
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogenation of organic compounds using continuous flow and microreactor technology
David
Cantillo
ab and
C. Oliver
Kappe
*ab
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010, Graz, Austria. E-mail: oliver.kappe@uni-graz.at
bResearch Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
First published on 16th November 2016
Abstract
The halogenation of organic substrates is one the most important transformations in organic synthesis. The most straightforward, inexpensive and atom economic halogenations involve the use of elemental halogens (X2) or hydrogen halides (HX). However, X2 and HX reagents are highly reactive, toxic and corrosive materials. Halogenations using these reagents are usually very fast and exothermic reactions, in which selectivity issues occur. Using continuous flow chemistry halogenations involving X2 and HX can be performed in a safe and controllable manner. Reagents can be accurately dosed even for gas/liquid reactions, and exotherms are easily controlled. Hazardous chemicals can be readily quenched in line avoiding any undesired exposures and significantly enhancing the process safety.
 David Cantillo | David Cantillo obtained his PhD from the University of Extremadura, Spain in 2011. His PhD focused on the experimental and theoretical study of 1,3-dipolar cycloadditions of mesoionic compounds. In 2011 he joined the Kappe Group at the University of Graz as a postdoctoral associate. Since this time his research interests and experience involve computational chemistry, flow photochemistry, photoredox catalysis, flow chemistry in extreme process windows and multistep API synthesis. |
 C. Oliver Kappe | C. Oliver Kappe is Professor of Chemistry at the University of Graz (Austria) and Key Researcher at the Research Center Pharmaceutical Engineering GmbH (RCPE). He studied at the University of Graz with Professor Gert Kollenz. After postdoctoral research with Professor Curt Wentrup at the University of Queensland and with Professor Albert Padwa at Emory University, he moved back to the University of Graz in 1996 to start his independent academic career. In 1999 he became Associate Professor and in 2011 was appointed Full Professor for “Technology of Organic Synthesis”. His research interests involve continuous-flow chemistry, API manufacturing, and process intensification technologies. Since 2011 he has been Editor-in-Chief of the Journal of Flow Chemistry. |
Introduction
The introduction of one or more halogen atoms into organic molecules is arguably one of the most important and often required transformations in organic synthesis.1,2 Halogenated compounds constitute ca. 20% of the repertory of active pharmaceutical ingredients (APIs), and 30% of current agrochemicals.3 Halogenated compounds also find applications as dyes, flame retardants, imaging agents in medical diagnosis, and in materials science.1–3 The significance of halogenated compounds is increasing rapidly. Thus, ca. 80% of newly developed agrochemicals during the first decade of the 21st century contained one or more halogen atoms.3 Notably, 7 out of the top-10 best-selling drugs in the US in 2014 were halogenated compounds (some examples are shown in Fig. 1).4 Almost 5000 naturally occurring organic halogenated compounds have been identified so far.5 The beneficial effects of a carbon–halogen bonds within the structure of organic compounds, such as increased durability, stability towards biodegradation and oxidation, and higher biological activity and membrane permeability, are shared by synthetic APIs and natural compound.6,7 A notable difference is the relative distribution of the halogen elements among synthetic and naturally occurring organic compounds (Fig. 2). While chlorinated and brominated derivatives predominate in natural metabolites, with a relatively small number of compounds containing fluorine and iodine, synthetic APIs and agrochemicals with fluorine and chlorine are more abundant.3,8 Despite the high incidence of chlorine and fluorine atoms in the final structure of APIs, brominations and iodinations are very often carried out for the generation of synthetic intermediates.9 Halogenated building blocks have seen increased relevance owing to the development of cross-coupling chemistry over the past four decades.10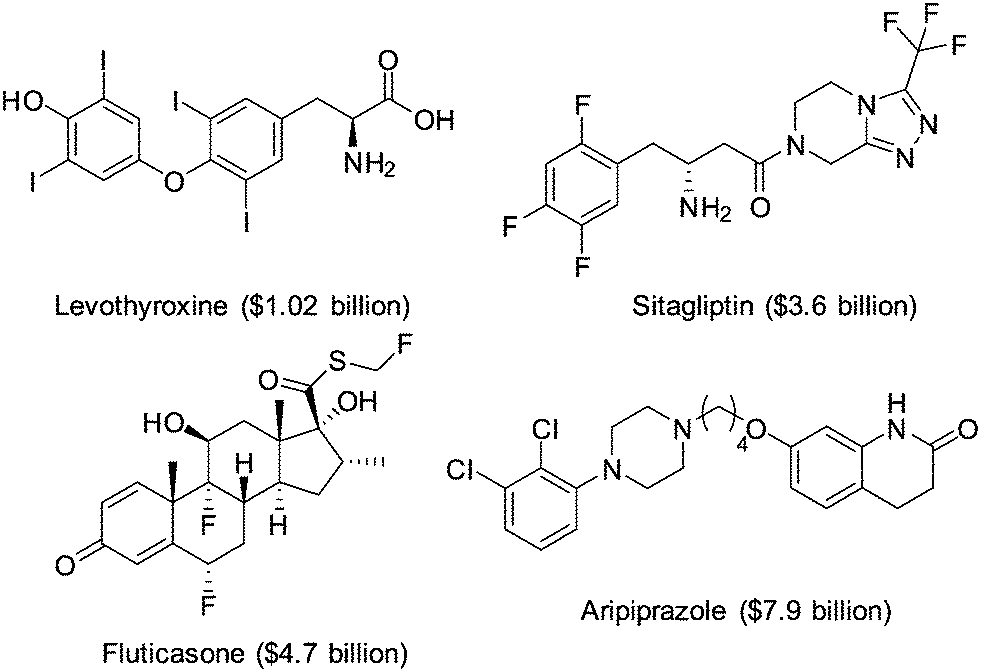 | ||
| Fig. 1 Examples of halogen compounds ranked among the top-10 selling drugs in the US in 2014. Sales are shown in brackets. | ||
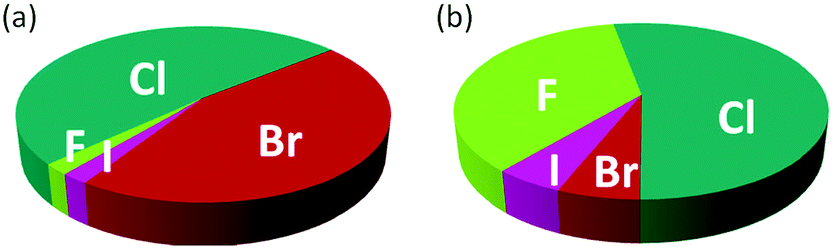 | ||
| Fig. 2 Distribution of halogens within the structure of naturally occurring compounds (a) and synthetic APIs and agrochemicals (b). | ||
Unlike biochemical processes that use relatively harmless anionic species (e.g. Cl−, Br−) as halogen sources for the halogenation of organic compounds, normally via an oxyhalogenation pathway,11 organic chemists usually employ highly reactive, toxic, and corrosive reagents to carry out halogenation reactions.1,2 Elemental halogens (X2) and hydrogen halides (HX) are the most convenient halogenating agents from the viewpoint of atom economy and simplicity. However, the highly corrosive and toxic nature of these reagents, as well as their high reactivity, are important drawbacks for their use in organic synthesis. Many examples of less reactive, easy to handle halogenating agents have been developed to avoid the use of X2 and HX reagents.12 Popular examples are N-bromosuccinimide (NBS) as a substituent of Br2, or the N-fluorobenzenesulfonimide (NFSI) and diethylaminosulfur trifluoride (DAST) fluorinating agents. Yet, preparation of these reagents ultimately requires the use of the corresponding elemental halogen or other highly reactive species.
During the past decade continuous flow- and microreactor technology have been shown to be powerful tools for the safe and controllable use of hazardous or highly reactive reagents in organic synthesis.13–16 The small channel dimensions characteristic of microreactors ensure a very high heat exchange efficiency that suppresses the formation of hotspots, temperature gradients or accumulation of heat.17 Thus, high reaction selectivity and enhanced safety can be achieved even for very fast and highly exothermic reactions. The excellent heat and mass transfer characteristics of microreactors, together with the fact that the reaction is resolved along the length of the reaction channel, enables a precise control of the residence time of intermediates or products by a thermal or chemical quench of the solution.18 Highly reactive, toxic, or explosive intermediates can be generated in situ and consumed within the reactor channel by combining multiple reagent streams. Thus, syntheses that were previously “forbidden” due to safety reasons or even reactions simply not possible in batch are possible to handle in a safe and controllable manner.16
Not surprisingly, during the past years the above mentioned benefits of continuous flow and microreactor technology have been applied to many halogenation reactions, were toxic and highly reactive reagents are usually utilized. Halogenations, especially fluorinations and chlorinations, where selectivity is a major issue in batch have especially benefited from the exceptional heat and mass transfer achievable under continuous flow conditions. Improved mixing and mass transfer efficiency in gas/liquid biphasic reactions utilizing F2 or Cl2 as halogenation reagents, or photochemical halogenations where the small channel dimensions of flow reactors ensure intense and uniform irradiation, are examples where translating a process from batch to flow substantially improves reaction efficiency and selectivity, as well as process safety.
Review articles focusing entirely on fluorinations in flow have been published in the past years.19,20 An early survey of the literature on fluorinations, chlorinations, and brominations in microreactors covering the literature until 2004 was published by the Hessel group.21 The current review analyzes the literature on continuous flow halogenations published during the past two decades, essentially from the flourish of microreactor technology at the beginning of the 2000s. Examples in which continuous flow technology has been successfully applied to halogenation reactions difficult to perform under classical batch conditions are described, emphasizing those cases dealing with highly reactive reagents or very exothermic reactions where batch protocols would be practically “forbidden” on industrial scale.
Fluorination
Organofluorine compounds have become one of the most important and common class of substances among pharmaceuticals and agrochemicals (cf.Fig. 2b).22 Decoration of an organic compound with fluorine atoms typically results in a significant enhancement of its biological properties.23 Increased bioavailability, lipophilicity, and metabolic stability otherwise difficult to obtain are typically acquired by the compounds after the introduction of one or more fluorine groups.23 The growing interest in this area has motivated intense research efforts for the development of novel catalytic methods and greener and more selective fluorinating agents.24 Elemental fluorine (F2) and hydrogen fluoride HF are among the most difficult to handle reagents in organic synthesis.25 F2 is the most powerful oxidant known, and both HF and F2 are highly corrosive and toxic compounds. Even some N-fluoro derivatives that are generally easier to handle are highly reactive, hazardous compounds. Fluorination reactions, especially those using F2 directly as fluorine source, are very exothermic and difficult to control. Lack of reaction selectivity is a common drawback. Not surprisingly, many research groups have studied the fluorination of organic materials using microreactor technology. The work in this area is extensive, and several reviews on the topic have been published.19,20Diethylaminosulfur trifluoride (DAST) is a widely utilized nucleophilic fluorinating agent. Its relatively high stability (it can be stored under inert atmosphere in the freezer for prolonged periods) and liquid form makes this reagent a convenient alternative for the lab-scale fluorination of alcohols, aldehydes and ketones. However, the use of DAST on large scale is very limited due to its propensity to detonate at temperatures above 90 °C. Under continuous flow conditions, the reaction exotherm can be readily dissipated and DAST fluorinations can be safely performed and scaled as demonstrated by the groups of Seeberger26 and Ley.27 In both cases the substrate and reagent in CH2Cl2 solution were mixed and reacted using fluoropolymer tubing as residence time unit, thus avoiding any degradation or corrosion of the reactor. At 70 °C several alcohols, carboxylic acids, and aldehydes were converted into the corresponding fluorinated derivatives after a residence time of 16 min (Scheme 1a).26 Unreacted DAST and the HF generated as side product were quenched inline using aqueous NaHCO3 before the reactor output. Contact with HF was therefore totally avoided, increasing the safety of the procedure. An interesting application of this methodology was reported by Bayer Schering Pharma for the geminal difluorination of 17-keto steroid 1 (Scheme 1b).28 The flow process could be performed safely with a productivity of 5–10 kg per day of the desired difluoro-derivative 2.
