
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of unprotected amino acids in metal-free tandem radical cyclization reactions: divergent synthesis of 6-alkyl/acyl phenanthridines†
Shi-Chao Lu a,
Hong-Shuang Li*b,
Ya-Ling Gonga,
Xiao-Lei Wanga,
Fu-Rong Lib,
Fei Lib,
Gui-Yun Duanb and
Shu Xu*a
a,
Hong-Shuang Li*b,
Ya-Ling Gonga,
Xiao-Lei Wanga,
Fu-Rong Lib,
Fei Lib,
Gui-Yun Duanb and
Shu Xu*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Beijing Key Laboratory of Active Substance Discovery and Drugability Evaluation, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, No. 1 Xiannongtan Street, Beijing 100050, PR China. E-mail: xushu@imm.ac.cn
bSchool of Pharmaceutical Sciences, Taishan Medical University, 619 Changcheng Road, Taian 271016, PR China. E-mail: lihongshuang8625@163.com
First published on 11th December 2017
Abstract
We reported the first example of the construction of C–C bonds using unprotected amino acids as stable alkyl/acyl radical precursors under metal-free conditions. This novel, environmentally friendly, and one-pot procedure was successfully applied to the radical alkylation or acylation/cyclization of isocyanides, which selectively affords 6-alkyl or acyl phenanthridines, depending on the substituent pattern of amino acid side chain groups.
Introduction
As an important building block, amino acids present the advantages of high stability, abundant natural resources, structural diversity and low cost, which make them ideal synthons for chemical synthesis.1 Although numerous methods for the introduction of amino acids into diverse organic structures via peptide coupling have been described,2 the application of amino acids in C–C bond-forming reactions is far less studied. In this context, synthetic utilization of α-aminoalkyl radicals generated by metal- or photo-catalyzed radical decarboxylation of N-protected amino acids has been achieved for the construction of C–C bonds (Scheme 1a and b). These reactions involved cross-coupling,3 conjugate additions,4 and Minisci reactions with electron-deficient heteroarenes.5 However, the strategy for the generation of the alkyl radicals from unprotected amino acids was seldom applied to molecular transformations due to their tendency for rapid oxidation of unprotected α-aminoalkyl radicals. Very recently, Baxter successfully realized a silver-catalyzed heterocycle C–H alkylation reaction using unprotected amino acids as stable radical precursors (Scheme 1c).6 This transformation was believed to undergo in situ aldehyde formation, namely Strecker degradation,7 followed by a Minisci-type radical decarbonylation/alkylation process. While radical alkylation reactions via aldehyde decarbonylation are known,8 aliphatic aldehydes are redox-sensitive liquids and usually require cold storage and special operations. In contrast, the resulting alkyl radicals via the multistep oxidative degradation of amino acids showed better selectivity and reactivity.6 However, all of the reactions involving the use of amine acids as radical precursors reported so far require a toxic transition metal catalytic system. In view of this, the development of a green reaction system for the construction of C–C bonds using amino acids as stable radical precursors is highly desirable and challenging. | ||
| Scheme 1 Amino acids as a source of alkyl radicals for C–C bond-forming reactions. | ||
Recently, the radical oxidative addition/cyclization of 2-isocyanobiphenyls with different radical precursors to synthesize functionalized phenanthridines, which are widely found in various natural products and possess a wide range of biological activities,9 have been extensively investigated.10 Our group has recently also reported a radical cascade decarboxylation/cyclization of 2-isocyanobiphenyls with carboxylic acids to afford 6-alkyl/aryl phenanthridines.10d Although these methods have their own specific applications, they still suffer from one or more limitations, such as a limited reaction scope, harsh reaction conditions, long reaction times, and the use of transition-metal catalysts. Therefore, it is still necessary to develop more radical precursors for practical, general, and environmentally benign methods to realize the addition/cyclization of 2-isocyanobiphenyls. To the best of our knowledge, amino acids have not been used as a source of radicals for construction of heterocycles. Herein, we develop a novel strategy that utilizes unprotected amino acids as inexpensive alkyl/acyl radical precursors for the addition/cyclization of 2-isocyanobiphenyls under metal-free conditions in aqueous solution, which provides a simple and general protocol for the divergent synthesis of 6-alkyl/acyl phenanthridines (Scheme 1d).
