075 (0.6 equiv.) | 100 | 56 | 14.8 |
| 6 | 0.075 (0.6 equiv.) | 120 | 50 | 13.2 |
| 7 | 0.10 (0.8 equiv.) | 80 | 59 | 15.6 |
| 8 | 0.10 (0.8 equiv.) | 100 | 56 | 14.8 |
| 9 | 0.10 (0.8 equiv.) | 120 | 45 | 10.6 |
| 10 | 0.20 (1.6 equiv.) | 80 | 50 | 13.2 |
| 11 | 0.20 (1.6 equiv.) | 100 | 48 | 12.7 |
| 12 | 0.20 (1.6 equiv.) | 120 | 45 | 11.9 |
Starting from 5.0 mmol of complex 6 (0.125 mol L−1), 20 mmol of vanillin (5) (0.50 mol L−1) and 2.0 mmol of n-BuNH2 (0.05 mol L−1) and varying the temperature from 80 °C to 120 °C, curcumin (1) was obtained in 40–50% yield (entries 1–3, Table 1) with the best result at 80 °C (STY = 15.6 g per day).
By using 3.0 mmol of n-BuNH2 (0.075 mol L−1), yields are slightly improved (50–59%), again with the best result at 80 °C (entries 4 and 5, Table 1). n-BuNH2 concentrations up to 0.20 mol L−1 gave similar or lower yields (entries 7–12, Table 1), showing the best concentration of the base to be 0.075 mol L−1 (0.6 equiv. with respect to complex 6) in these first conditions.
After establishing the suitable concentration of the base, different vanillin (5) concentrations were evaluated from 0.50 mol L−1 (4 equiv.) to 1.0 mol L−1 (8 equiv.) in the same temperature range (Table 2). From these results, we conclude that 0.88 mol L−1 is the best concentration for producing 1 (69% yield and an STY of 18.3 g per day, entry 7, Table 2).
| Entrya | Vanillin (5) (mol L−1) | T (°C) | Curcumin yield (%) | STY (g per day) |
|---|---|---|---|---|
| a n-BuNH2 conc. of 0.075 mol L−1 (0.6 equiv.). | ||||
| 1 | 0.50 (4 equiv.) | 80 | 59 | 15.6 |
| 2 | 0.50 (4 equiv.) | 100 | 56 | 14.8 |
| 3 | 0.50 (4 equiv.) | 120 | 40 | 10.6 |
| 4 | 0.75 (6 equiv.) | 80 | 64 | 16.9 |
| 5 | 0.75 (6 equiv.) | 100 | 61 | 16.1 |
| 6 | 0.75 (6 equiv.) | 120 | 55 | 14.5 |
| 7 | 0.88 (7.05 equiv.) | 80 | 69 | 18.3 |
| 8 | 0.88 (7.05 equiv.) | 100 | 60 | 15.9 |
| 9 | 0.88 (7.05 equiv.) | 120 | 55 | 14.5 |
| 10 | 1.00 (8 equiv.) | 80 | 69 | 18.3 |
| 11 | 1.00 (8 equiv.) | 100 | 65 | 17.2 |
| 12 | 1.00 (8 equiv.) | 120 | 55 | 14.5 |
Furthermore, varying the flow rates from 0.2 mL min−1 (100 min) to 0.4 mL min−1 (50 min), and keeping the same previous optimized conditions (entry 7, Table 2), yields ranged from 45 to 69% (Table 3) with the best result at 0.3 mL min−1 (entry 2, Table 3) giving an STY of 24.1 g per day and a shorter residence time (66.6 min).
Integrating operations 2 and 3
The integration of operations 2 and 3 (Scheme 2) was performed under optimized conditions for curcumin (1) synthesis (the same conditions, see Table 3, entry 2). The output of operation 2 was connected to a Schott® flask (100 mL) maintained at 50 °C and with magnetic stirring. In addition, simultaneous 1.0 mol L−1 HCl pumping at 0.9 mL min−1 was established (Pump 3, Scheme 2). After reaction termination, the organic phase was separated and evaporated under reduced pressure to give curcumin (1) in 62% yield after recrystallization from ethanol:water (1:1).