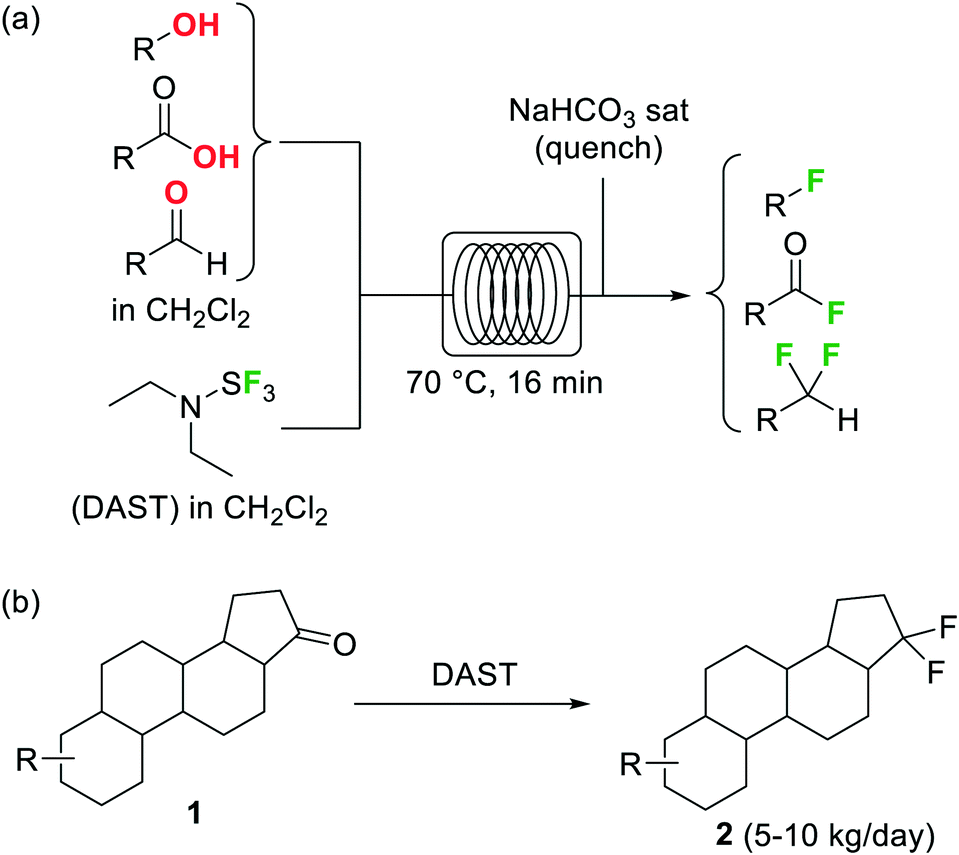 | ||
| Scheme 1 Deoxyfluorination reactions using DAST in a microreactor. | ||
Halogenation reactions, in which careful temperature control is typically required, perform well in continuous flow devices owing to their enhanced heat transfer. An illustrative example described by the Ley group is the diastereoselective electrophilic fluorination of the lithium enolate of carbonyl compound 3 with N-fluorobenzenesulfonimide (NFSI) (Scheme 2).29 The batch process required cooling of all reagents before addition at −78 °C. Simple addition of NFSI at room temperature to a −78 °C solution of the lithium enolate intermediate resulted in <50% conversion. Using a cryo-flow reactor at −60 °C, 88% conversion for the desired α-fluorocarbonyl product 4 (62% isolated) was achieved. Formation of the enolate from the substrate and LiHMDS could be performed at room temperature under continuous flow conditions.
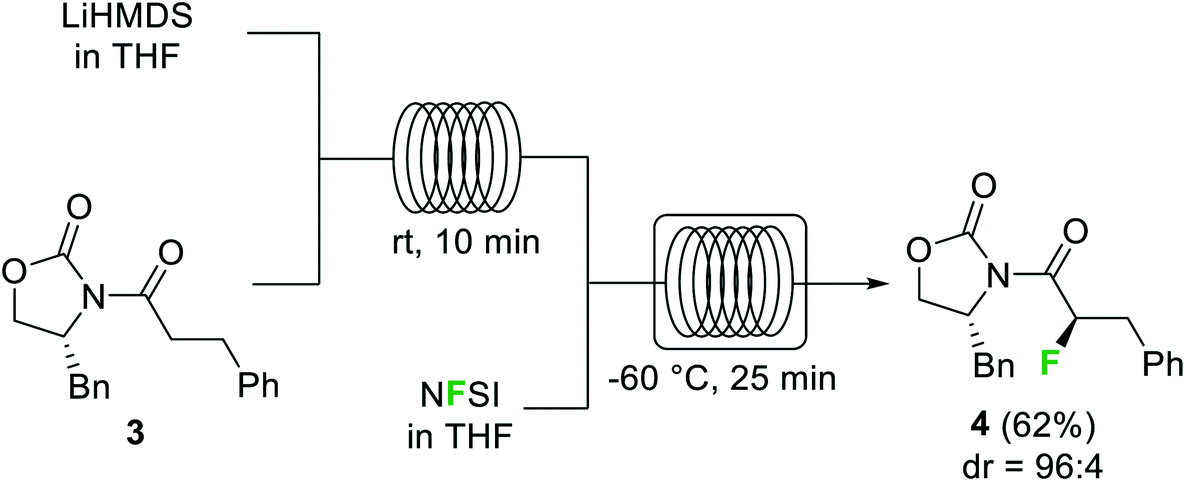 | ||
| Scheme 2 α-Fluorination of carbonyls using NFSI in a cryo-flow reactor. | ||
Photochemical fluorination reactions, especially trifluoromethylations, have gained popularity during the past 5 years.25 Light induced reactions are significantly enhanced under continuous flow conditions due to the intense and uniform irradiation achieved within the capillary tubing in a flow reactor.30 In this context, various continuous flow trifluoromethylation protocols have been described.31 The direct fluorination of benzylic compounds 5 (Scheme 3) using Selectfluor as fluorine source and xanthone as catalyst using a photochemical flow reactor has recently been reported.32 The reactor consisted of a black-light (365 nm) compact fluorescent lamp (CFL) as light source and transparent fluorinated ethylene propylene (FEP) tubing. The combination of xanthone with black light irradiation resulted in a very fast fluorination process. Several benzyl fluorinated compounds were synthetized using this method, including biologically relevant examples 6a and 6b (Scheme 3).
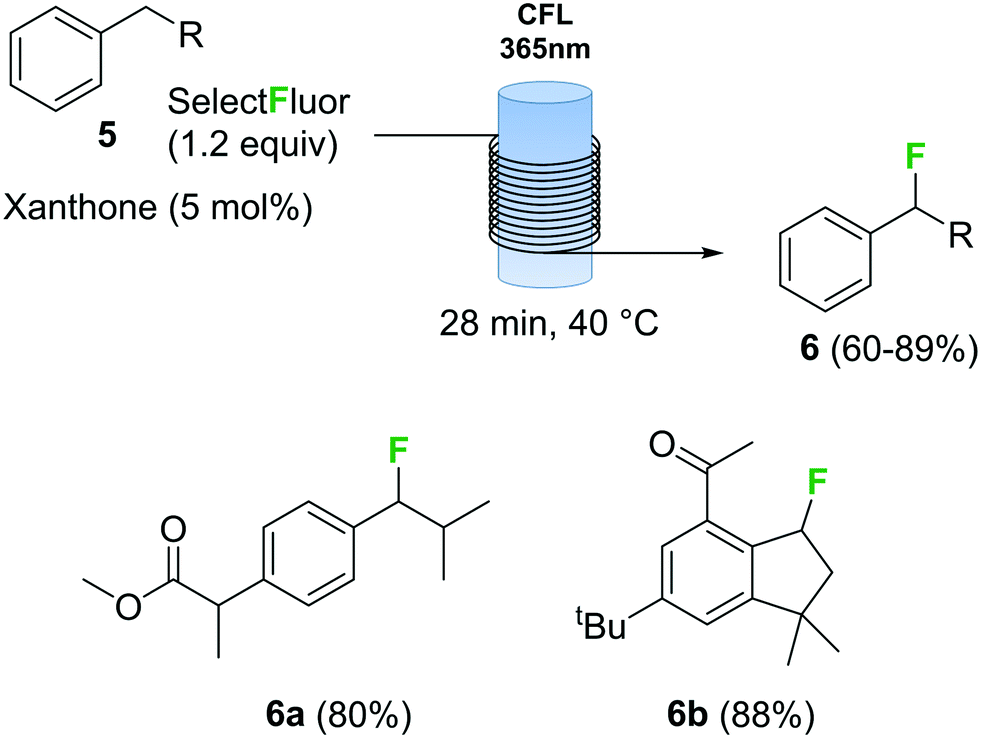 | ||
| Scheme 3 Continuous flow photochemical fluorination of benzylic compounds with Selectfluor. | ||
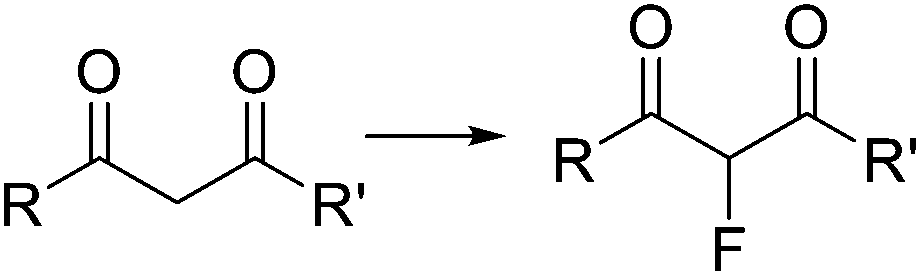
Fluorination of organic compounds using F2 as reagent is the preferred choice from the viewpoint of simplicity and atom economy. However, the extremely high reactivity of this reagent, in addition to the problems associated with its highly corrosive nature, are limiting factors for its implementation in organic synthesis. From the dawn of microreactor technology at the beginning of this century F2 fluorination of organic compounds has been approached by many research groups, including the groups of Hessel, Jensen, and the extensive contributions by Chambers/Sanford (see ref. 23 and references therein).33–38 In the pioneering work of Chambers and Spink, a microreactor fabricated from a block of nickel (or copper) specifically designed to perform fluorination reactions with elemental fluorine was described.36 The reactor showed excellent performance for the fluorination of β-dicarbonyl compounds (Scheme 4a). Scale-out of the microreactor using a simple numbering up strategy could be demonstrated for the same reaction using three reactors in parallel (Scheme 4b).37 In a more recent publication the microreactor was applied for the two-step synthesis of 4-fluoropyrazole derivatives 7 (Scheme 4c).38 After fluorination of the β-dicarbonyl compound, an organic hydrazine was introduced into the system through a T-mixer to generate the desired fluorinated pyrazoles in good yields.