Results and discussion
In our initial studies, we selected 2-isocyano-1,1′-biphenyl (1a) and L-tert-leucine (2a) as model substrates to investigate the reaction conditions. Inspired by our previous research on decarboxylative cross-coupling reactions, the model reaction was carried out in CH3CN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 80 °C in the presence of 3.0 equiv. of K2S2O8 and 1.5 equiv. of K2CO3. To our delight, the desired product 3a was obtained in a 36% yield. In view of the fact that the addition of 20 mol% of AgNO3 cannot improve the yield (Table 1, entry 1), we decided to explore the metal-free oxidative radical cyclization for the construction of 6-alkyl phenanthridines. A survey of the reaction parameters, such as the oxidants, bases, solvents and temperature, were conducted. Increasing the temperature to 100 °C gave good conversion to the expected product (Table 1, entry 2). Further investigation on the bases, such as Na2CO3, K3PO4, Cs2CO3, and NaOAc, revealed that K2CO3 was the best choice (Table 1, entries 3–6). Subsequently, we attempted to examine the effect of different solvent systems on the model reaction. Among the solvents screened, water-miscible solvents (Table 1, entries 7–10) as well as water (Table 1, entry 11) were not effective for this transformation. In addition, the reaction was also not carried out in biphasic mixtures (DCE/H2O) (Table 1, entry 12). It was observed that increasing the ratio of CH3CN to H2O had a positive effect on the yield of 3a (Table 1, entries 13 and 14). When the ratio was 5:1, the reaction can be achieved in a 78% yield. However, 98% of the reactant 1a was recovered while using CH3CN as the sole solvent (Table 1, entry 15). Similarly, other anhydrous organic solvents such as DMF, DMSO, or THF were not effective for this conversion. Further investigation showed that both Na2S2O8 and (NH4)2S2O8 negatively affected the reaction (Table 1, entries 16 and 17). Meanwhile, the decreasing the amount of K2S2O8 led to a low yield (Table 1, entry 18). The control experiment indicated that K2S2O8 was necessary for the transformation (Table 1, entry 19). The optimal conditions were finally determined to be 2.0 equiv. of amino acid, 2.0 equiv. of K2CO3 and 4.0 equiv. of K2S2O8 in CH3CN/H2O (v/v = 5/1) under Ar at 100 °C for 1 h (Table 1, entry 14).
:1) at 80 °C in the presence of 3.0 equiv. of K2S2O8 and 1.5 equiv. of K2CO3. To our delight, the desired product 3a was obtained in a 36% yield. In view of the fact that the addition of 20 mol% of AgNO3 cannot improve the yield (Table 1, entry 1), we decided to explore the metal-free oxidative radical cyclization for the construction of 6-alkyl phenanthridines. A survey of the reaction parameters, such as the oxidants, bases, solvents and temperature, were conducted. Increasing the temperature to 100 °C gave good conversion to the expected product (Table 1, entry 2). Further investigation on the bases, such as Na2CO3, K3PO4, Cs2CO3, and NaOAc, revealed that K2CO3 was the best choice (Table 1, entries 3–6). Subsequently, we attempted to examine the effect of different solvent systems on the model reaction. Among the solvents screened, water-miscible solvents (Table 1, entries 7–10) as well as water (Table 1, entry 11) were not effective for this transformation. In addition, the reaction was also not carried out in biphasic mixtures (DCE/H2O) (Table 1, entry 12). It was observed that increasing the ratio of CH3CN to H2O had a positive effect on the yield of 3a (Table 1, entries 13 and 14). When the ratio was 5:1, the reaction can be achieved in a 78% yield. However, 98% of the reactant 1a was recovered while using CH3CN as the sole solvent (Table 1, entry 15). Similarly, other anhydrous organic solvents such as DMF, DMSO, or THF were not effective for this conversion. Further investigation showed that both Na2S2O8 and (NH4)2S2O8 negatively affected the reaction (Table 1, entries 16 and 17). Meanwhile, the decreasing the amount of K2S2O8 led to a low yield (Table 1, entry 18). The control experiment indicated that K2S2O8 was necessary for the transformation (Table 1, entry 19). The optimal conditions were finally determined to be 2.0 equiv. of amino acid, 2.0 equiv. of K2CO3 and 4.0 equiv. of K2S2O8 in CH3CN/H2O (v/v = 5/1) under Ar at 100 °C for 1 h (Table 1, entry 14).