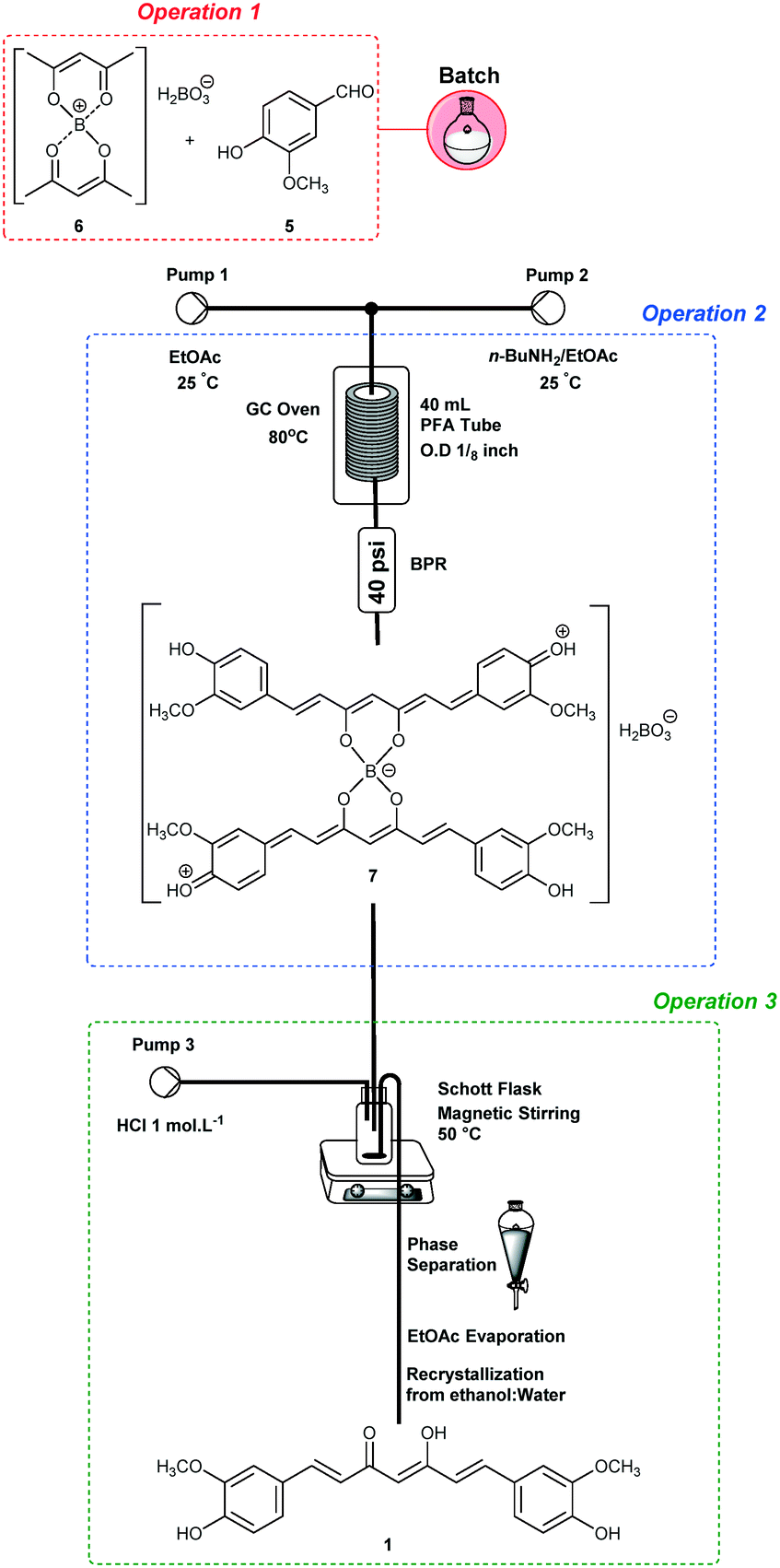 | ||
| Scheme 2 Integration of operations 2 and 3. | ||
Having optimized the critical operation for the end-to-end synthesis of curcumin (1), we have decided to scale-up46,47 the protocol (operations 2 + 3) starting from 16.0 mmol of complex 6 under the optimized conditions (Table 3, entry 2). Thus, curcumin (1) was obtained on a 7.50 g scale (63% yield) after 7 h of process intensification (STY = 25.5 g per day).
It is important to mention that during the optimizations of operation 2, we have checked the reproducibility under all the optimized conditions in at least two experiments, obtaining very good comparison (variations of no more than ±3%).