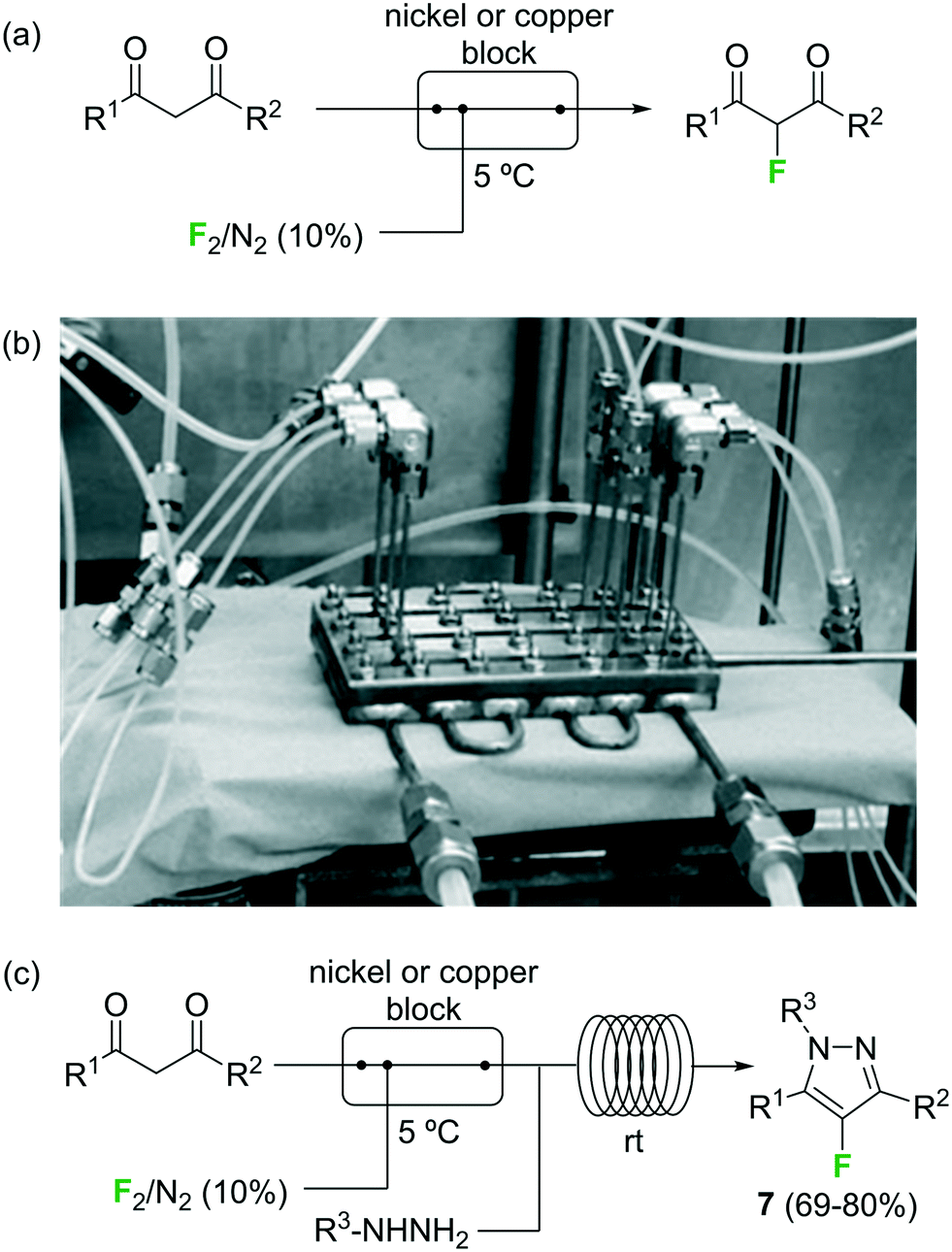 | ||
| Scheme 4 Reactions with elemental fluorine. Image 4b reproduced with permission from ref. 37: R. D. Chambers, M. A. Fox, D. Holling, T. Nakano, T. Okazoe and G. Sandford, Lab Chip, 2005, 5, 191–198. Copyright© 2005, Royal Society of Chemistry. | ||
Large scale fluorination of a 1,3-dicarbonyl compound commercialized by Axyntis was developed by scientist of the MEPI (Maison Européene des Procédés Innovants, France) using a sintered silicon carbide (SiC) reactor (Fig. 3).39 The excellent heat transfer capacity and corrosion resistance of the SiC material is perfectly suited for the highly exothermic reaction on a large scale. Using a ca. 2 M solution of the substrate in MeCN/AcOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and F2/N2 in a gas/liquid flow regime excellent conversions were obtained resulting in a productivity for the desired product of approximately 200 g h−1.
:1) and F2/N2 in a gas/liquid flow regime excellent conversions were obtained resulting in a productivity for the desired product of approximately 200 g h−1.
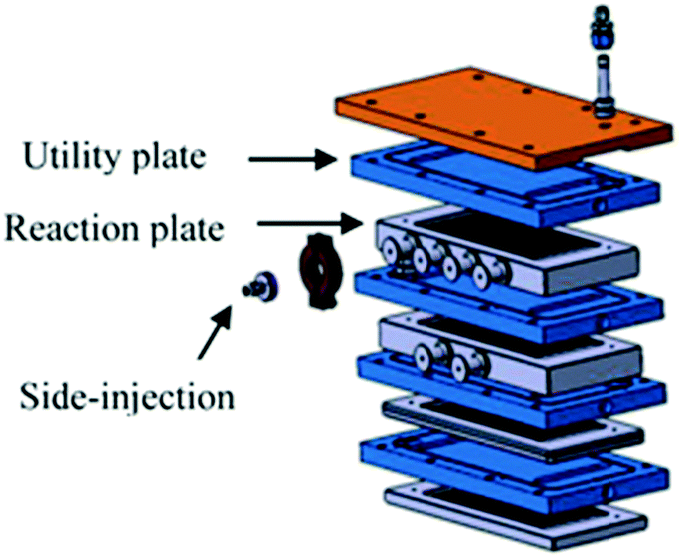 | ||
| Fig. 3 Continuous flow SiC reactor. Reproduced with permission from ref. 39: S. Elgue, A. Conte, C. Gourdon and Y. Bastard, Chim. Oggi, 2012, 30(4), 18–21. Copyright© 2012 Teknoscienze Publisher. | ||
Chlorination
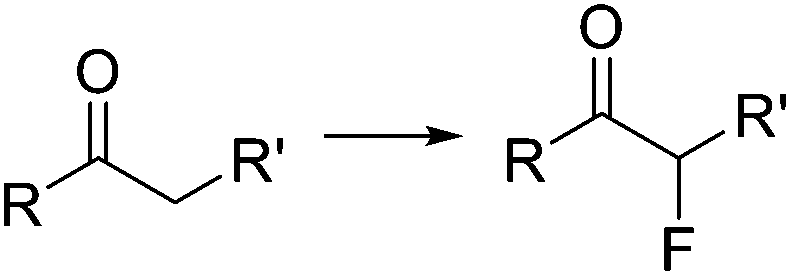
The importance of chlorinated organic compounds in the field of agrochemicals and pharmaceuticals is comparable to that for organofluorines regarding improved pharmacological properties. In addition, chlorinated compounds are useful building blocks for the generation of more complex structures, as well as for the preparation of plastic materials or dyes.40One of the most widely utilized procedures for the generation of organochlorines is the transformation of alcohols into the corresponding chlorides by substitution of the OH group. Chlorinating reagents such as SOCl2 or PCl5 are often used for this reaction. The greenest alternative for these chlorodehydroxylation reactions is the use of HCl as reagent, as demonstrated by the group of Hessel.41 Using directly anhydrous HCl gas, the neat alcohols were converted into the corresponding chlorides at 120 °C and 10 bar pressure (Scheme 5). Special precautions had to be taken to keep the HCl dosing part of the system anhydrous, as moisture in the HCl produced significant damage of metal parts such as the mass flow controller. In a previous work the use of aqueous concentrated HCl instead of the gas had been demonstrated as a suitable alternative,42 although a larger excess of HCl (3 equiv.) was required.
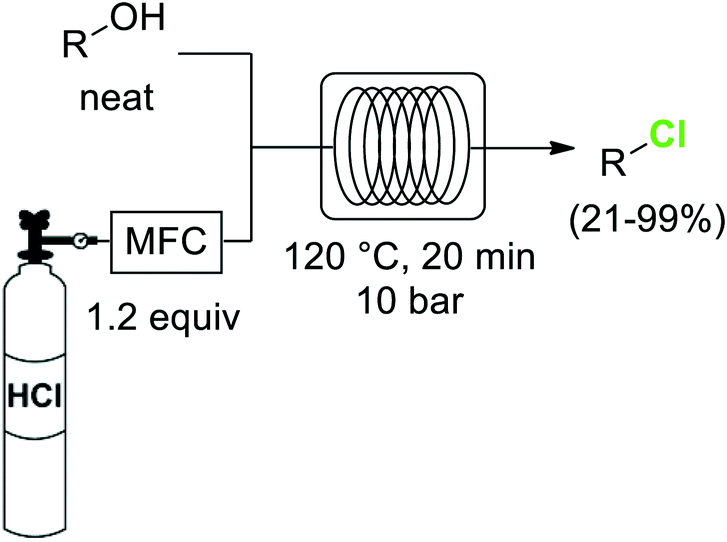 | ||
| Scheme 5 Conversion of alcohols to chlorides using HCl gas. | ||
Elemental chlorine (Cl2) is a powerful and atom-economic chlorinating agent for organic compounds, as well as one of the cheapest oxidizing agents available.43,44 Despite the importance of Cl2 as an oxidizing and chlorinating agent in organic synthesis, its extremely high reactivity limits its use in many instances, as undesired overreactions and exotherms typically occur. The efficient mixing and enhanced mass transfer achieved under continuous flow conditions minimize these problems. Moreover, the small channel dimensions in a microreactor are a key advantage in the case of photochlorination reactions, where intense and uniform irradiation of the reaction mixture is required. This was demonstrated by the pioneering work of the group of Jähnisch,45 where a falling film microreactor was used to carry out the gas/liquid reaction of toluene-2,3-diiocyanate (8) with Cl2 under UV irradiation (Scheme 6a). The falling film reactor was equipped with a quartz window (Scheme 6b) and a 1000 W xenon lamp (190–250 nm). Compared to batch mode higher selectivity for the desired benzyl chloride 9 was obtained. The space-time yield achieved in flow (400 mol L−1 h−1) was also much higher than in batch (1.3 mol L−1 h−1) due to the small reaction volume and short residence time required. A similar strategy was used by the group of Ryu for the radical chlorination of cycloalkanes.46 The microreactor consisted of stainless steel microchannels covered by a Pyrex glass window, that was irradiated with a 15 W black light CFL. The neat cycloalkanes were introduced into the reactor using a syringe pump. A moderate yield (20%) was obtained for the generation of chlorocyclohexane. Alternatively, SO2Cl2 was used as chlorine source.46 In this case a solution of the reagent in the corresponding cycloalkane was directly pumped through the microreactor (Scheme 6c) under the same conditions. Plug flow derived from the release of SO2 could be suppressed by installing a back pressure regulator at the end of the flow setup.