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Base (equiv.) | Solvent (v/v) | Yieldb |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), K2S2O8 (0.8 mmol), K2CO3 (0.4 mmol), in 6.0 mL of solvent at 100 °C for 60 min under Ar.b Isolated yield.c At 80 °C.d 20 mol% of AgNO3 was used as a catalyst.e At 90 °C.f At 110 °C. | ||||
| 1c | K2S2O8 (3.0) | K2CO3 (1.5) | CH3CN/H2O (1:1) |
36 (38)d |
| 2 | K2S2O8 (4.0) | K2CO3 (2.0) | CH3CN/H2O (1:1) |
62 (54)e (60)f |
| 3 | K2S2O8 (4.0) | Na2CO3 (2.0) | CH3CN/H2O (1:1) |
53 |
| 4 | K2S2O8 (4.0) | K3PO4 (2.0) | CH3CN/H2O (1:1) |
42 |
| 5 | K2S2O8 (4.0) | Cs2CO3 (2.0) | CH3CN/H2O (1:1) |
46 |
| 6 | K2S2O8 (4.0) | NaOAc (2.0) | CH3CN/H2O (1:1) |
27 |
| 7 | K2S2O8 (4.0) | K2CO3 (2.0) | DMF/H2O (1:1) |
0 |
| 8 | K2S2O8 (4.0) | K2CO3 (2.0) | Dioxane/H2O (1:1) |
0 |
| 9 | K2S2O8 (4.0) | K2CO3 (2.0) | DMSO/H2O (1:1) |
12 |
| 10 | K2S2O8 (4.0) | K2CO3 (2.0) | Acetone/H2O (1:1) |
0 |
| 11 | K2S2O8 (4.0) | K2CO3 (2.0) | H2O | 0 |
| 12 | K2S2O8 (4.0) | K2CO3 (2.0) | DCE/H2O (1:1) |
0 |
| 13 | K2S2O8 (4.0) | K2CO3 (2.0) | CH3CN/H2O (3:1) |
72 |
| 14 | K2S2O8 (4.0) | K2CO3 (2.0) | CH3CN/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1) |
78 |
| 15 | K2S2O8 (4.0) | K2CO3 (2.0) | CH3CN | 0 |
| 16 | Na2S2O8 (4.0) | K2CO3 (2.0) | CH3CN/H2O (5:1) |
42 |
| 17 | (NH4)2S2O8 (4.0) | K2CO3 (2.0) | CH3CN/H2O (5:1) |
30 |
| 18 | K2S2O8 (2.0) | K2CO3 (2.0) | CH3CN/H2O (5:1) |
28 |
| 19 | — | K2CO3 (2.0) | CH3CN/H2O (5:1) |
0 |

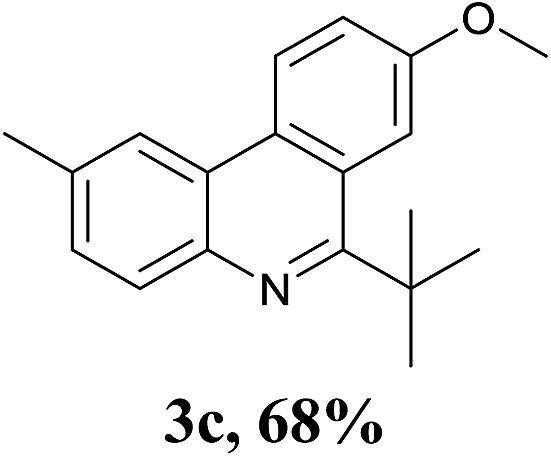
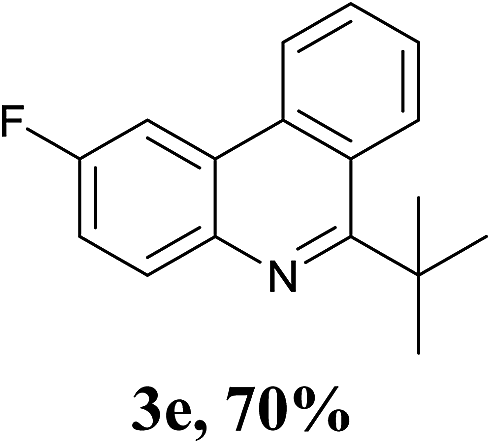