Integrating operations 1, 2 and 3 under continuous flow conditions
The end-to-end continuous flow synthesis of curcumin (1) was possible by the integration of operations 1, 2 and 3 (Scheme 3), starting from the optimized conditions (Table 3, entry 2).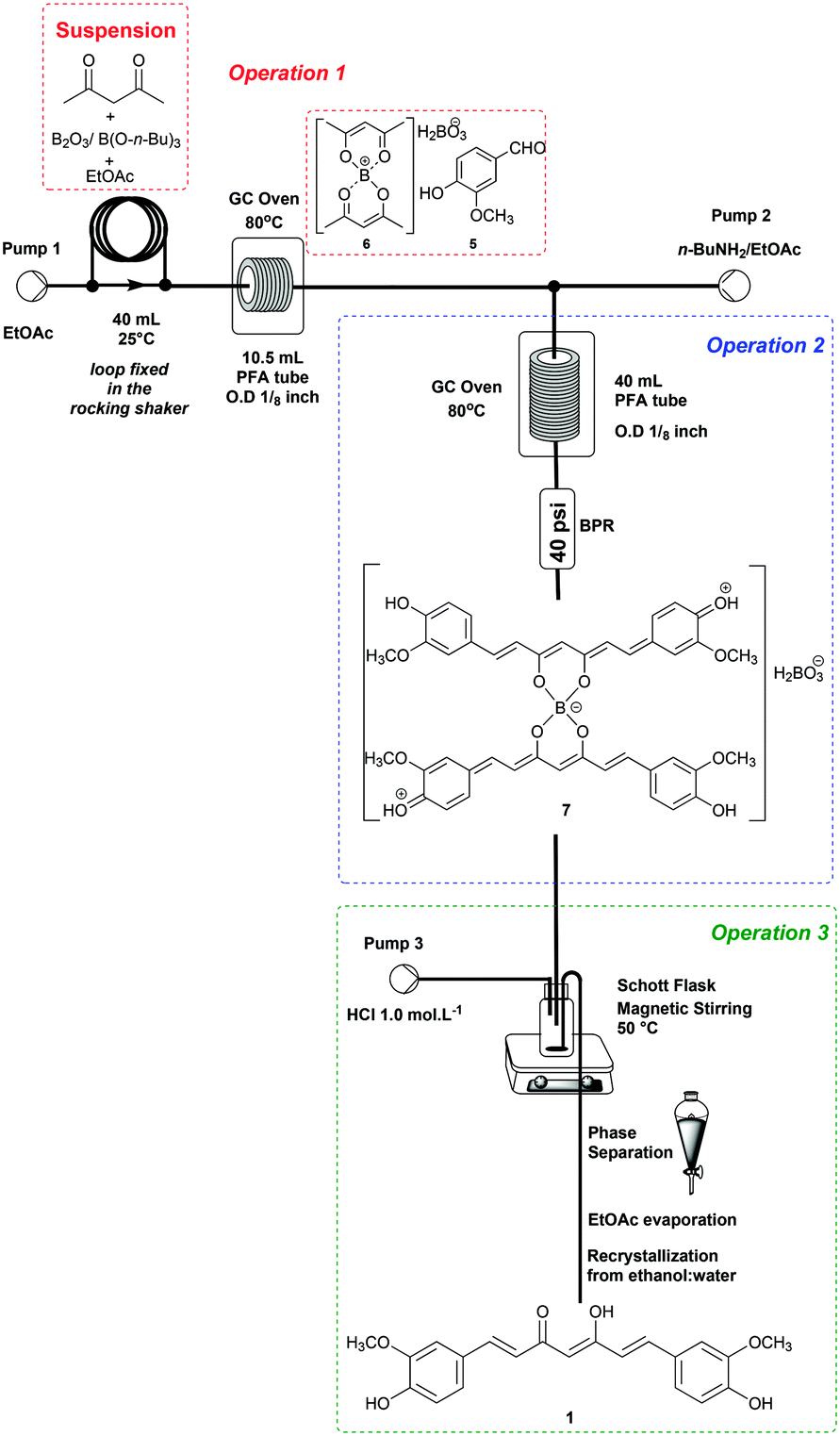 | ||
| Scheme 3 End-to-end synthesis of curcumin (1) – telescoped process. | ||
Thus, a suspension containing B2O3 (5.0 mmol)/B(O-n-Bu)3 (40 mmol), acetylacetone (4) (10 mmol), and vanillin (5) (35.2 mmol) in EtOAc was loaded into a 40 mL PTFE loop module fixed in a rocking shaker. Operation 1 was performed by pumping EtOAc at 0.3 mL min−1 directly into the loop module, which was connected to a tubular reactor (10.5 mL) at 80 °C. The first generated complex 6 encountered 3.0 equiv. of n-BuNH2 solution (0.075 mol L−1) at 0.3 mL min−1 proceeding as optimized for operations 2 and 3.
Curcumin (1) was obtained on a 0.94 g scale after recrystallization from ethanol:water (1:1) (51% yield, STY = 20.5 g per day), and the reproducibility was checked twice, obtaining 48 and 53% yields. Despite the successful end-to-end process established for 1, the yield/STY obtained from these fully integrated processes (51%, 20.5 g per day) are lower than when compared to the trial and error optimized conditions achieved for combined process 1 (in batch), and both processes 2 and 3 (continuously) (60%, 24.1 g per day). This decrease is probably due to the difficulty of keeping the slurry from operation 1 well mixed. In addition, another weakness of this process is the continuous scale limited by the loop size.