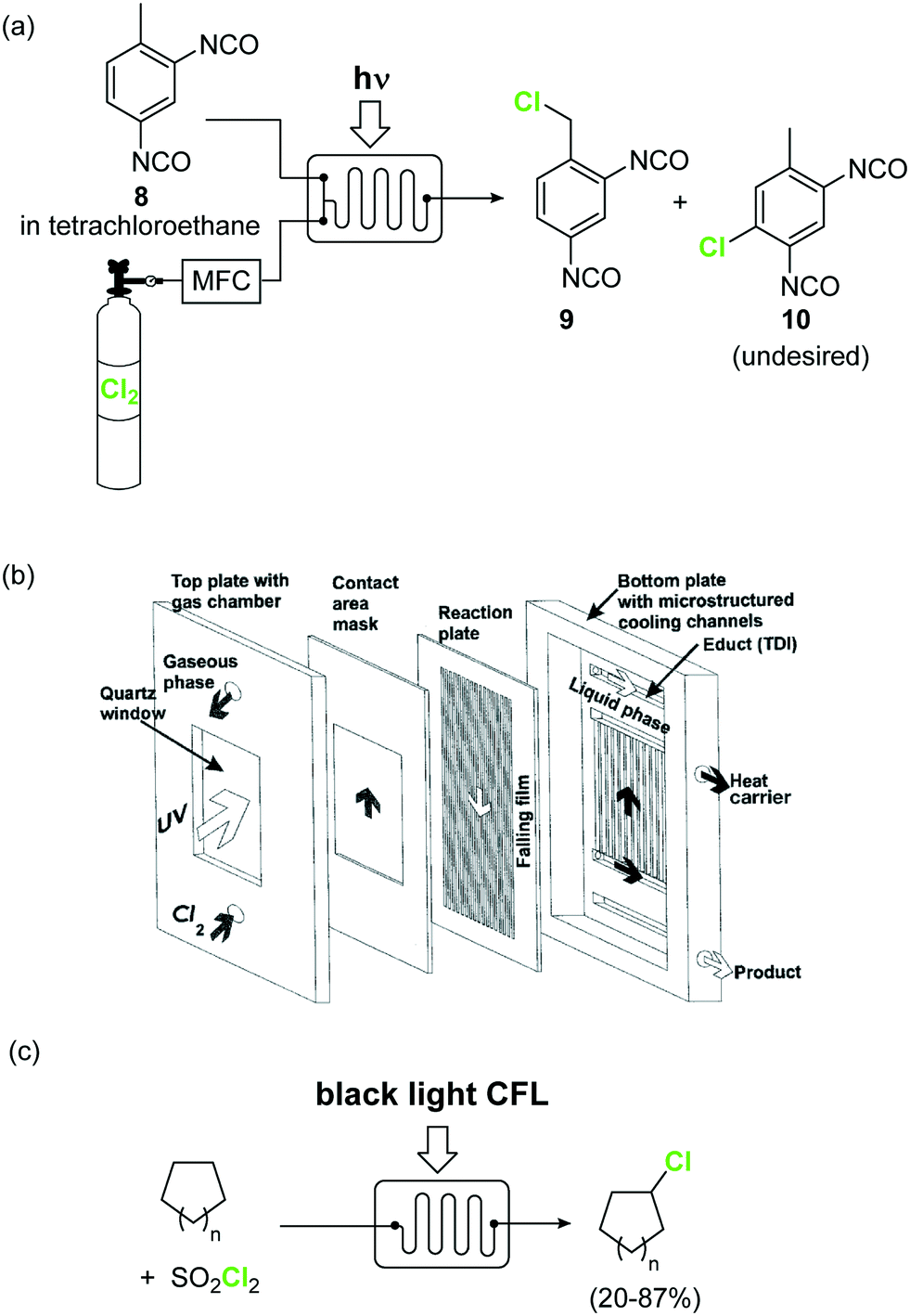 | ||
| Scheme 6 Photochemical chlorinations using Cl2 gas. Image 6b reproduced with permission from ref. 45: H. Ehrich, D. Linke, K. Morgenschweis, M. Baerns and K. Jähnisch, Chimia, 2002, 56, 647–653. Copyright© 2002 Swiss Chemical Society. | ||
Apart from the problems associated with the high reactivity of Cl2 towards organic compounds, handling and storage of Cl2 gas cylinders in organic synthesis labs and production facilities is an important safety issue on his own.47 Use, handling, and transportation regulations for this substance are strictly regulated. Sites where Cl2 gas is used normally need to be isolated, and the personnel specifically equipped and trained for the handling of chlorine.47 To solve this problems a continuous flow generator of Cl2 based on the reaction of NaOCl and HCl was developed.48 The simple continuous flow setup consisted of three peristaltic pumps (Vapourtec E-Series) that pumped aqueous solutions of NaOCl and HCl, in addition to CHCl3 (Scheme 7). Upon mixing of the aqueous solutions, Cl2 gas was rapidly formed and extracted to the CHCl3 phase. A hydrophobic membrane based continuous liquid–liquid separator was then used to separate the organic phase containing Cl2 in solution from the aqueous phase. A yield of 90% based on NaOCl was obtained.48 The chlorine generator was tested for several organic reactions, including the selective oxidation of primary alcohols using the chlorine–pyridine complex, and the chlorination of silanes and benzylic compounds. For the photochemical chlorination of benzylic compound the Cl2 solution in CHCl3 obtained from the membrane separator was mixed with the neat toluene derivative (Scheme 7) before entering a photoreactor equipped with a Hg lamp (150 W). After a residence time in the photoreactor of ca. 15 min the reaction mixture was quenched at the reactor output with isopropanol. Excellent yields were obtained for several benzyl chlorides. NaOCl has also been used directly for the continuous chlorination of amines.49
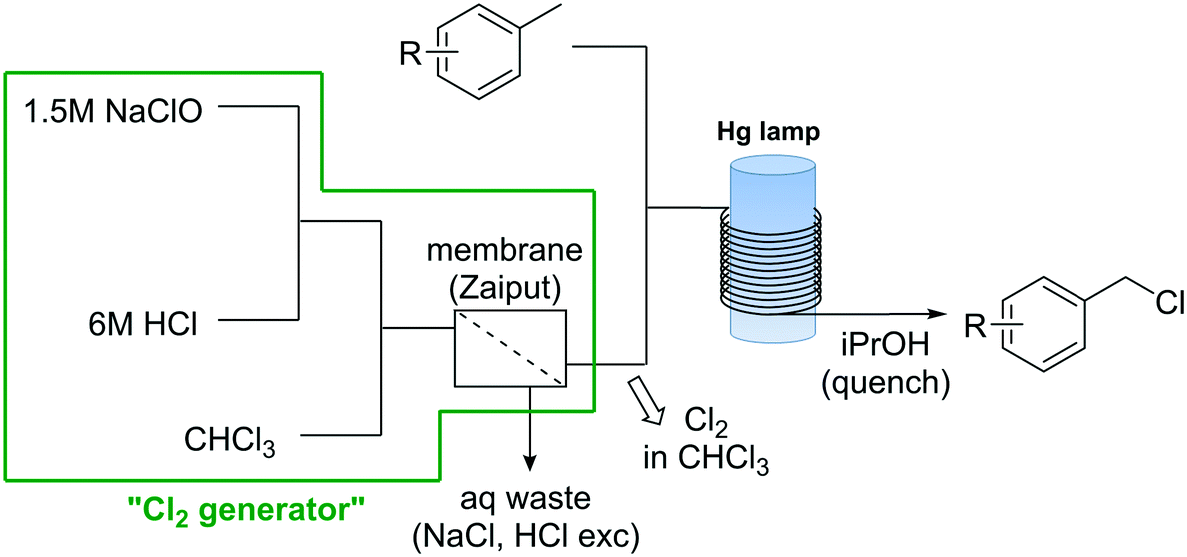 | ||
| Scheme 7 Photochemical chlorination of toluene derivatives with in situ generated Cl2. | ||
Bromination
Bromination of organic compounds is one of the most important transformations in organic synthesis. In addition to the many uses of brominated compounds as dyes, flame-retardants, and active pharmaceutical ingredients, organobromides serve as essential building blocks for the preparation of complex molecules.50 The good leaving group character of the bromide anion has made alkyl bromides a classical source of electrophilic synthons. Aryl bromides have also become highly important building blocks since the development of cross-coupling chemistry.Introduction of bromine into organic molecules can be accomplished using elemental bromine (Br2). Br2 is an extremely corrosive and toxic reagent in both liquid and vapor form.51 Many other easy to handle brominating agents have been described,50N-bromosuccinimide being the most popular example. An early example of bromination using liquid Br2 in a microreactor was provided by the group of Hessel (Scheme 8a).52 The neat substrate was mixed with liquid Br2 using a micromixer, before entering a residence time unit which consisted of PTFE tubing. Solvent-free bromination of thiophene to produce the 2,5-dibromo derivative 11 (Scheme 8a) could be performed in less than one second with high selectivity and yields up to 86%. As the reaction is extremely fast (<1 s) important mixing effects were found on the reaction selectivity. Thus a selectivity of 88% was achieved using a micromixer with an internal active mixing geometry, while using a normal T-junction only 72% selectivity for the desired 2,5-dibromothiophene was obtained. Similar approaches have been used for the α-bromination of acetophenone53 and the 1,2-dibromination of alkenes.54 Catalytic electrophilic bromination with Br2 and in situ generated FeBr3 was performed in continuous flow for the preparation of 2,4,5-trifluorobromobenzene 13 (Scheme 8b),55 an important intermediate for the synthesis of peptides and fluorescent reagents. The FeBr3 catalyst was generated by passing the Br2 solution through a PTFE tubing (i.d. 4 mm) packed with iron Dixon rings (0.69 × 0.69 mm, 0.2 mm diameter). The FeBr3 amount was controlled by the packed bed temperature and flow rate. At 70 °C and a flow rate of 0.2 ml min−1 a concentration corresponding to 10 mol% FeBr3 was achieved. The solution was then mixed with the substrate using a T-junction before entering a reactor (FEP tubing) at 70 °C. After 4 min residence time the reaction mixture was quenched with aqueous Na2S2O3. A yield of 78% was obtained for the desired compound 13.55 Elemental bromine was used for the in situ generation of KOBr, a useful reagent for the bromination of methylsulfones by the group of Stevens (Scheme 8c).56 KOBr was rapidly generated upon mixing of neat Br2 with aqueous 5 M KOH. The resulting solution of KOBr in water was pumped and mixed with a solution of the substrate in toluene, generating a liquid/liquid segmented flow. Tetrabutylammonium bromide (TBAB) was used as ion exchange catalyst to enhance the reaction in the biphasic system.56 After 3 min residence time in a glass chip reactor at 85 °C quantitative yield and a productivity of >50 g per day for the desired tribromomethyl sulfones 14 was obtained.