With the optimized conditions in hand, the scope and limitations of this tandem reaction were then investigated (Table 2). A variety of 2-isocyanobiphenyls containing an electron-withdrawing or electron-donating substituent were reacted with L-tert-leucine 2a efficiently to afford the corresponding 6-alkyl phenanthridines in moderate to good yields (3a–h). Among them, the cyclization of the meta-substituted substrate preferably took place at the less sterically hindered position (3g and 3h). In addition, heterocyclic 2-isocyanobiphenyl was also favored in this system and afforded the corresponding product in a 42% yield (3i). On the other hand, the oxidative cyclization of other unprotected amino acids bearing secondary alkyl substituents (such as valine, isoleucine, and cyclohexylglycine) performed well with a series of 2-isocyanobiphenyls (3j–o). It should be noted that this process was limited to simple alkyl-bearing amino acids, while other amino acids with polar side chains, such as asparagine, glutamine, and lysine, failed to afford the corresponding products under the optimized conditions.


















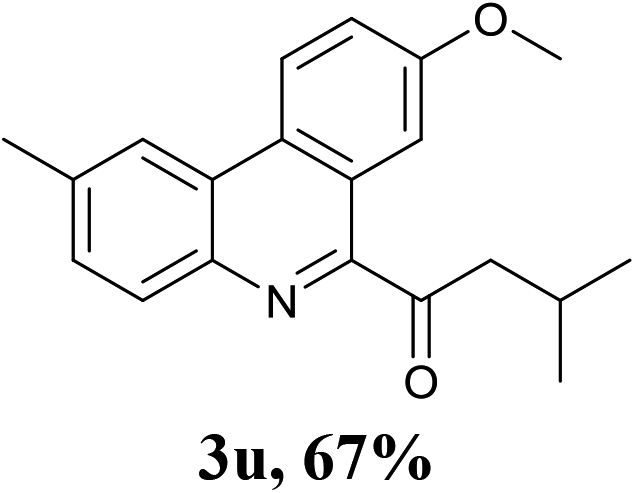
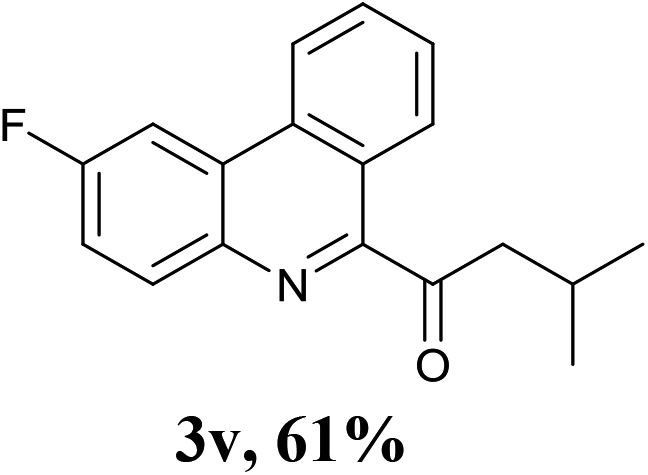

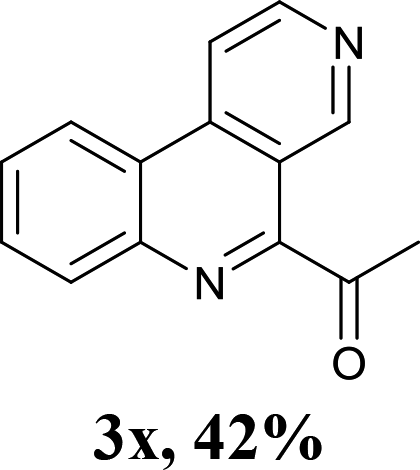
Encouraged by the above results, we then investigated the reactivity of amino acids bearing primary side chains (such as alanine, leucine, and homophenyl alanine) toward 2-isocyanobiphenyls. Interestingly, the green system afforded 6-acyl phenanthridines as main products instead of 6-alkyl phenanthridines with moderate to good yields (3p–x). These results could be attributed