Expanding the processes to curcuminoids 2 and 3
We also carried out the synthesis of bis-demethoxycurcumin (2) using the previously optimized reaction conditions (entry 2, Table 3) or integration of the three processes. For the synthesis of 2, the aldehyde source was changed to 4-hydroxybenzaldehyde (10). Both protocols with operation 1 in batch and the others under continuous flow conditions or fully telescoped in flow (end-to-end) are successful, obtaining bis-demethoxycurcumin (2) in 60% (0.96 g and STY: 20.6 g per day) and 45% (0.69 g and STY: 15.0 g per day) yields, respectively.For the synthesis of non-symmetrical demethoxycurcumin (3), one additional step is necessary for preparing intermediate 9 (Scheme 4). The mono-condensation of symmetrical diketones such as 4 requires major control, since mixtures are frequently obtained. However, by using continuous flow conditions, it was possible to control this step carefully, and improved results were obtained compared to batch procedures. Thus, operation 1 was performed in batch, starting from 14.4 mmol of acetylacetone (4) and B2O3 (10.8 mmol)/B(O-n-Bu)3 (13.2 mmol) in EtOAc (25 mL), and vanillin (6.0 mmol) was then added before the pumping began (Pump 1). n-BuNH2 (3.0 mmol in 30 mL of EtOAc) was also pumped (Pump 2) varying the temperature and flow rates (Table 4).
 | ||
| Scheme 4 Synthesis of intermediate 9. | ||
| Entry | Residence time (min) | T (°C) | Intermediate 9 yield (%) | STY (g per day) |
|---|---|---|---|---|
| 1 | 66.6 | 80 | 64 | 13.0 |
| 2 | 66.6 | 100 | 66 | 13.5 |
| 3 | 66.6 | 120 | 55 | 16.6 |
| 4 | 44.4 | 80 | 56 | 17.1 |
| 5 | 44.4 | 100 | 59 | 18.0 |
| 6 | 44.4 | 120 | 56 | 17.1 |
| 7 | 33.3 | 80 | 45 | 18.2 |
| 8 | 33.3 | 100 | 48 | 19.5 |
| 9 | 33.3 | 120 | 45 | 18.2 |
Keeping the flow rates at 0.3 mL min−1 for both pumps 1 and 2, the yields of 9 ranged from 55 to 66% (entries 1–3, Table 4), with 66% yield at 100 °C. By increasing the flow rates to 0.45 mL min−1, intermediate 9 was obtained in 59% yield at 100 °C (entry 5, Table 4). At 0.6 mL min−1, no improved results were obtained (entries 7–9, Table 4).
Hence, we suggest that the best flow rates are 0.9 mL min−1 (residence time of 44.4 min) since a better ratio yield/STY, as well as lower amounts of by-products, were obtained. The preparation of intermediate 9 was also performed end-to-end in 50% yield using the PTFE loop module (see schemes in the ESI†).
For the synthesis of demethoxycurcumin (3) (Scheme 5), operation 1 was performed in batch using intermediate 9 (4.2 mmol) and B2O3 (2.2 mmol)/B(O-n-Bu)3 (8.4 mmol) in EtOAc (28 mL), and p-hydroxybenzaldehyde (8.4 mmol) was added before pumping (Pump 1, 0.3 mL min−1). A solution of n-BuNH2 (1.40 mmol in 30 mL of EtOAc) was pumped (Pump 2) at 0.3 mL min−1 and the reactor temperature was maintained at 80 °C. Demethoxycurcumin (3) was isolated after column chromatography in 30% yield (0.28 g, STY = 6.2 g per day).