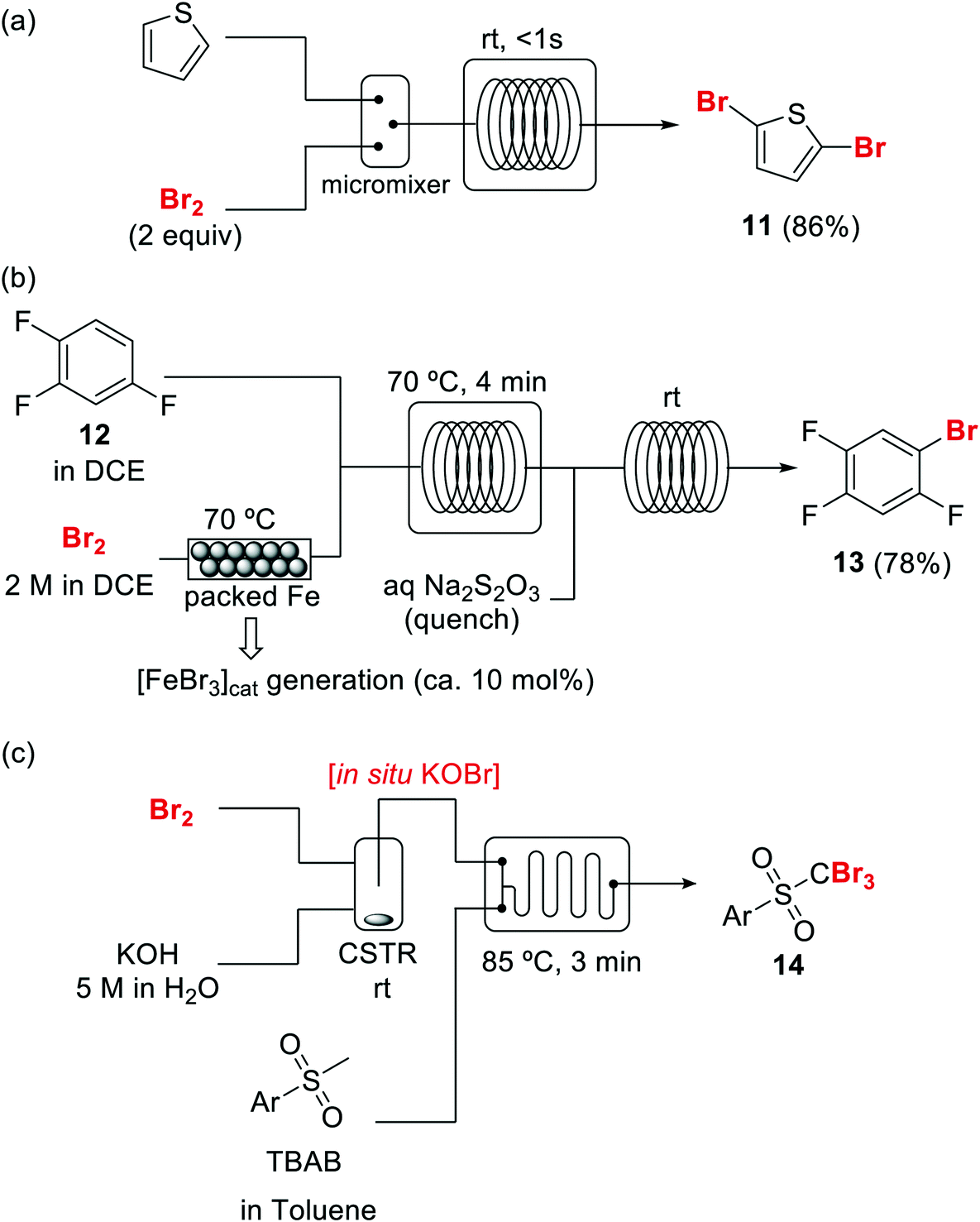 | ||
| Scheme 8 Brominations with elemental bromine in flow (DCE = dichloroethane, CSTR = continuous stirred tank reactor). | ||
Photochemical brominations using elemental bromine can be significantly accelerated in a microreactor, as demonstrated by the group of Ryu.57 Using a glass chip microreactor irradiated with a 360 nm LED (1.95 W) lamp a series of benzylic compounds could be brominated within seconds to minutes at room temperature (Scheme 9a). A single feed containing the substrate and Br2 (1.2 equiv.) dissolved in CCl4 was introduced into the photoreactor using a syringe pump. The reaction mixture was quenched at the reaction output by collection over an aqueous solution of Na2S2O3. Excellent selectivity for the monobrominated product was obtained. A similar approach using sun light to irradiate the flow reactor was reported by In and Park.58 Photochemical benzylic brominations with NBS in continuous flow have also been described.59,60 A green protocol using acetonitrile as solvent instead of chlorinated compounds such as CCl4 was achieved using a simple flow photochemical reactor built with transparent FEP tubing wrapped around a household CFL (Scheme 9b).59 With only 1.05 equiv. NBS a wide variety of benzylic compounds was converted to their corresponding bromides, with good to excellent yields being obtained in all cases. A throughput of 180 mmol h−1 was achieved. The same principle was used for the flow photochemical bromination to produce the 5-bromoethylpyrimidine 11, a precursor of Rosuvastatin, in a collaboration between scientist of Sandoz and the University of Ljubljana.60 Compared to batch mode, lower levels of side products and shortened reaction times were obtained. An important drawback of the use of NBS as brominating agent is the formation of equivalent amounts of succinimide as side product. Apart from complicating the isolation of the brominated products and formation of undesired waste, the presence of succinimide can create problems of clogging under continuous flow conditions, as it is insoluble in many organic solvents. An elegant approach to avoid these problems consists of extracting the succinimide into an aqueous phase using a liquid/liquid segmented flow regime, as demonstrated by O′Brien and Cooper.61 This method was applied to the bromination of enaminones 16 (Scheme 9c). An aqueous solution of Na2S2O3 and K2CO3 was mixed with the reaction mixture stream in dichloromethane. The excess of NBS was quenched and succiminide was extracted to the aqueous phase which could be conveniently separated using a liquid/liquid separator. The resulting organic phase contained the desired bromide 17 which could be isolated in good to excellent yields by simply evaporating the organic solvent. No traces of succinimide could be detected in the isolated products by 1H NMR.61
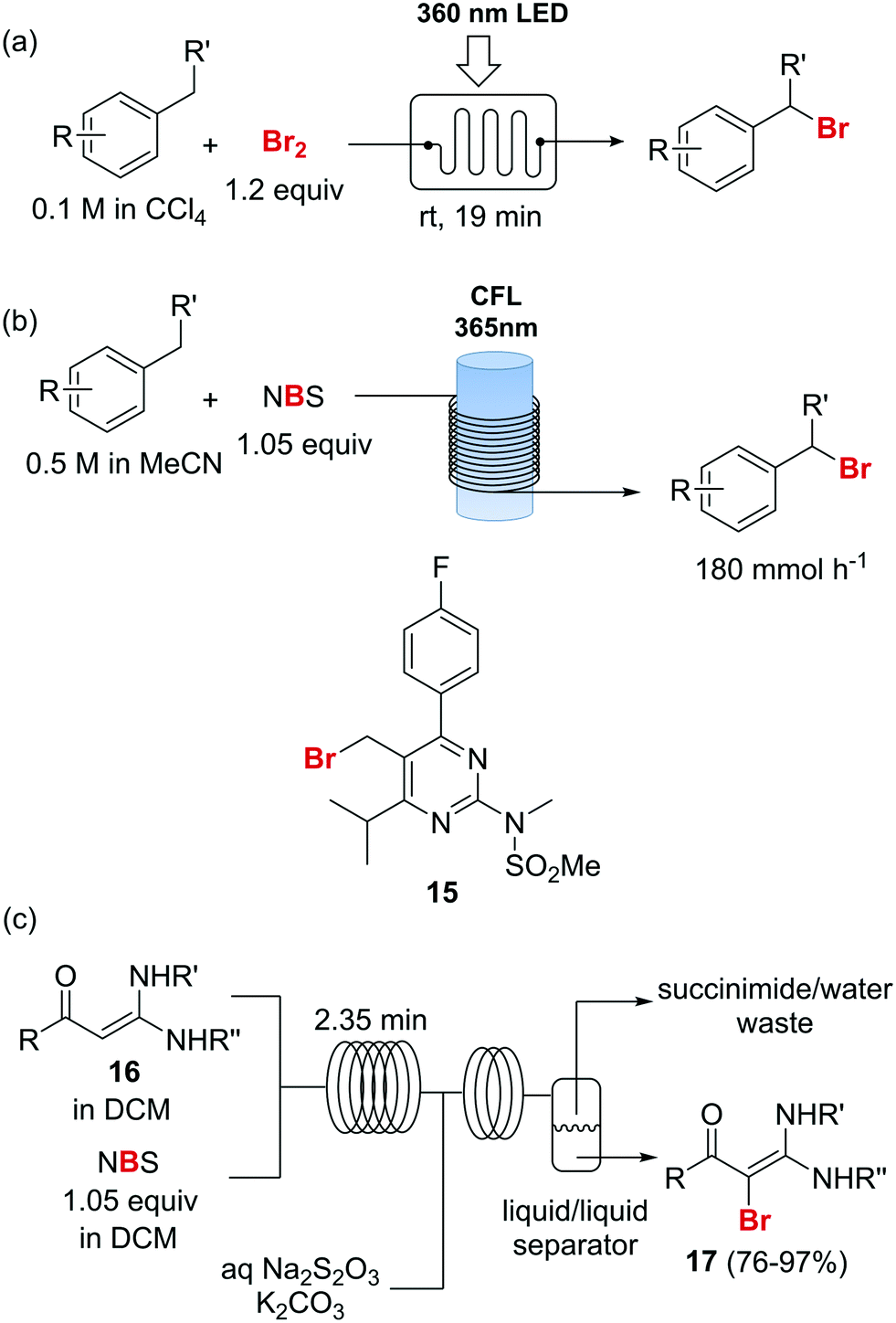 | ||
| Scheme 9 Photochemical brominations and use of N-bromosuccinimide. | ||
The preparation of vicinal 1,2-bromine azide derivatives can be achieved by treating olefins with bromine azide (BrN3). However, BrN3 is an extremely toxic and explosive compound, and its use in organic synthesis constitutes a very challenging task.62 A continuous flow protocol for the safe generation and use of BrN3 that enabled the preparation of vicinal 1,2-bromiazides was recently developed.63 The hazardous reagent was generated in situ using NaBr and NaN3 as bromine and azide sources, and Oxone as oxidizing agent. The continuous flow setup was based on a three-feed approach (Scheme 10). Two of the feeds contained aqueous solutions of NaBr and NaN3, and Oxone, respectively. A third feed contained the substrate 18 in an organic solvent (EtOAc or CH2Cl2). Upon mixing of the three feeds, a liquid/liquid segmented flow was created. The BrN3 rapidly formed in the aqueous phase was extracted into the organic phase containing the substrate. 1,2-Addition to the olefin was enhanced by irradiation with a black-light CFL (max. 365 nm) in continuous flow. The desired 1,2-bromine azides 19 were obtained with excellent purity, and could be isolated by simply separating the organic phase and evaporating the solvent. Good to excellent yields were obtained. A similar procedure was subsequently developed for the generation of chlorine azide (ClN3) and its addition to olefins.64