to the stability of the alkyl radical species by decarbonylation of the corresponding acyl radicals.8g Although some groups have reported their efforts for the synthesis of 6-acyl phenanthridines through the reactions of 2-isocyanobiphenyls with corresponding radical precursors including aromatic aldehydes,11 potassium oxophenylacetate,12 and benzylic alcohols13 under iron- or silver-catalyzed radical conditions, the construction of 6-aliphatic acyl phenanthridines remains elusive. In view of this, this strategy afforded a novel, environmentally friendly and complementary approach to produce 6-acyl phenanthridines.
To gather some insights into the mechanism of this reaction, (2,2,6,6-tetramethylpiperdin-1-yl)oxyl (TEMPO, 2.0 equiv.) and 2,6-di-tert-butyl-p-cresol (BHT) as radical scavengers were exposed separately to the standard reaction condition (Scheme 2a). As a consequence, the reaction was completely inhibited, which could indicate that this transformation involves radical intermediates. Then, we chose isobutyraldehyde as a radical precursor to test the radical mechanism (Scheme 2b). The results showed the expected 3n could be obtained in a 35% yield under the same reaction conditions. By contrast, the multistep degradation of amino acids to generate alkyl radicals offer better reactivity.
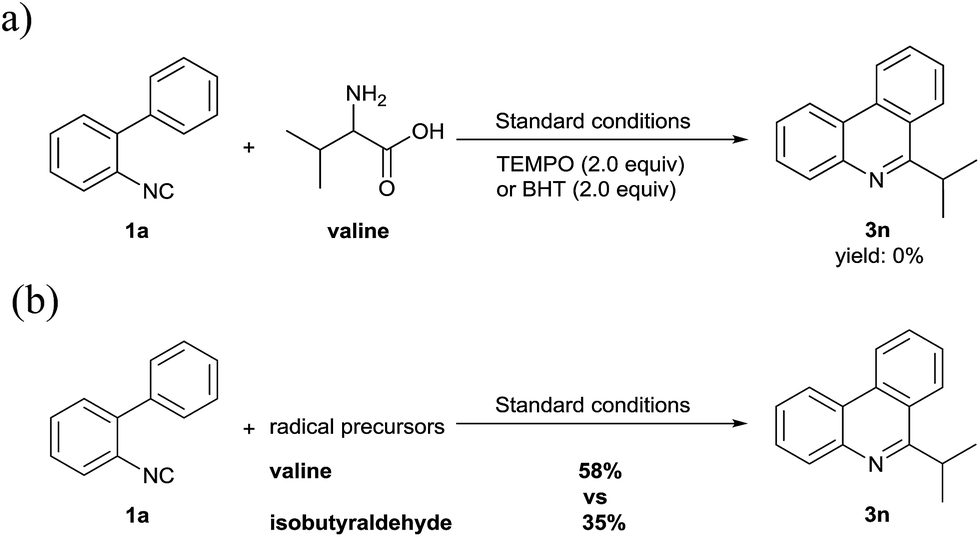 | ||
| Scheme 2 Control experiments for preliminary mechanism study. | ||