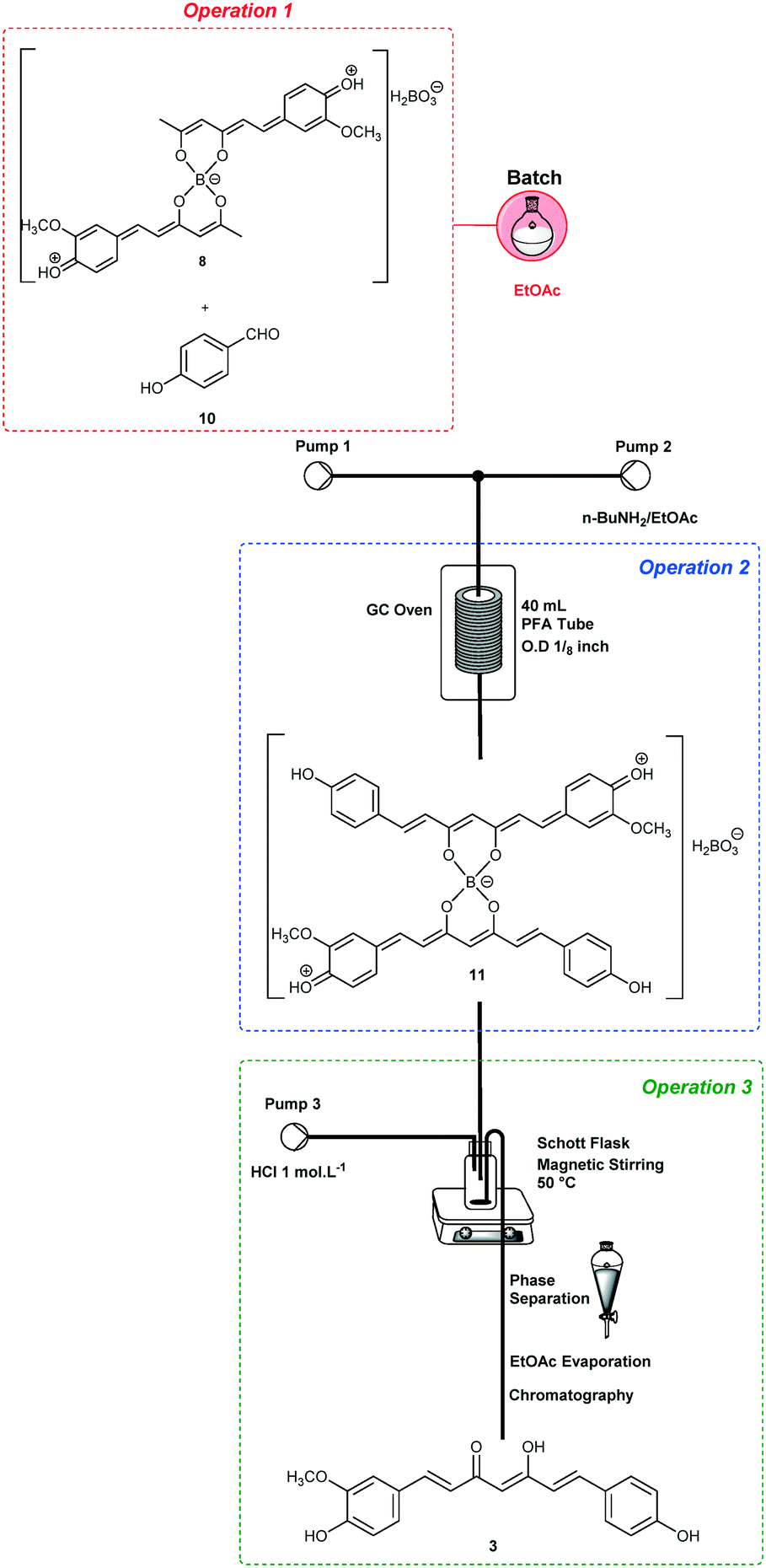 | ||
| Scheme 5 Synthesis of demethoxycurcumin (3). | ||
Attempts to improve the yields of this process were performed by using excesses of p-hydroxybenzaldehyde (10), different reactor temperatures and different amounts of base, but without success.
The end-to-end synthesis of curcuminoid 3 was also tested, but in this case with less success (15% yield). Clogging was observed even after optimizations, making this procedure less adaptable to the end-to-end continuous flow synthesis of 3.
Comparison between fully-batch and integrated protocols
Classical batch procedures were carried out to synthesize curcumin (1) and its natural analogues 2 and 3 for comparison. The integrated protocols developed here offer advantages over the batch-only procedures, such as (I) optimization of the workspace, (II) reduction of large quantities of glassware in a wide range of sizes, (III) shorter reaction times, (IV) reproducibility, (V) process intensification and better STY in a safer manner, (VI) exact control of residence time, (VII) energy saving, and most importantly, (VIII) lower amounts of by-products and (IX) highly pure curcuminoids obtained only by crystallisation.Curcumin (1) was prepared in batch starting from 10 mmol of acetylacetone (4) and adequate amounts of the other reagents, in order to compare with the integrated conditions (entry 2, Table 3). The three operations in batch were performed in 4.5 h disregarding work-up times, and curcumin (1) was obtained in 69% yield (2.5 g, STY: 12.0 g per day). This is the largest scale reported in batch in the literature,45,48 and a 1 L round bottom flask is necessary for this procedure. Compared to the optimized integrated conditions, we observed a clear advantage (60% yield, STY: 24.1 g per day) in a well-controlled process. Similarly, curcuminoid 2 was prepared in 66% yield (2.0 g, STY: 9.6 g per day) using a batch-only protocol against the integrated protocol which gave 2 in 60% yield (0.96 g, STY: 20.6 g per day). Also, an improved result was achieved for the preparation of intermediate 9, which was obtained in 40% yield (0.47 g, STY: 7.5 g per day) in a batch-only protocol against 59% yield (0.69 g, STY: 22.0 g per day) using the integrated process. In this last case, since temperature control is crucial for only one condensation reaction, the flow conditions are significant for the selectivity. We emphasize that all the purifications performed after the integrated processes are simple since low amounts of by-products remain.