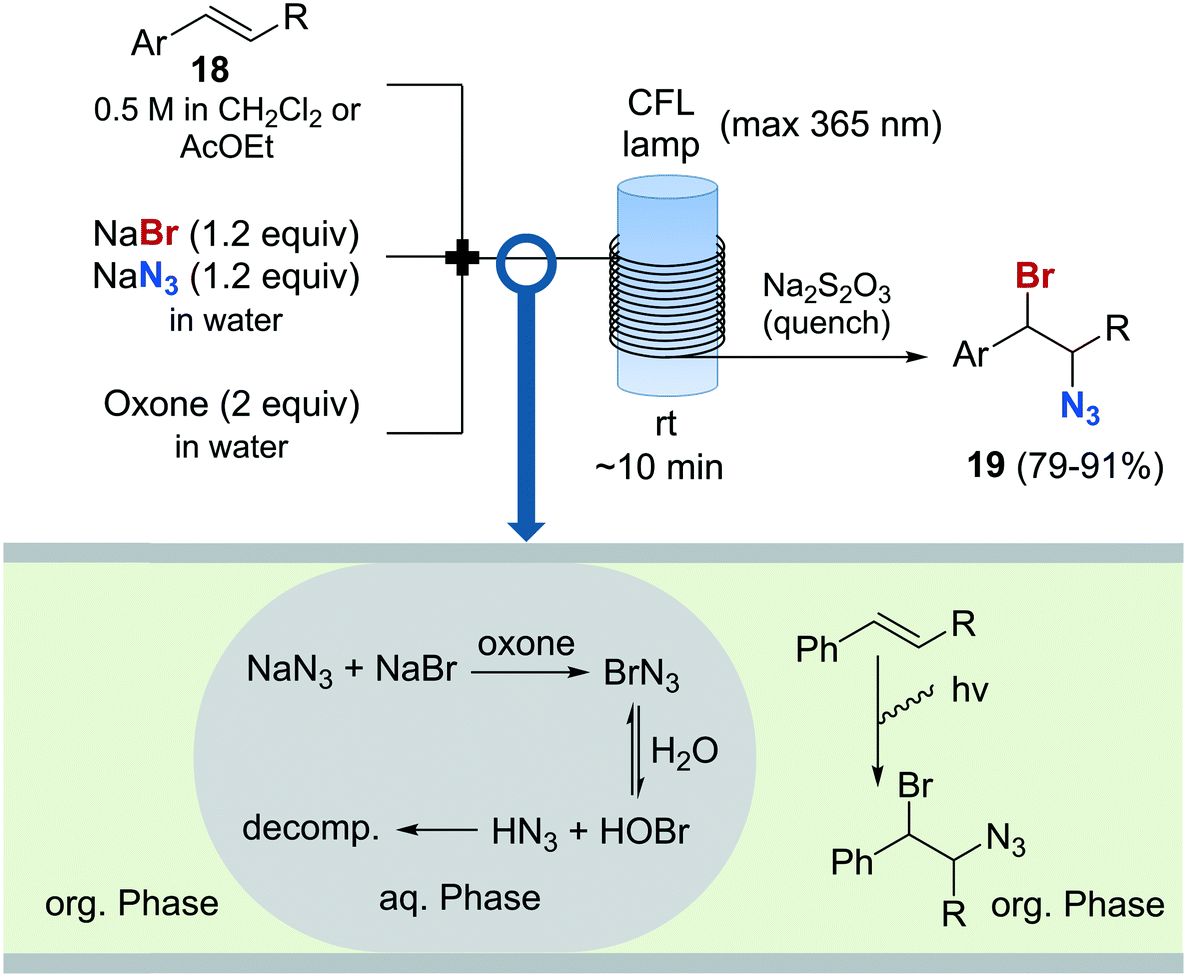 | ||
| Scheme 10 Generation and use of bromine azide and its addition to olefins. | ||
Iodination
Organoiodine compounds are uncommon in nature and not very frequently found in pharmaceuticals and agrochemicals (cf.Fig. 2). Until 2014, 184 natural iodinated compounds have been described.65 Yet, essential iodinated organic compounds such as thyroid hormones thyroxine T4 and triiodothyronine are produced by living organisms. Levothyroxine, a synthetic thyroid hormone used for the treatment of hypothyroidism, is one of the best selling drugs worldwide. Undoubtedly, iodination of organic compounds is most important for the generation of intermediates and useful building blocks for the preparation of more complex structures.Iodination of organic materials using microreactor technology was first described by the group of Yoshida.66 Highly reactive I+ species were generated by electrolysis of I2 and used for the electrophilic iodination of methoxybenzenes. The extremely fast reaction gave a 5:2 mixture of mono- and di-iodinated products in batch. The poor selectivity was ascribed to the inefficient mixing. Using a micromixer significantly improved the selectivities for the desired monoiodinated compounds (Scheme 11). Thus, a solution of the substrate in MeCN was mixed with the solution of I+ in MeCN generated electrochemically using a micromixer with 50 μm channel width cooled at 0 °C, before entering a microtube reactor to provide residence time. Using a flow rate of 3.0 mL for each reactant feed a selectivity of 95% for the monoiodinated compound was obtained. The effect of the flow rates on the reaction selectivity was very significant, decreasing when lower flow rates were applied due to the less efficient mixing (Scheme 11b).
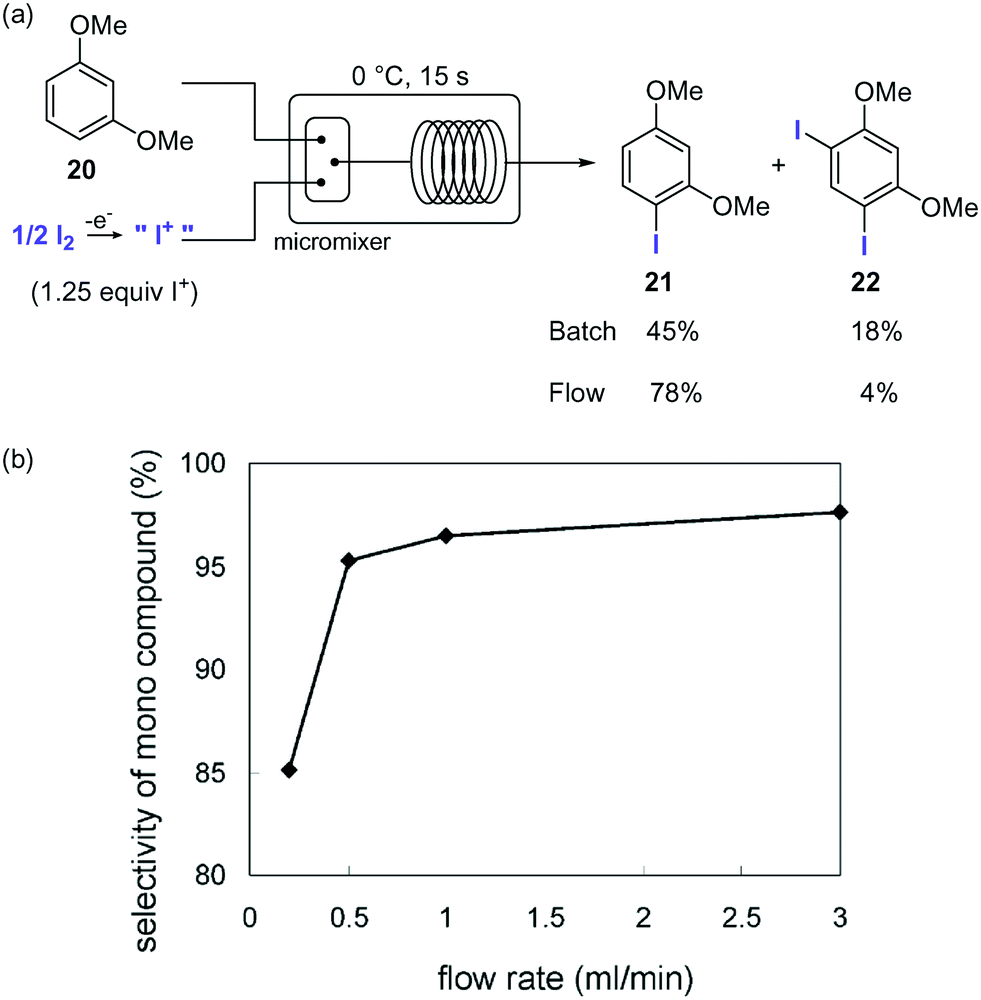 | ||
| Scheme 11 Iodination of aromatics with electrochemically generated I+. | ||
Elemental iodine (I2) has also been employed as a reagent for the direct iodination of aromatic compounds, in particular for the preparation of several 3-iodoindoles of type 24 (Scheme 12a).67 The simple flow setup consisted of two reagent feeds, containing the substrate and Et3N as base in DMF, and a solution of 1.1 equiv. I2 in DMF. The feeds were mixed using a T-mixer and reacted in a residence time unit made of 1 mm i.d. PTFE tubing. After a residence time of only 1 min at room temperature the reaction mixture was quenched with an aqueous solution of Na2S2O3 and NH4OH. Very good isolated yields were obtained for several 3-iodoindoles.67
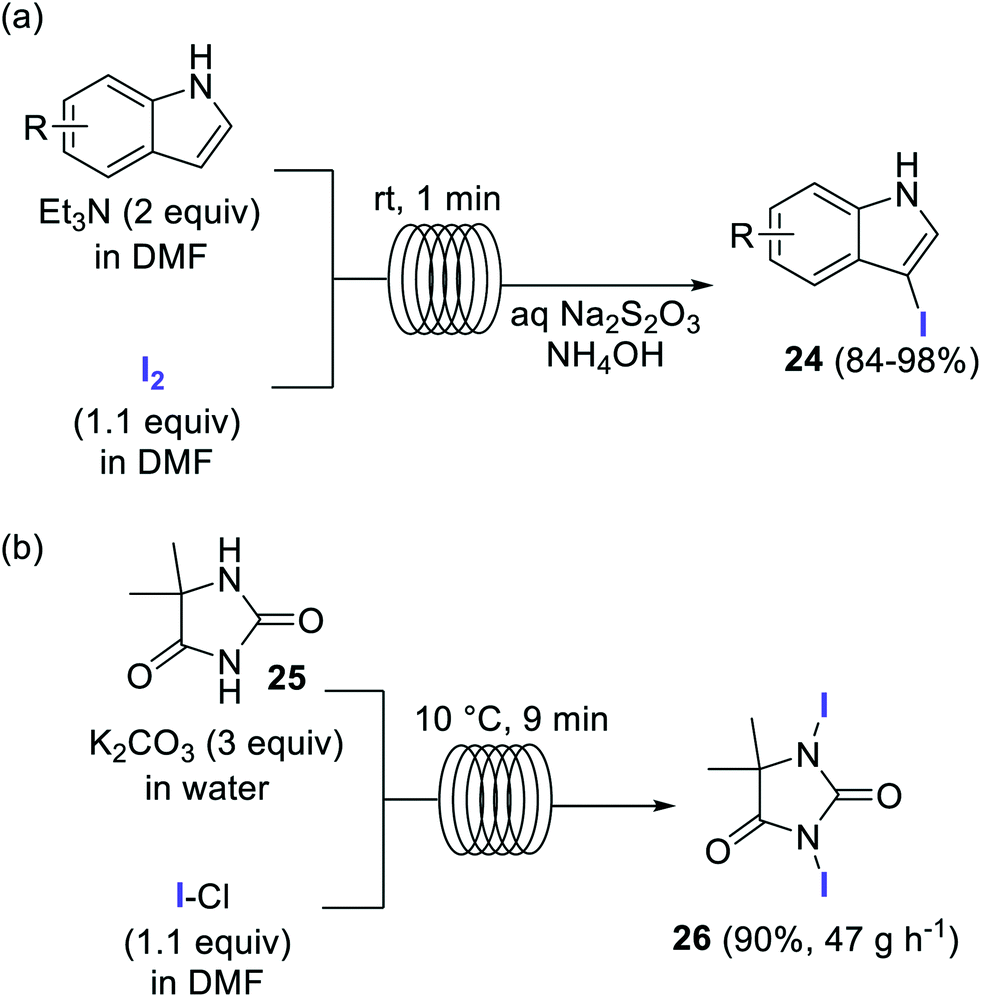 | ||
| Scheme 12 Iodination of organic compounds using I2 and ICl as halogen source. | ||
Iodine monochloride (ICl) is an excellent source of I+ frequently used for the iodination of aromatic compounds.68 Multi-100 gram scale preparation of the iodinating agent 26 was accomplished in a flow reactor using ICl as iodine source (Scheme 12b).69,70 A biphasic system comprising an aqueous solution of the substrate and K2CO3, and ICl in EtOAc was introduced into a tubular reactor at 10 °C. After a residence time of 9 min the slurry obtained from the reactor output was directly filtered using a semicontinuous filtration approach. The reactor was operated for 8 h with a yield of 90% for the pure product (375 g), which corresponds to a productivity of 47 g h−1.