On the basis of this observation and the literature evidence,6,10 a proposed mechanism is shown in Scheme 3. First, the single-electron oxidative decarboxylation of amino acid anion I affords a radical II in the presence of sulfate anion radicals generated through homolytic cleavage of K2S2O8. Second, II is rapidly oxidized to the corresponding iminium species III and is converted to aldehyde IV in the presence of water. A sulfate anion radical abstracts the aldehyde hydrogen atom to provide the acyl radical V, which undergoes radical decarbonylation to yield the alkyl radical VI. Subsequently, the radical VI adds to the isocyanide 1a to provide the imidoyl radical VII, which undergoes an intramolecular radical cyclization to form the intermediate VIII. Finally, the intermediate VIII is then further oxidized to form the corresponding carbocation, which can be converted to the desired product 3 by losing a proton. In the case of 1° substituted amino acids, acyl radical V may be trapped by the isocyanide 1a to form the acylated product 4 in a similar manner.
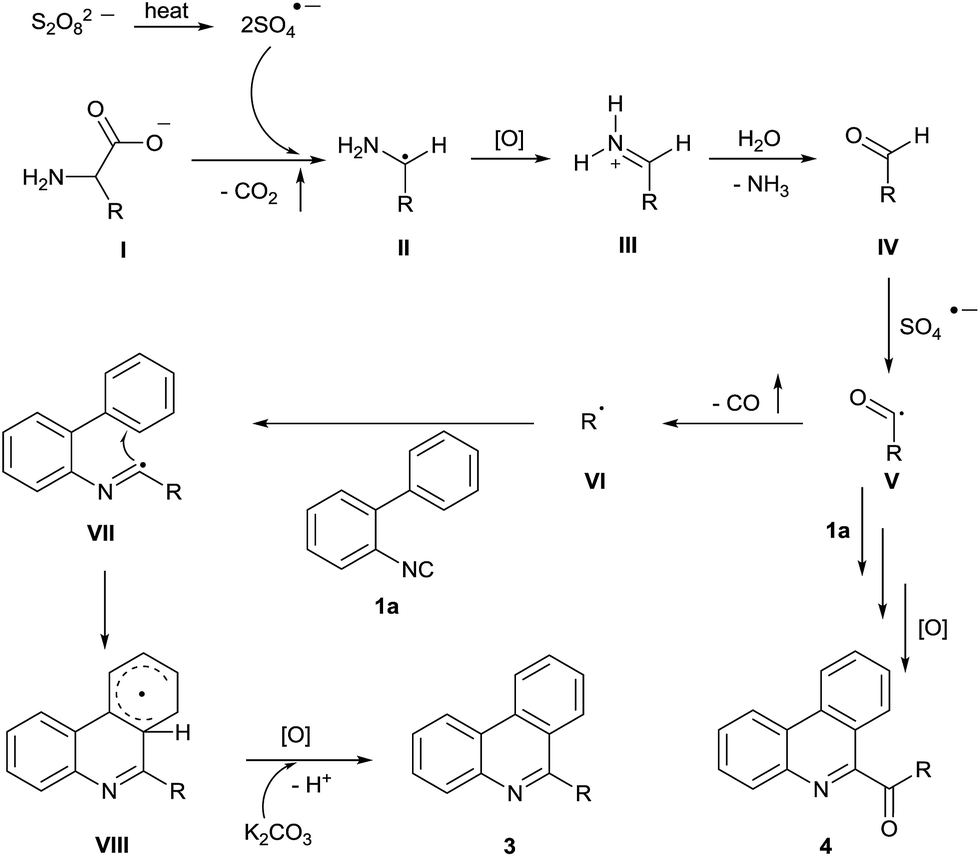 | ||
| Scheme 3 Postulated reaction pathway. | ||
Experimental section
General procedure for synthesis of 6-alkyl/acyl phenanthridines
A 25 mL oven-dried sealed tube was charged with 2-isocyanobiphenyls (1, 0.20 mmol, 1.0 equiv.), amine acids (2, 0.40 mmol, 2.0 equiv.), K2CO3 (55 mg, 0.40 mmol, 2.0 equiv.) and K2S2O8 (216 mg, 0.80 mmol, 4.0 equiv.) in CH3CN/H2O (6.0 mL, v/v = 5:1). The tube was sealed and heated at 100 °C for 1 h under an Ar atmosphere. After completion of the reaction, the reaction mixture was added water (5 mL), and then extracted with dichloromethane (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 100:1) to afford 3.