To summarize the achieved results, we have compiled the yields of both batch plus continuous flow conditions and fully assisted by continuous flow processes, where it is possible to see the superiority of the integrated protocol with operation 1 in batch and operations 2 and 3 in flow, over the three operations under continuous flow. This opens up possibilities for future numbering up approaches and therefore applications in industry after checking the techno-economic and commercial viability of these protocols on larger scales.49–51
Conclusions
In this study, we have shown that integration of batch and flow processes are in some cases better than the end-to-end flow process. Of course, this is a specific case which involves the formation of slurry in the first step for the synthesis of curcuminoids, but the recent literature reinforces the idea about integration for better results. Our results contribute by showing that continuous flow and batch procedures improve the results for scalability, safety and other benefits, changing the way we perform chemical syntheses (Table 5).| Methodologies | Curcumin (1) | Bis-demethoxycurcumin (2) | Demethoxycurcumin (3) | Intermediate (9) | ||||
|---|---|---|---|---|---|---|---|---|
| Yield (%) | STY (g per day) | Yield (%) | STY (g per day) | Yield (%) | STY (g per day) | Yield (%) | STY (g per day) | |
| Batch + continuous flow | 60 | 24.1 | 60 | 20.6 | 30 | 6.2 | 59 | 18.0 |
| Continuous flow | 51 | 20.4 | 45 | 15.0 | 15 | 3.1 | 50 | 15.1 |
Experimental
General methods
Starting materials were purchased from Sigma-Aldrich and were used without further purification. Curcumin (1) and its derivatives 2 and 3 were obtained based in the batch procedures described in the literature.10,45,52 For thin layer chromatography (TLC), we used 60 F254 (0.25 mm) silica gel. When necessary, column chromatography using 70–230 mesh silica gel was performed for product purifications.1H NMR (400 MHz) spectra were recorded in a Bruker Avance 400 spectrometer at 25 °C, using DMSO-d6. Chemical shifts are quoted in ppm relative to tetramethylsilane (TMS) using the residual solvent peak as a reference. The following notations are used for the 1H NMR spectral splitting patterns: singlet (s), doublet (d), triplet (t) and multiplet (m), and the coupling constants (J) are given in Hertz.
All continuous flow experiments were carried out using an Asia Syringe Pump with blue syringes (500 μL and 1000 μL), a PFA in-house engineered reactor and a back-pressure regulator at 40 psi.
Synthesis of curcuminoids 1 and 2 using the integrated protocols
:water, curcumin (1) was obtained as an orange solid, and bis-demethoxycurcumin (2) was obtained as a reddish orange solid.
Curcumin (1): 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.84 (s, 6H), 6.06 (s, 1H), 6.74 (d, J = 15.8 Hz, 2H), 6.81 (d, J = 8.2 Hz, 2H), 7.14 (m, 2H), 7.32 (s, 2H), 7.53 (d, J = 15.8 Hz), 9.66 (s, 2H), 16.40 (s, 1H). m.p. 180–181 °C (lit. 181–182 °C).53 Elemental analysis calcd (%) for C21H20O6: C 68.47, H 5.47; found: C 68.20, H 5.08.