Conclusions
Continuous flow chemistry offers unique opportunities to expand the array of synthetic methods available to the organic chemist. Chemistries and reagents classically “forbidden” under batch conditions, especially on a large scale due to safety reasons, become feasible using continuous flow and microreactor technology. Very fast and exothermic reactions and transformation that require highly reactive, toxic, or corrosive reagents are the prototype examples in which moving from classical batch reactor to flow is the best solution.71 As described in this review, halogenations are excellent examples of reactions that clearly benefit from the use of flow reactors. The most inexpensive, simple and atom economic halogenation procedures involve the use of highly reactive X2 and HX species. Organic chemists have traditionally searched for alternative, less reactive and less corrosive halogenating agents to carry out this type of transformations. Since the advent of flow chemistry numerous examples of highly reactive reagents such as F2 or Cl2 have been successfully utilized for the synthesis of organohalogens in a safe and controllable manner. Table 1 summarizes the reagents, target reactions, and typical experimental conditions for the continuous flow halogenations described herein. Further advantages of continuous flow halogenations arise for reaction involving gas/liquid biphasic mixtures, or photochemical halogenations that require light irradiation to proceed. Gas/liquid reactions are enhanced in continuous flow owing to the enhanced mass transfer and the fast mixing achieved. Thus, reactions involving the use of F2 or Cl2 gas gain in efficiency and selectivity as the reagents are dosed rapidly and homogeneously to the reaction mixture. Photochemical reactions are accelerated when glass chips or transparent tubing are used in flow, as very intense and uniform irradiation can be achieved. This has been demonstrated by several research groups for photochemical chlorinations with Cl2 and brominations with Br2 and NBS. Key to the advances accomplished in this field has been the implementation of microreactors built of corrosion resistant materials such as SiC or fluoropolymers, which allow the use of extremely aggressive halogenation reagents such as HF or F2. The application of these principles to other components of the flow reactors and equipment, such as pumping systems or back-pressure regulators is crucial to enable this technology to succeed on large scale preparations. Yet, a significant hazard often is the handling and storage of large quantities of toxic and corrosive halogenation reagents, even for their use in continuous flow. For that reason, the development of procedures for the continuous generation of halogenation reagents for their use in situ “on-demand” will be of interest in the future.15,48| Halogen source | Transformation | Conditions | Yields (%) | References | |
|---|---|---|---|---|---|
| F | F2/N2 |
|
0–5 °C | 60–80% | 33–35 |
|
0–10 °C | 70–80% | 35, 36, 38, 39 | ||
| DAST |
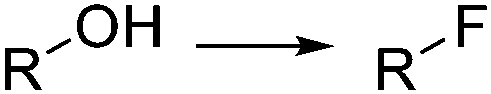
|
70 °C, 15 min | 60–90% | 26, 27 | |

|
70 °C, 15 min | 70–90% | 26, 27 | ||
| NFSI |
|
−60 °C, 20 min | 50–60% | 29 | |
| Selectfluor |
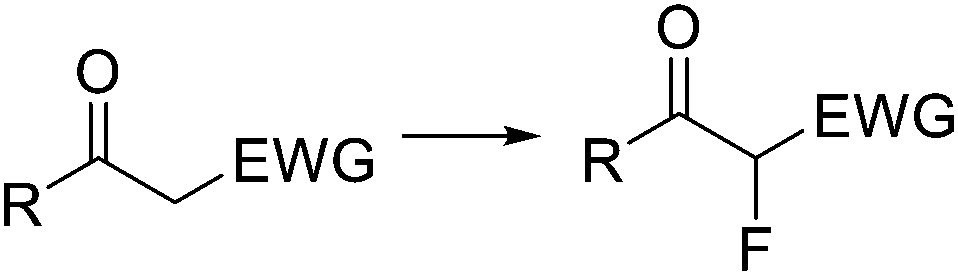
|
100–120 °C, 27 min | 80–90% | 27b | |

|
hν, 25–60 °C, 30 min | 60–90% | 32 | ||
| Cl | Cl2 |
|
hν, 15 min | 80–99% | 45, 48 |

|
rt, 10 min | 96–99% | 48 | ||

|
rt, 15 min | 40–50% | 48 | ||
|
hν, 19 min | 20% | 46 | ||
| SO2Cl2 |
|
hν, 19 min | 20–87% | 46 | |
| NaOCl |

|
rt, 20 min | 84–99% | 49 | |
| HCl |
|
120 °C, 20 min | 21–99% | 41, 42 | |
| Br | Br2 |
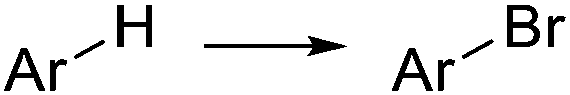
|
rt, 1 s–10 min | 80–90% | 52, 53 |
|
rt, 19 min | 60–99% | 58, 59 | ||
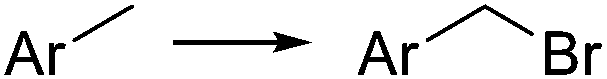
|
rt, 10 s–8 min | 60–80% | 58 | ||
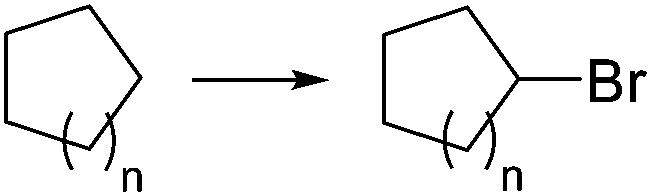
|
rt, 2 min | 80–90% | 55 | ||
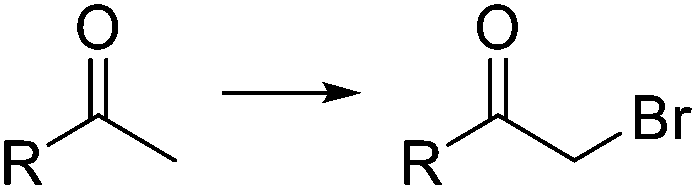
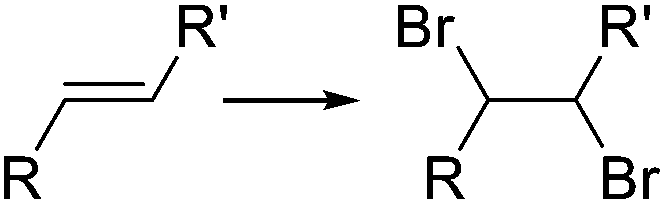
|
20 °C, 1 min | 99% | 54 | ||
| NBS |
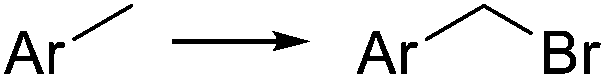
|
hν, 0–60 °C, 15 min | 70–90% | 60, 61 | |
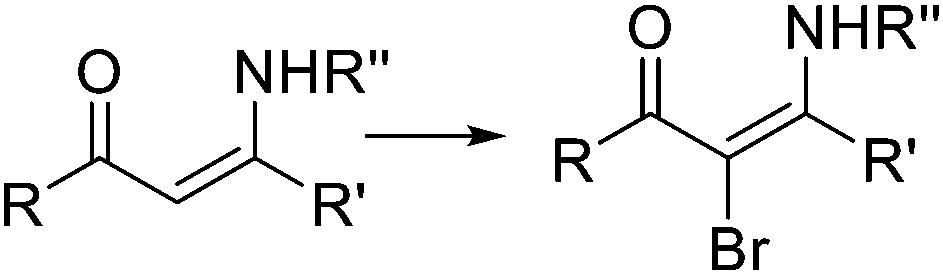
|
rt, 2 min | 80–90% | 62 | ||
| KBrO |
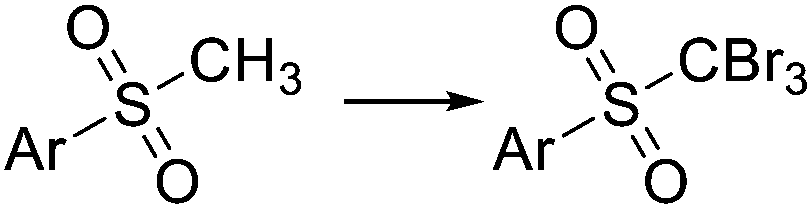
|
85 °C, 3 min | 50–99% | 57 | |
| I | I2 |
|
rt, <1 min | 80–99% | 66, 67 |
| ICl |

|
10 °C, 9 min | 90% | 69 |
The development of the technology necessary for continuous flow synthesis during the past years, with a growing number of commercial equipment available, has enabled a very rapid growth of this area of research. A variety of rather creative chemistries have been explored in flow mode and, more importantly, many chemical reactions and reagents that had been “forbidden” or “forgotten” within the toolbox of organic synthesis due to safety reasons have been revisited with the application of continuous flow technology. Flow chemistry has demonstrated to be able to unconstrain chemical syntheses involving highly reactive reagents or very exothermic reactions, such as halogenations. Further innovations should be based in chemistries specifically developed to be performed in continuous flow.