Conclusions
In conclusion, we have demonstrated a novel protocol utilizing unprotected amino acids as stable radical sources under metal-free conditions. The method is able to efficiently functionalize 2-isocyanobiphenyls by virtue of the corresponding amino acids, which presents the major advantages of environmentally benign character, a broad substrate scope, and readily available starting materials. Further studies of the reaction mechanism and the extension of the substrate scope are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is financially supported by the National Natural Science Foundation of China (21602256, 21502234 and 21672262), the Beijing Natural Science Foundation (2164074), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-3-009 and 2017-I2M-2-004), the Research Funds from State Key Laboratory of Bioactive Substance and Function of Natural Medicines (GTZB201404), the Fundamental Research Funds for CAMS/PUMC (2016RC350004), the Natural Science Foundation of Shandong Province (ZR2015BL006, ZR2013HM036) and the Project of Shandong Province Higher Educational Science and Technology Program (J13LM01).Notes and references
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS PubMed.
- For a recent review on peptide couplings, see: A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed.
- (a) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (c) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed.
- (a) S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728 CrossRef CAS PubMed; (b) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef CAS PubMed.
- (a) D. G. M. Shore, K. A. Wasik, J. P. Lyssikatos and A. A. Estrada, Tetrahedron Lett., 2015, 56, 4063 CrossRef CAS; (b) C. J. Cowden, Org. Lett., 2003, 5, 4497 CrossRef CAS PubMed.
- D. N. Mai and R. D. Baxter, Org. Lett., 2016, 18, 3738 CrossRef CAS PubMed.
- (a) O. Nashalian and V. A. Yaylayan, J. Agric. Food Chem., 2014, 62, 8518 CrossRef CAS PubMed; (b) G. P. Rizzi, Food Rev. Int., 2008, 24, 416 CrossRef CAS; (c) Y. Zelechonok and R. B. Silverman, J. Org. Chem., 1992, 57, 5787 CrossRef CAS; (d) A. Schonberg and R. Moubacher, Chem. Rev., 1952, 50, 261 CrossRef CAS.
- For radical-based alkylations from aldehydes, see: (a) C. Pan, Y. Chen, S. Song, L. Li and J. Yu, J. Org. Chem., 2016, 81, 12065 CrossRef CAS PubMed; (b) L. Yang, W. Lu, W. Zhou and F. Zhang, Green Chem., 2016, 18, 2941 RSC; (c) S. Paul and J. Guin, Chem.–Eur. J., 2015, 21, 17618 CrossRef CAS PubMed; (d) R. J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055 CrossRef CAS. for radical-based acylations from aldehydes, see: (e) J. Y. Chen, M. Wan, J. Hua, Y. Sun, Z. Lv, W. Li and L. Liu, Org. Biomol. Chem., 2015, 13, 11561 RSC; (f) Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS PubMed; (g) P. Cheng, Z. Qing, S. Liu, W. Liu, H. Xie and J. Zeng, Tetrahedron Lett., 2014, 55, 6647 CrossRef CAS; (h) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS PubMed; (i) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- (a) L. Sripada, J. A. Teske and A. Deiters, Org. Biomol. Chem., 2008, 6, 263 RSC; (b) O. B. Abdel-Halim, T. Morikawa, S. Ando, H. Matsuda and M. Yoshikawa, J. Nat. Prod., 2004, 67, 1119 CrossRef CAS PubMed; (c) T. Nakanishi, A. Masuda, M. Suwa, Y. Akiyama, N. Hoshino-Abe and M. Suzuki, Bioorg. Med. Chem. Lett., 2000, 10, 2321 CrossRef CAS PubMed; (d) T. Ishikawa, Med. Res. Rev., 2001, 21, 61 CrossRef CAS PubMed; (e) S. Simeon, J. L. Rios and A. Villar, Pharmazie, 1989, 44, 593 CAS; (f) S. D. Phillips and R. N. J. Castle, Heterocycl. Chem., 1981, 18, 223 CrossRef CAS.
- For some selected recent examples of preparation of phenanthridines, see: (a) W. Song, P. Yan, D. Shen, Z. Chen, X. Zeng and G. Zhong, J. Org. Chem., 2017, 82, 4444 CrossRef CAS PubMed; (b) S. Feng, T. Li, C. Du, P. Chen, D. Song, J. Li, X. Xie and X. She, Chem. Commun., 2017, 53, 4585 RSC; (c) P. Xiao, J. Rong, C. Ni, J. Guo, X. Li, D. Chen and J. Hu, Org. Lett., 2016, 18, 5912 CrossRef CAS PubMed; (d) S. Lu, Y. Gong and D. Zhou, J. Org. Chem., 2015, 80, 9339 Search PubMed; (e) J. J. Cao, T. H. Zhu, S. Y. Wang, Z. Y. Gu, X. Wang and S. J. Ji, Chem. Commun., 2014, 50, 6439 RSC; (f) L. Wang, W. X. Sha, Q. Dai, X. M. Feng, W. T. Wu, H. B. Peng, B. Chen and J. Cheng, Org. Lett., 2014, 16, 2088 CrossRef CAS PubMed; (g) Z. J. Li, F. H. Fan, J. Yang and Z. Q. Liu, Org. Lett., 2014, 16, 3396 CrossRef CAS PubMed; (h) W. X. Sha, J. T. Yu, Y. Jiang, H. T. Yang and J. Cheng, Chem. Commun., 2014, 50, 9179 RSC; (i) J. J. Cao, X. Wang, S. Y. Wang and S. J. Ji, Chem. Commun., 2014, 50, 12892 RSC; (j) L. J. Gu, C. Jin, J. Y. Liu, H. Y. Ding and B. M. Fan, Chem. Commun., 2014, 50, 4643 RSC; (k) Z. H. Xia, J. B. Huang, Y. M. He, J. J. Zhao, J. Lei and Q. Zhu, Org. Lett., 2014, 16, 2546 CrossRef CAS PubMed; (l) H. Jiang, Y. Z. Cheng, R. Z. Wang, M. M. Zheng, Y. Zhang and S. Y. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS PubMed; (m) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed. for recent reviews see: (n) B. Song and B. Xu, Chem. Soc. Rev., 2017, 46, 1103 RSC; (o) J. Lei, J. Huang and Q. Zhu, Org. Biomol. Chem., 2016, 14, 2593 RSC.
- D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef CAS PubMed.
- J. Liu, C. Fan, H. Yin, C. Qin, G. Zhang, X. Zhang, H. Yi and A. Lei, Chem. Commun., 2014, 50, 2145 RSC.
- Z. Nie, Q. Ding and Y. Peng, Tetrahedron, 2016, 72, 8350 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General considerations, experimental procedures, spectroscopic data. See DOI: 10.1039/c7ra12318c |
| This journal is © The Royal Society of Chemistry 2017 |