Bis-demethoxycurcumin (2): 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.04 (s, 1H), 6.67 (d, J = 15.8 Hz, 2H), 6.81 (d, J = 8.7 Hz, 4H), 7.53–7.58 (m, 6H), 10.05 (s, broad, 2H), 16.39 (s, broad, 1H). m.p. 229–230 °C (lit. 231–232 °C).54
Synthesis of intermediate 9 using the integrated protocols
Operation 2
The previous mixture was pumped at 0.45 mL min−1 through a T-piece where it encountered a solution of n-BuNH2 in EtOAc pumped at 0.45 mL min−1 (3.0 mmol in 30 mL of EtOAc) before passing through the temperature-controlled PFA coil reactor (40 mL).:EtOAc (1.5:1) as the eluent. Compound 9 was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.12 (s, 3H), 3.82 (s, 3H), 5.84 (s, 1H), 6.62 (d, J = 15.8 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 8.2 Hz, 1H), 7.29 (s, 1H), 7.47 (d, J = 15.8 Hz, 1H), 9.77 (s, broad, 1H). m.p. 143–144 °C (lit. 146–147 °C).55
Synthesis of curcuminoid 3 using the integrated protocols
:EtOAc (1.5:1) as the eluent. Curcuminoid 3 was obtained as a dark orange solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.84 (s, 3H), 6.05 (s, 1H), 6.67 (d, J = 15.8 Hz, 1H), 6.74 (d, J = 15.8 Hz, 1H), 6.81 (d, 3H), 7.16 (m, 1H), 7.32 (s, 1H), 7.52–7.58 (m, 4H), 9.65 (s, broad, 1H), 10.05 (s, broad, 1H), 16.40 (s, broad, 1H). m.p. 172–173 °C (lit. 175–176 °C).54
Acknowledgements
The authors would like to thank the São Paulo Research Foundation FAPESP (2015/21110-4, 2015/11801-0 2013/07276-1, and 2011/13993-2) for financial support, and also CAPES and CNPq for fellowships.References
- B. B. Aggarwal, C. Sundaram and N. Malani, Adv. Exp. Med. Biol., 2007, 595, 1–75 CrossRef PubMed.
- T. Esatbeyoglu, P. Huebbe, I. M. A. Ernst, D. Chin, A. E. Wagner and G. Rimbach, Angew. Chem., Int. Ed., 2012, 51, 5308–5332 CrossRef CAS PubMed.
- W. M. Weber, L. A. Hunsaker, S. F. Abcouwer, L. M. Deck and D. L. Vander Jagt, Bioorg. Med. Chem., 2005, 13, 3811–3820 CrossRef CAS PubMed.
- P. Anand, S. G. Thomas, A. B. Kunnumakkara, C. Sundaram, K. B. Harikumar, B. Sung, S. T. Tharakan, K. Misra, I. K. Priyadarsini, K. N. Rajasekharan and B. B. Aggarwal, Biochem. Pharmacol., 2008, 76, 1590–1611 CrossRef CAS PubMed.
- N. Handler, W. Jaeger, H. Puschacher, K. Leisser and T. Erker, Chem. Pharm. Bull., 2007, 55, 64–71 CrossRef CAS PubMed.
- G. K. Jayaprakasha, P. S. Negi, C. Anandharamakrishnan and K. K. Sakariah, Z. Naturforsch., C: J. Biosci., 2001, 56, 40–44 CAS.
- L. N. Dovigo, J. C. Carmello, C. A. de Souza Costa, C. E. Vergani, I. L. Brunetti, V. S. Bagnato and A. C. Pavarina, Med. Mycol., 2013, 51, 243–251 CrossRef CAS PubMed.
- M. Sagnou, K. P. Mitsopoulou, G. Koliopoulos, M. Pelecanou, E. A. Couladouros and A. Michaelakis, Acta Trop., 2012, 123, 190–195 CrossRef CAS PubMed.
- N. Tortik, A. Spaeth and K. Plaetzer, Photochem. Photobiol. Sci., 2014, 13, 1402–1409 CAS.
- H. Pabon, Recl. Trav. Chim. Pays-Bas, 1964, 83, 379 CrossRef CAS.
- A. Kirschning, W. Solodenko and K. Mennecke, Chem. - Eur. J., 2006, 12, 5972–5990 CrossRef CAS PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138 CrossRef CAS PubMed.
- J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- J. Hartwig, J. B. Metternich, N. Nikbin, A. Kirschning and S. V. Ley, Org. Biomol. Chem., 2014, 12, 3611–3615 CAS.
- P. F. Carneiro, B. Gutmann, R. O. M. A. De Souza and C. O. Kappe, ACS Sustainable Chem. Eng., 2015, 3, 3445–3453 CrossRef CAS.
- T. Razzaq and C. O. Kappe, Chem. - Asian J., 2010, 5, 1274–1289 CAS.
- K. T. de Oliveira, L. Z. Miller and D. T. McQuade, RSC Adv., 2016, 6, 12717–12725 RSC.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- F. J. Strauss, D. Cantillo, J. Guerra and C. O. Kappe, React. Chem. Eng., 2016, 1, 472–476 CAS.
- P. B. Momo, B. S. Bellete, T. J. Brocksom, R. O. M. A. de Souza and K. T. de Oliveira, RSC Adv., 2015, 5, 84350–84355 RSC.
- R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144–148 CrossRef CAS PubMed.
- B. J. Reizman, Y.-M. Wang, S. L. Buchwald and K. F. Jensen, React. Chem. Eng., 2016, 1, 658–666 CAS.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- J. A. Glaser, Clean Technol. Environ. Policy, 2013, 15, 205–211 CrossRef.
- K. Gilmore, D. Kopetzki, J. W. Lee, Z. Horváth, D. T. McQuade, A. Seidel-Morgenstern and P. Seeberger, Chem. Commun., 2014, 50, 12652–12655 RSC.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- F. R. Carrel, K. Geyer, J. D. C. Codée and P. H. Seeberger, Org. Lett., 2007, 9, 2285–2288 CrossRef CAS PubMed.
- M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, ChemBioChem, 2014, 15, 713–720 CrossRef CAS PubMed.
- T. Broom, M. Hughes, B. G. Szczepankiewicz, K. Ace, B. Hagger, G. Lacking, R. Chima, G. Marchbank, G. Alford, P. Evans, C. Cunningham, J. C. Roberts, R. B. Perni, M. Berry, A. Rutter and S. A. Watson, Org. Process Res. Dev., 2014, 18, 1354–1359 CrossRef CAS.
- D. O. Nogueira, S. P. de Souza, R. A. C. Leão, L. S. M. Miranda and R. O. M. A. de Souza, RSC Adv., 2015, 5, 20945–20950 RSC.
- F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708–3722 RSC.
- G. C. O. Silva, J. R. Correa, M. O. Rodrigues, H. G. O. Alvim, B. C. Guido, C. C. Gatto, K. A. Wanderley, M. Fioramonte, F. C. Gozzo, R. O. M. A. de Souza and B. A. D. Neto, RSC Adv., 2015, 5, 48506–48515 RSC.
- V. D. Pinho, B. Gutmann, L. S. M. Miranda, R. O. M. A. de Souza and C. O. Kappe, J. Org. Chem., 2014, 79, 1555–1562 CrossRef CAS PubMed.
- D. E. Fitzpatrick and S. V. Ley, React. Chem. Eng., 2016, 1, 629–635 CAS.
- R. De Oliveira, A. S. De Miranda, B. Reichart, T. Glasnov, C. O. Kappe, R. C. Simon, W. Kroutil, L. S. M. Miranda, I. C. R. Leal and R. O. M. A. De Souza, J. Mol. Catal. B: Enzym., 2014, 104, 101–107 CrossRef.
- M. Baumann and I. R. Baxendale, J. Org. Chem., 2015, 80, 10806–10816 CrossRef CAS PubMed.
- B. Gutmann, U. Weigl, D. P. Cox and C. O. Kappe, Chem. - Eur. J., 2016, 22, 10393–10398 CrossRef CAS PubMed.
- C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed.
- N. H. Park, T. J. Senter and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 1–6 Search PubMed.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS PubMed.
- S. Newton, C. F. Carter, C. M. Pearson, L. de C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRef CAS PubMed.
- GrandViewResearchInc, http://www.grandviewresearch.com/industry-analysis/turmeric-extract-curcumin-market, 2016 (Accesed 13-11-2017).
- W. Wichitnithad, U. Nimmannit, S. Wacharasindhu and P. Rojsitthisak, Molecules, 2011, 16, 1888–1900 CrossRef CAS PubMed.
- K. F. Jensen, B. J. Reizman and S. G. Newman, Lab Chip, 2014, 14, 3206–3212 RSC.
- Y. Su, K. Kuijpers, V. Hessel and T. Noël, React. Chem. Eng., 2016, 1, 73–81 CAS.
- W. Buadonpri, W. Wichitnithad, P. Rojsitthisak and P. Towiwat, J. Health Res., 2009, 23, 11–16 CAS.
- H. G. Jolliffe and D. I. Gerogiorgis, Comput. Chem. Eng., 2016, 91, 269–288 CrossRef CAS.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- S. D. Schaber, D. I. Gerogiorgis, R. Ramachandran, J. M. B. Evans, P. I. Barton and B. L. Trout, Ind. Eng. Chem. Res., 2011, 50, 10083–10092 CrossRef CAS.
- A. Pal, B. Sung, B. A. Bhanu Prasad, P. T. Schuber, S. Prasad, B. B. Aggarwal and W. G. Bornmann, Bioorg. Med. Chem., 2014, 22, 435–439 CrossRef CAS PubMed.
- J. Y. Feng, Z. Q. Liu and J. Agric, Food Chem., 2009, 57, 11041–11046 CrossRef CAS PubMed.
- G. K. Jayaprakasha, L. J. M. Rao and K. K. Sakariah, J. Agric. Food Chem., 2002, 50, 3668–3672 CrossRef CAS PubMed.
- L. Lin, Q. Shi, A. K. Nyarko, K. F. Bastow, C. C. Wu, C. Y. Su, C. C. Y. Shih and K. H. Lee, J. Med. Chem., 2006, 49, 3963–3972 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00207b |
| This journal is © The Royal Society of Chemistry 2017 |