Acknowledgements
The authors gratefully acknowledge Dr. Bernhard Gutmann for proofreading the manuscript.References
- M. Hudlicky and T. Hudlicky, Formation of carbon-halogen bonds, in The Chemistry of Functional Groups, Supplement D, ed. S. Patai and Z. Rappoport, John Wiley and Sons Ltd., New Jersey, 1983 Search PubMed.
- Y. Sasson, Formation of carbon–halogen bonds (Cl, Br, I), in PATAI's Chemistry of Functional Groups, John Wiley and Sons Ltd, 2009 Search PubMed.
- L. N. Herrera-Rodriguez, F. Khan, K. T. Robins and H.-P. Meyer, Chim. Oggi, 2011, 29(4), 31–33 CAS.
- T. Brown, The 10 Most-Prescribed and Top-Selling Medications, WebMD News Archive, May 8, 2015, http://www.webmd.com/news/20150508/most-prescribed-top-selling-drugs Search PubMed.
- W.-j. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 CrossRef CAS PubMed.
- L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed.
- C. S. Neumann, D. Galonić Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99–109 CrossRef CAS PubMed.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS PubMed.
- A. deMeijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- (a) C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed.
- H. Veisi and R. Ghorbani-Vaghei, Tetrahedron, 2010, 76, 7445–7463 CrossRef.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- P. Poechlauer, S. Braune, B. Dielemans, B. Kaptein, R. Obermüller and M. Thathagar, Chim. Oggi, 2013, 30(4), 51–54 Search PubMed.
- B. Gutmann and C. O. Kappe, Chim. Oggi, 2015, 33(3), 18–24 CAS.
- (a) Flow Chemistry, ed. F. Darvas, V. Hessel and G. Dorman, De Gruyter, Berlin, 2014 Search PubMed; (b) Microreactors in Preparative Chemistry, ed. W. Reschetilowski, Wiley-VCH, Weinheim, 2013 Search PubMed; (c) Microreactors in Organic Synthesis and Catalysis, ed. T. Wirth, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (d) Handbook of Micro Reactors, ed. V. Hessel, J. C. Schouten, A. Renken, Y. Wang and J.-i. Yoshida, Wiley-VCH, Weinheim, 2009 Search PubMed.
- J.-i. Yoshida, Flash Chemistry: Fast Organic Synthesis in Microsystems, Wiley, Chichester, 2008 Search PubMed.
- (a) H. Amii, A. Nagaki and J.-i. Yoshida, Beilstein J. Org. Chem., 2013, 9, 2793–2802 CrossRef PubMed; (b) C. B. McPake and G. Sandford, Org. Process Res. Dev., 2012, 16, 844–851 CrossRef CAS.
- T. H. Rehm, Chem. Eng. Technol., 2016, 39, 66–80 CrossRef.
- P. Löb, H. Löwe and V. Hessel, J. Fluorine Chem., 2004, 125, 1677–1694 CrossRef.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–309 RSC; (b) S. Purser, P. R. Moore, S. Swallow and V. Governeur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- (a) S. P. Kotun, Fluorine, in e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rf003; (b) K. G. Davenport, Hydrogen Fluoride in e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rh037.
- T. Gustafsson, R. Gilmour and P. H. Seeberger, Chem. Commun., 2008, 3022–3024 RSC.
- (a) M. Baumann, I. R. Baxendale and S. V. Ley, Synlett, 2008, 2111–2114 CAS; (b) M. Baumann, I. R. Baxendale, L. J. Martin and S. V. Ley, Tetrahedron, 2009, 65, 6611–6625 CrossRef CAS.
- U. Budde, Statuskolloquium Mikroverfahrenstechnik, Deutsche Bundesstiftung Umwelt, Osnabrück, Germany, 2007 Search PubMed.
- K. Nakayame, D. L. Browne, I. R. Baxendale and S. V. Ley, Synlett, 2013, 24, 1298–1302 CrossRef.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- (a) N. J. W. Straathof, H. P. L. Gemoets, X. Wang, J. C. Schouten, V. Hessel and T. Noël, ChemSusChem, 2014, 7, 1612–1617 CrossRef CAS PubMed; (b) D. Cantillo, O. de Frutos, J. A. Rincón, C. Mateos and C. O. Kappe, Org. Lett., 2014, 16, 896–899 CrossRef CAS PubMed; (c) X. Zhang, P. Huang, Y. Li and C. Duan, Org. Biomol. Chem., 2015, 13, 10917–10922 RSC; (d) J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- D. Cantillo, O. de Frutos, J. A. Rincón, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 8486–8490 CrossRef CAS PubMed.
- N. de Mas, A. Günther, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2003, 42, 698 CrossRef CAS.
- K. Jähnisch, M. Baerns, V. Hessel, W. Ehrfeld, V. Haverkamp, H. Löwe, C. Wille and A. Guber, J. Fluorine Chem., 2000, 105, 117 CrossRef.
- C. B. McPake and G. Sandford, Org. Process Res. Dev., 2012, 16, 844 CrossRef CAS.
- R. D. Chambers and R. C. H. Spink, Chem. Commun., 1999, 883 RSC.
- R. D. Chambers, M. A. Fox, D. Holling, T. Nakano, T. Okazoe and G. Sandford, Lab Chip, 2005, 5, 191–198 RSC.
- J. R. Breen, G. Sandford, D. S. Yufit, J. A. K. Howard, J. Fray and B. Patel, Beilstein J. Org. Chem., 2011, 7, 1048 CrossRef CAS PubMed.
- S. Elgue, A. Conte, C. Gourdon and Y. Bastard, Chim. Oggi, 2012, 30(4), 18–21 CAS.
- K. Naumann, J. Prakt. Chem., 1999, 340, 450 Search PubMed.
- S. Borukhova, T. Noël and V. Hessel, Org. Process Res. Dev., 2016, 20, 568–573 CrossRef CAS.
- B. Reichart, G. Tekautz and C. O. Kappe, Org. Process Res. Dev., 2013, 17, 152–157 CrossRef CAS.
- P. Schmittinger, T. Florkiewicz, L. C. Curlin, B. Lüke, R. Scannell, T. Navin, E. Zelfel and R. Bartsch, Chlorine in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2001 Search PubMed.
- V. Cornel, Chlorine, in e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rp071.
- H. Ehrich, D. Linke, K. Morgenschweis, M. Baerns and K. Jähnisch, Chimia, 2002, 56, 647–653 CrossRef CAS.
- H. Matsubara, Y. Hino, M. Tokizane and I. Ryu, Chem. Eng. J., 2011, 167, 567–571 CrossRef CAS.
- The Chlorine Manual, 6th Edition, The Chlorine Institute, Inc., Washington, D. C., 2001 Search PubMed.
- F. Strauss, D. Cantillo, J. Guerra and C. O. Kappe, React. Chem. Eng., 2016, 1, 472–476 CAS.
- A. J. Blacker and K. E. Jolley, Beilstein J. Org. Chem., 2015, 11, 2408–2417 CrossRef CAS PubMed.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed.
- R. R. Goehring, Bromine in e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rb256.
- P. Löb, V. Hessel, H. Klefenz, H. Löwe and K. Mazanek, Lett. Org. Chem., 2005, 2, 767–779 CrossRef.
- R. Becker, S. A. M. W. van den Brock, P. J. Nieuwland, K. Koch and F. P. J. R. Rutjes, J. Flow Chem., 2012, 2, 87–91 CrossRef CAS.
- T. Fukuyama, Md. T. Rahman, N. Kamata, M. Tokizane, Y. Fukuda and I. Ryu, J. Flow. Chem., 2013, 3, 4–6 CrossRef CAS.
- Q. Deng, R. Shen, R. Ding and L. Zhang, Chem. Eng. Technol., 2016, 39, 1445–1450 CrossRef CAS.
- F. E. A. Van Waes, S. Seghers, W. Dermaut, B. Cappuyns and C. V. Stevens, J. Flow Chem., 2014, 4, 118–124 CrossRef CAS.
- Y. Manabe, Y. Kitawaki, M. Nagasaki, K. Fukase, H. Matsubara, Y. Hino, T. Fukuyama and I. Ryu, Chem. – Eur. J., 2014, 20, 12750–12753 CrossRef CAS PubMed.
- Y. J. Kim, M. J. Jeong, J. E. Kim, I. In and C. P. Park, Aust. J. Chem., 2015, 68, 1653–1656 CrossRef CAS.
- D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 223–229 CrossRef CAS PubMed.
- D. Šterk, M. Jukič and Z. Časar, Org. Process Res. Dev., 2013, 17, 145–151 CrossRef.
- M. O'Brien and D. Cooper, Synlett, 2016, 27, 164–168 CrossRef.
- I. C. Tornieporth-Oetting and T. M. Klapötke, Angew. Chem., Int. Ed. Engl., 1995, 34, 511–520 CrossRef CAS.
- D. Cantillo, B. Gutmann and C. O. Kappe, Org. Biomol. Chem., 2016, 14, 853–857 CAS.
- B. Leforestier and M. Vögtle, Synlett, 2016, 27, 1957–1962 CrossRef CAS.
- L. Wang, X. Zhou, M. Fredimoses, S. Liao and Y. Liu, RSC Adv., 2014, 4, 57350–57376 RSC.
- K. Midorikawa, S. Suga and J.-i. Yoshida, Chem. Commun., 2006, 3794–3796 RSC.
- J. D'Attoma, G. Cozien, P. L. Brun, Y. Robin, S. Bostyn, F. Buron and S. Routier, ChemistrySelect, 2016, 3, 338–342 CrossRef.
- R. G. Brisbois, R. A. Wanke, K. A. Stubbs, R. V. Stick and U. Ellervik, Iodine Monochloride in e-EROS Encyclopedia of Reagents for Organic Synthesis, 2013, DOI:10.1002/047084289X.ri014.pub3.
- M. Ferreri, A. Drageset, C. Gambarotti and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 379–386 CAS.
- D. R. Snead and T. F. Jamison, ICl has also been used to promote an 1,2-aryl migration via iodinated intermediates in continuous flow, Angew. Chem., Int. Ed., 2015, 54, 983–987 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |