
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retracted Article: Vitexin mitigates myocardial ischemia reperfusion-induced damage by inhibiting excessive autophagy to suppress apoptosis via the PI3K/Akt/mTOR signaling cascade
Zhaobin Tang†
a,
Lei Yang†b and
Xuesong Zhang *c
*c
aDepartment of Cardiology, The First Hospital of Zibo, Shandong 255200, China
bDepartment of Cardiology, Hospital of Laiwu Steel Group, Shandong 271100, China
cDepartment of Cardiology, Liaocheng People's Hospital, 67# Dong Chang West Road, Liaocheng, Shandong 252000, China. E-mail: zhangxuesonglc@163.com; Tel: +86-635-8276199
First published on 15th December 2017
Abstract
Myocardial ischemia reperfusion (MI/R) injury is reported to induce apoptosis and autophagy of myocardial cells and contribute to adverse cardiovascular outcomes. Vitexin, a flavonoids compound, exhibited anti-inflammatory, anti-oxidative and antitumor effects in various studies. Here, we investigated the cardioprotective effect and underlying mechanism of vitexin treatment in MI/R injury in vivo and in vitro. We found that vitexin decreased the high cell apoptosis rate caused by MI/R injury significantly in a dose-dependent manner. The expression of cleaved Caspase-3 and Bax was down-regulated and the expression of Bcl-2 was up-regulated by vitexin compared with MI/R model group, indicating that vitexin suppressed apoptosis of rat cardiomyocytes H9c2 cells in MI/R injury. The concentrations of LDH, CK and MDA were decreased while the concentration of SOD was increased by vitexin during MI/R injury. Moreover, vitexin also suppressed autophagy of H9c2 cells in MI/R injury by down-regulating the expression of Beclin1 and LC3I/II while up-regulating the expression of p62. Activation of autophagy by Rapa significantly increased cleaved Caspase-3, Bcl-2 expression and the concentration of LDH, CK, MDA while decreased the concentration of SOD, indicating that vitexin inhibited autophagy to suppress apoptosis of H9c2 cells in MI/R injury. The co-treatment of PI3K inhibitor LY294002 reversed the inhibition on apoptosis and autophagy by vitexin, suggesting that vitexin inhibited apoptosis and autophagy to ameliorate MI/R injury through activation of PI3K/Akt/mTOR signaling pathway. We also constructed MI/R animal model using C57BL/6 mice to investigate the cardioprotective effects of vitexin in vivo. We observed that vitexin mitigated MI/R injury by decreasing the concentrations of LDH, CK and MDA, increasing the concentration of SOD, and counteracting the irregular pathological changes induced by MI/R injury dose-dependently. Vitexin also suppressed apoptosis and autophagy in myocardium cells of C57BL/6 mice in MI/R injury via activation of PI3K/Akt/mTOR signaling pathway. In conclusion, vitexin mitigated MI/R injury by inhibiting apoptosis and excessive autophagy via the PI3K/Akt/mTOR signaling pathway.
Introduction
Cardiovascular diseases, including myocardial infarction, and ischemic heart disease, are common diseases which cause high mortality worldwide.1 Myocardial ischemia is generally caused by a lack of coronary blood flow to the myocardium.2 During myocardial ischemia, insufficient blood supply often leads to severe and irreversible injury of myocardial cells. In order to limit myocardial infarct size and reduce mortality, rapid restoration of blood flow to the ischemia myocardium, namely reperfusion, is the most effective treatment to improve the clinical outcome.3 However, reperfusion itself can cause additional cardiac damage and complications including paradoxical increases in infarct size and induction of arrhythmia. This is known as myocardial ischemia reperfusion (MI/R) injury.4 Nowadays, incidence of MI/R injury is increasing around the world. Therefore, examining new and effective methods to reduce MI/R injury has become an area of intense research.Numerous studies demonstrated that MI/R injury induces various types of cell death including necrosis, apoptosis and autophagy.5 Apoptosis and autophagy are two main types of programmed cell death during MI/R injury. Apoptosis induced by MI/R injury increases the severity of cardiomyocyte injury.6 Myocardial cells apoptosis during MI/R injury causes irreversible damage to cardiac function. Autophagy is a mechanism of cellular degradation through lysosomes and is reported to coordinate with apoptosis to promote cell death.7 Under physiologic conditions, autophagy maintains at a low level and degrades damaged mitochondria to protect cells from oxidative injury. However, under pathologic conditions including MI/R, hypoxia and mitochondrial dysfunction, excessive autophagy is emerged and contributes to organ dysfunction.8 Research showed that autophagy is a “double-edged sword” in MI/R injury: during ischemia, a slight increase of autophagy inhibited apoptosis and promoted cell survival, but an excessive increase of autophagy during the reperfusion period contributed to MI/R injury.9 Therefore, suppressing excessive autophagy may protect myocardial cells against MI/R injury.
Vitexin (apigenin-8-C-D-glucopyranoside), which belongs to flavonoids compounds, is isolated from the leaf of Crataegus pinnatifida Bunge.10 Vitexin is reported to reduce blood pressure and exerts anti-inflammation effect.11 Wang's study revealed that vitexin protected brain against ischemia/reperfusion injury via regulating MAPK and apoptosis signaling pathways.10 Vitexin is also reported to engender antitumor effects through the regulation of the apoptosis-related gene expression of p53 and Bcl-2. Previous researches indicated that vitexin had a protective effect on acute MI/R injury and myocardial cells stuffed hypoxia reoxygenation.12,13 However, whether vitexin mitigated MI/R injury through inhibiting autophagy was still unclear and needed further studied in depth.
In this present study, we explored the protective effects of vitexin on MI/R injury and the potential mechanism. Our data indicated that the underlying molecular mechanisms of vitexin's cardioprotective effects were related to the inhibition of excessive autophagy via activating the PI3K/Akt/mTOR signaling pathway.
Materials and methods
Drugs and reagents
Vitexin (structure shown in Fig. 1A), obtained from Selleck Inc (USA), was dissolved by DMSO (Sigma, St. Louis, MO, USA) and stored at −20 °C. DMEM medium and FBS were purchased from Gibco (Gibco, Rockville, MD). MTT, DAPI and the Annexin V-FITC/PI Staining Apoptosis Detection Kit were purchased from Sigma-Aldrich. The total lactate dehydrogenase (LDH), Creatine Kinase (CK), superoxide dismutase (SOD) and malondialdehyde (MDA) detection kits were bought from Nanjing Jian-cheng Co., Ltd. PI3K inhibitor LY294002 was purchased from Selleck Inc (USA). Antibodies for cleaved Caspase-3, Bax, Bcl-2, Beclin1, p62, LC3I/II, p-Akt, mTOR, GAPDH and HRP-conjugated secondary were purchased from Cell Signaling Technology Inc (USA) or Abcam. Colorimetric TUNEL Apoptosis Assay Kit was purchased from Beyotime Institute of Biotechnology, Jiangsu, China. Rapamycin was purchased from Selleck Inc (USA). | ||
| Fig. 1 Vitexin doesn't have significant effect on the viability of H9c2 cells. (A) Structure of vitexin. (B) Effect of vitexin on the cell viability of H9c2 cells was detected by MTT assay. The bars showed means ± SD of three independent experiments. | ||
Cell culture and establishment of a stimulated MI/R model in H9c2 cells
H9c2 cells were purchased from ATCC (USA) and cultured in DMEM medium supplemented with 10% FBS at 37 °C with 5% CO2. In order to mimic MI/R induced injury in vitro, stimulated ischemia followed by reperfusion was performed in H9c2 cells according to the protocol described before.14 Cells were transferred to an ischemia-simulating buffer solution (137 mM NaCl, 12 mM KCl, 0.49 mM MgCl2, 0.9 mM CaCl2·2H2O, 4 mM HEPES and 20 mM sodium lactate), and then placed in a sealed hypoxia chamber (containing 95% N2 and 5% CO2) for 4 h at 37 °C to induce anoxia. Cells were treated for 4 h of simulated ischemia. Then, the medium was replaced by normal medium, and cells were incubated in a 5% CO2 incubator at 37 °C for 24 h to simulate reperfusion.H9c2 cells were divided into 7 groups randomly as follows: (1) control group: H9c2 cells without treatment; (2) MI/R model group: H9c2 cells received MI/R injury treatment mentioned above; (3) MI/R + vitexin 50 μM group: H9c2 cells received MI/R injury treatment and then treated with vitexin (50 μM) for 4 h; (4) MI/R + vitexin 100 μM group: H9c2 cells received MI/R injury treatment and then treated with vitexin (100 μM) for 4 h; (5) MI/R + vitexin 200 μM group: H9c2 cells received MI/R injury treatment and then treated with vitexin (200 μM) for 4 h; (6) MI/R + vitexin 200 μM + Rapamycin (Rapa) group: H9c2 cells received MI/R injury treatment and then treated with vitexin (200 μM) combined with Rapa (40 nM) for 4 h; (7) MI/R + vitexin 200 μM + LY294002 group: H9c2 cells received MI/R injury treatment and then treated with vitexin (200 μM) combined with LY294002 (10 μM) for 4 h.
Cell viability analysis
The effects of vitexin on H9c2 cells viability were assessed by MTT analysis. Cells were seeded into 96-well plates at the concentration of 4 × 104 cells per well. After 24 h of incubation at 37 °C, cells were treated with 0, 10, 20, 50, 100, 150 or 200 μM vitexin for 24 h. Then MTT was added in each well (5 mg mL−1) and incubated in the dark at 37 °C for 2 h. Absorbance was determined at a wavelength of 570 nm.Flow cytometric analysis for cell apoptosis
H9c2 cells in different groups were harvested, washed in PBS and resuspended in annexin-binding buffer. Then 5 μL of Annexin V-FIFC and 10 μL of PI were added to the suspension and cells were cultured at room temperature in the dark for 20 min. After DAPI staining, apoptotic cell death was analyzed by flow cytometry.Western-blot analysis
Protein samples were lysed with SDS-PAGE sample buffer. Proteins were separated by 10% or 12% SDS-PAGE and then transferred into PVDF membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were blocked with TBS containing 5% nonfat milk and then incubated with primary antibodies purchased from Cell Signaling Technology or Abcam at 4 °C overnight as following: cleaved Caspase-3 (Cell Signaling Technology, no. 9661, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000), Bax (Cell Signaling Technology, no. 2772, 1:1000), Bcl-2 (Abcam, ab59348, 1:1000), Beclin1 (Abcam, ab62557, 1:1000), p62 (Abcam, ab56416, 1:1000), LC3I/II (Cell Signaling Technology, no. 4108, 1:1000), p-Akt (Cell Signaling Technology, no. 4060, 1:2000), mTOR (Cell Signaling Technology, no. 2983, 1:1000) and GAPDH (Cell Signaling Technology, no. 5174, 1:2500). After washing with TBST, the membranes were incubated with HRP-conjugated anti-IgG (Cell Signaling Technology, no. 14708 or no. 14709, 1:5000) at room temperature for 1 h. Signal detection was performed using an ECL system (Millipore, Billerica MA, USA). Image quantifications were performed using Image J software.
:1000), Bax (Cell Signaling Technology, no. 2772, 1:1000), Bcl-2 (Abcam, ab59348, 1:1000), Beclin1 (Abcam, ab62557, 1:1000), p62 (Abcam, ab56416, 1:1000), LC3I/II (Cell Signaling Technology, no. 4108, 1:1000), p-Akt (Cell Signaling Technology, no. 4060, 1:2000), mTOR (Cell Signaling Technology, no. 2983, 1:1000) and GAPDH (Cell Signaling Technology, no. 5174, 1:2500). After washing with TBST, the membranes were incubated with HRP-conjugated anti-IgG (Cell Signaling Technology, no. 14708 or no. 14709, 1:5000) at room temperature for 1 h. Signal detection was performed using an ECL system (Millipore, Billerica MA, USA). Image quantifications were performed using Image J software.
Detection of LDH, CK, SOD and MDA
Culture medium of H9c2 cells in different groups were collected and centrifuged for 5 min at 1500g. Experiments were carried out using the corresponding commercial kits according to the manufacturer's instructions and detected respectively at 440 nm and 340 nm by spectrophotometry.Fluorescence microscopy
H9c2 cells were transfected with the GFP-LC3 plasmid using Lipofectamine 3000™ (Invitrogen) according to the manufacturer's guidelines. After H9c2 cells were treated with or without vitexin, the puncta formation of GFP-LC3 was determined under fluorescent microscopy (Olympus BX5) as the previous study described.15 Cells were considered to have accumulated autophagosomes when more than 5 puncta were counted.Animal experiments in vivo
Male, 6–7 weeks C57BL/6 mice were purchased from the experimental animal center of Shandong University. Animal experiments were performed according to the guidelines of the National Institute of Health (NIH). All animal studies were approved by Medical Ethics Committee of The First Hospital of Zibo.Coronary artery ligation method was used to establish MI/R injury model. Mice were anesthetized with sodium pentobarbital intraperitoneal injection (30 mg kg−1). Electrocardiograph (ECG) electrodes were subcutaneously connected in all limbs and continuously monitored throughout the experiment. Mice received endotracheal intubation and artificial ventilation. The chest was opened through a left thoracotomy in the fourth intercostal space to expose the heart. Left coronary artery was ligated by thread to produce ischemia and ECG showed ST-segment elevation after ligation. After 1 h of ischemia, the coronary artery was reperfused for 60 min by releasing the ligation. Mice were randomly divided into 5 groups: Sham group (operated with no ischemia), MI/R group, MI/R + vitexin 2 mg kg−1 group, MI/R + vitexin 4 mg kg−1 group, MI/R + vitexin 6 mg kg−1 group. The vitexin of different doses were given by intravenous injection in 20 minutes before coronary artery ligation, and the same amount of saline solution was given for Sham group and MI/R group.
Heart rates and myocardial infarct area detection
Heart rates (HR) were recorded by ECG. The myocardial infarct area was measured by Evans blue and tetrazoliumchloride (TTC) staining (Shanghai Chemical Reagent Co., China) as described previously.16Hematoxylin and eosin (HE) staining
Heart from mice were fixed in 4% paraformaldehyde for 24 h, dehydrated, embedded in paraffin and cut into 4 μm-thick sections for HE staining according to immunohistochemical examination described previously and the pathological change was observed under optical microscope (Olympus, Japan).17TUNEL assay
TUNEL assay was performed to detect cells apoptosis rates in hearts tissue. Heart sections were incubated in 3% H2O2 and then in the TUNEL reaction mixture. The sections were rinsed and visualized using DAB. Hematoxylin was used for counter-staining. The numbers of TUNEL-positive cells of 6 random fields were counted under light microscopy. The apoptosis index was calculated as the percent of TUNEL-positive cells relative to the total cells.Statistical analysis
The results were expressed as the mean ± standard deviation (SD) of independent experiments. Statistical analysis was performed with one-way analysis of variance (ANOVA). The difference was considered statistically significant at P < 0.05. All statistical analysis was performed with SPSS 19.0 and GraphPad Prism Version 5.0 softwares.Results
Vitexin doesn't have significant effect on the viability of H9c2 cells
The effects of vitexin on cell viability in rat cardiomyocytes H9c2 were examined in response to increasing concentrations of vitexin for 24 h. Along with the concentrations rising, vitexin didn't affect the viability of H9c2 cells significantly. Although in the concentrations of 150 μM and 200 μM, vitexin inhibited the viability of H9c2 cells slightly, there was no statistically significant difference existed for the P values of 150 μM and 200 μM were 0.092 and 0.076 respectively (Fig. 1B). Our results suggested that vitexin didn't have significant effect on the viability of H9c2 cells.Vitexin suppresses apoptosis of H9c2 cells in MI/R injury
H9c2 cells were used to construct stimulated MI/R injury model in vitro in this study. To detect whether vitexin inhibited apoptosis of cardiomyocytes during MI/R injury, cell apoptosis was determined by flow cytometry with Annexin V-FITC/PI Staining. Vitexin treatment suppressed the high cell apoptosis rates induced by stimulated MI/R injury in vitexin treated group compared with MI/R model group (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 2A and B). Besides, the higher the concentration of vitexin, the stronger the suppression effect on apoptosis.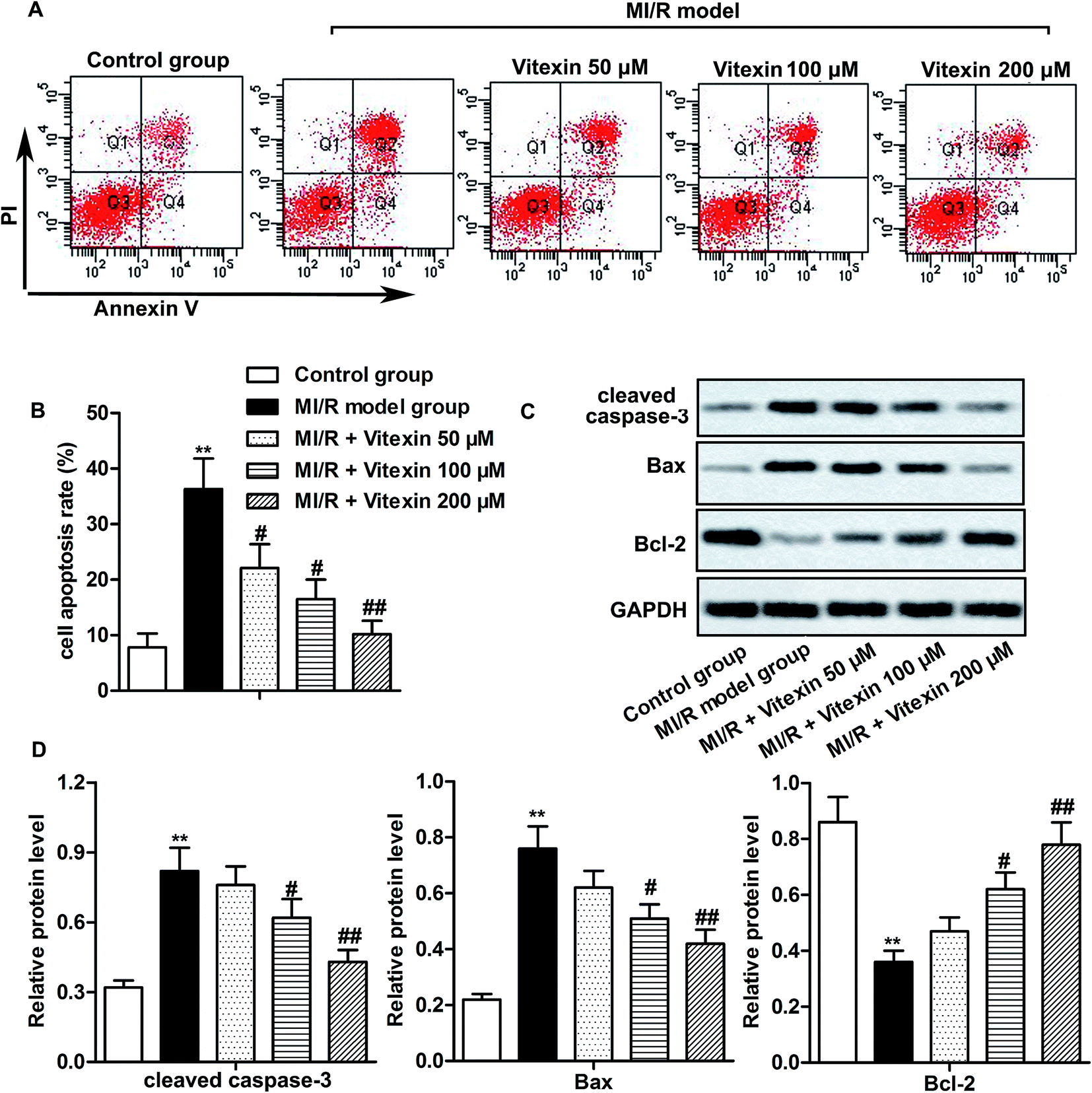 | ||
| Fig. 2 Vitexin suppresses apoptosis of H9c2 cells in MI/R injury. H9c2 cells were divided into control group, MI/R model group and MI/R + vitexin treated group (50 μM, 100 μM and 200 μM respectively). (A and B) Cell apoptosis rates of different groups were analyzed by flow cytometry. (C and D) Relative protein level of cleaved Caspase-3, Bax and Bcl-2 in different groups was measured by western blot. The bars showed means ± SD of three independent experiments. **P < 0.01 compared with control group, #P < 0.05, ##P < 0.01 compared with MI/R model group. | ||
To further evaluate the molecular mechanisms of suppressed apoptosis induced by vitexin, we explored the expression of apoptosis-related proteins in different groups by western blot. The high expression of cleaved Caspase-3 and Bax caused by stimulated MI/R injury was down-regulated by vitexin treatment, while the expression of anti-apoptosis protein Bcl-2 was up-regulated by vitexin in a dose-dependent manner (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 2C and D). Together, these results indicated that vitexin suppressed apoptosis of cardiomyocytes H9c2 in MI/R injury in a dose-dependent manner.
Effect of vitexin on the concentrations of LDH, CK, SOD and MDA in MI/R injury
The concentrations of LDH, CK, SOD and MDA of different groups were shown in Fig. 3. After MI/R, the cellular damage indicators of cardiomyocytes such as CK, LDH and oxidative parameter MDA were increased significantly in MI/R model group compared with control group, indicating that injury on H9c2 cells after MI/R leading to the leakage of LDH, CK and MDA. However, these parameters significantly decreased by vitexin treatment compared with MI/R model group. Moreover, vitexin treatment increased the low concentration of antioxidant enzyme SOD induced by MI/R in a dose-dependent manner (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 3A–D). Our data indicated that vitexin repaired myocardial lesion by down-regulating the concentrations of LDH, CK and MDA and up-regulating the concentration of SOD of H9c2 cells in MI/R injury. | ||
| Fig. 3 Effect of vitexin on the concentrations of LDH, CK, SOD and MDA in MI/R injury. H9c2 cells were divided into control group, MI/R model group and MI/R + vitexin treated group (50 μM, 100 μM and 200 μM respectively). (A–D) The concentrations of LDH, CK, SOD and MDA were measured by detection kits respectively. The bars showed means ± SD of three independent experiments. **P < 0.01 compared with control group, #P < 0.05, ##P < 0.01 compared with MI/R model group. | ||
Vitexin inhibits autophagy to suppress apoptosis of H9c2 cells in MI/R injury
Autophagy is reported to coordinate with apoptosis to promote cell death in MI/R injury.7 To investigate whether autophagy could be regulated by vitexin in H9c2 cells during MI/R injury, the expression of autophagy-related proteins were measured by western-blot. Relative protein level of Beclin1 and LC3 II/I was decreased while relative protein level of p62 was increased markedly by vitexin treatment compared with stimulated MI/R model group (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 4A–D). We further examined the localization of LC3 to autophagosome formation as assessed through GFP-LC3 expression. GFP-LC3 puncta formation observed as punctate dots of green fluorescence was significantly decreased in vitexin treated H9c2 cells compared with stimulated MI/R model group (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 4E and F).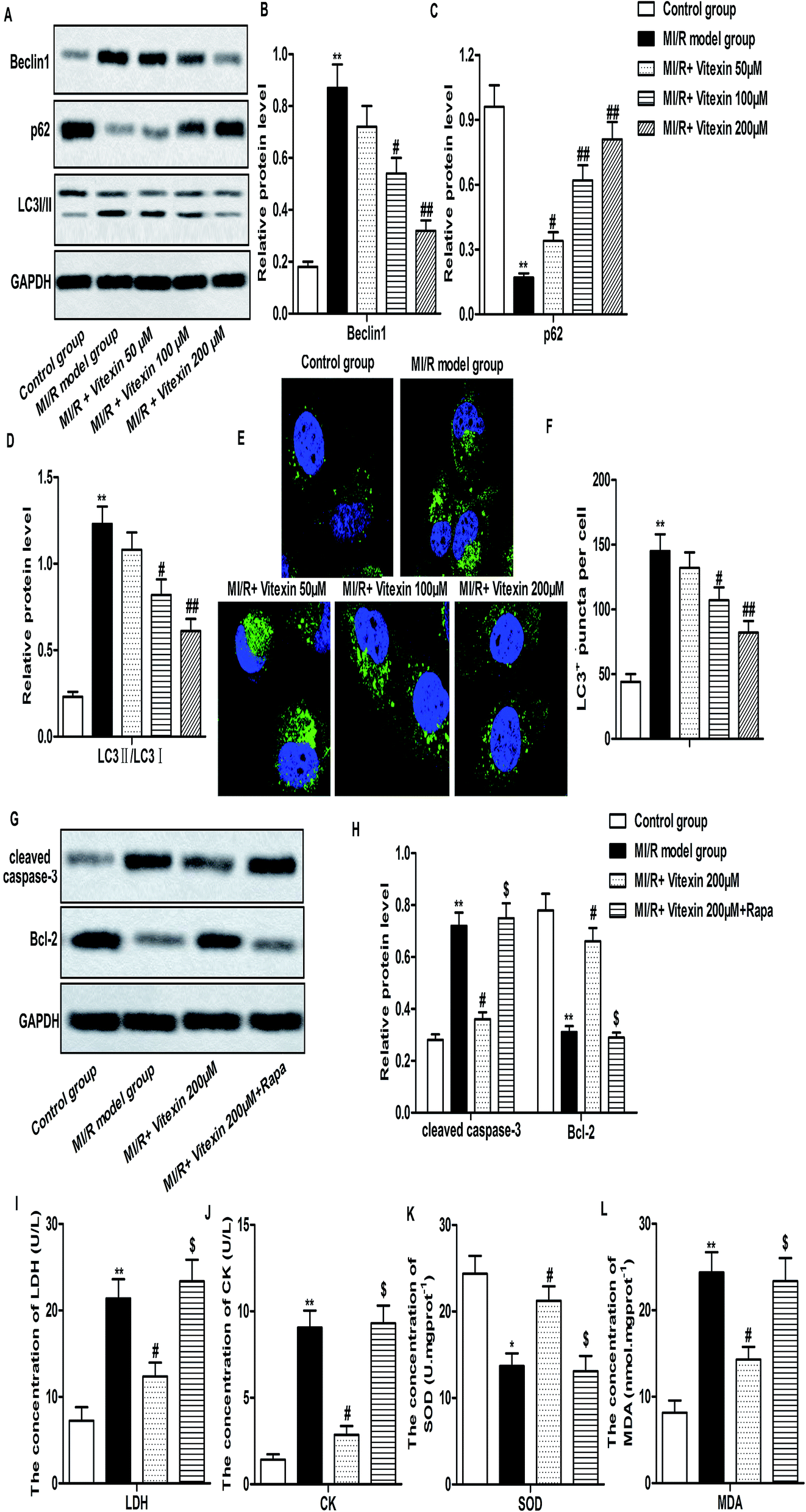 | ||
| Fig. 4 Vitexin inhibits autophagy to suppress apoptosis of H9c2 cells in MI/R injury. H9c2 cells were divided into control group, MI/R model group, MI/R + vitexin treated group (50 μM, 100 μM and 200 μM respectively) and MI/R + vitemin 200 μM + Rapa group. (A–D) Relative protein level of Beclin1, p62 and LC3I/II in different groups was measured by western blot. (E) Immunofluorescence of H9c2 cells as detected by the accumulation of autophagic vacuoles and quantified by the number of GFP-LC3 dots. Green dots represent autophagosomes. (F) The bars showed that the number of GFP-LC3 puncta per cell in H9c2 cells. (G and H) Relative protein level of cleaved Caspase-3 and Bcl-2 in different groups was measured by western blot. (I–L) The concentrations of LDH, CK, SOD and MDA were measured by detection kits respectively. The bars showed means ± SD of three independent experiments. **P < 0.01 compared with control group, #P < 0.05, ##P < 0.01 compared with MI/R model group, $P < 0.05 compared with MI/R + vitexin 200 μM group. | ||
In order to confirm the protective role of suppressed autophagy against vitexin-induced apoptosis, a specific autophagy activator rapamycin (Rapa) was used to treat H9c2 cells in our experiments. We observed that activation of autophagy by Rapa significantly increased the expression of cleaved Caspase-3, Bcl-2 and the concentration of LDH, CK, MDA while decreased the concentration of SOD compared with MI/R + vitexin 200 μM group (Fig. 4G–L, *P < 0.05, **P < 0.01, #P < 0.05, $P < 0.05). These results suggested that activation of autophagy counteracted the inhibitory effect of vitexin on apoptosis and myocardial injury. Taken together, results above demonstrated that vitexin inhibited autophagy to suppress apoptosis of H9c2 cells in MI/R injury.
Vitexin mitigates MI/R injury through activation of PI3K/Akt/mTOR signaling pathway
Autophagy is mediated by various signal pathways, most of which were related to the PI3K/Akt/mTOR signaling pathway.18 Here, we measured the expression of phosphorylated Akt (p-Akt) and mTOR in different groups by western blot. Our data showed that the low expression of p-Akt and mTOR induced by MI/R injury was up-regulated by vitexin treatment in a dose-dependent manner (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 5A–C).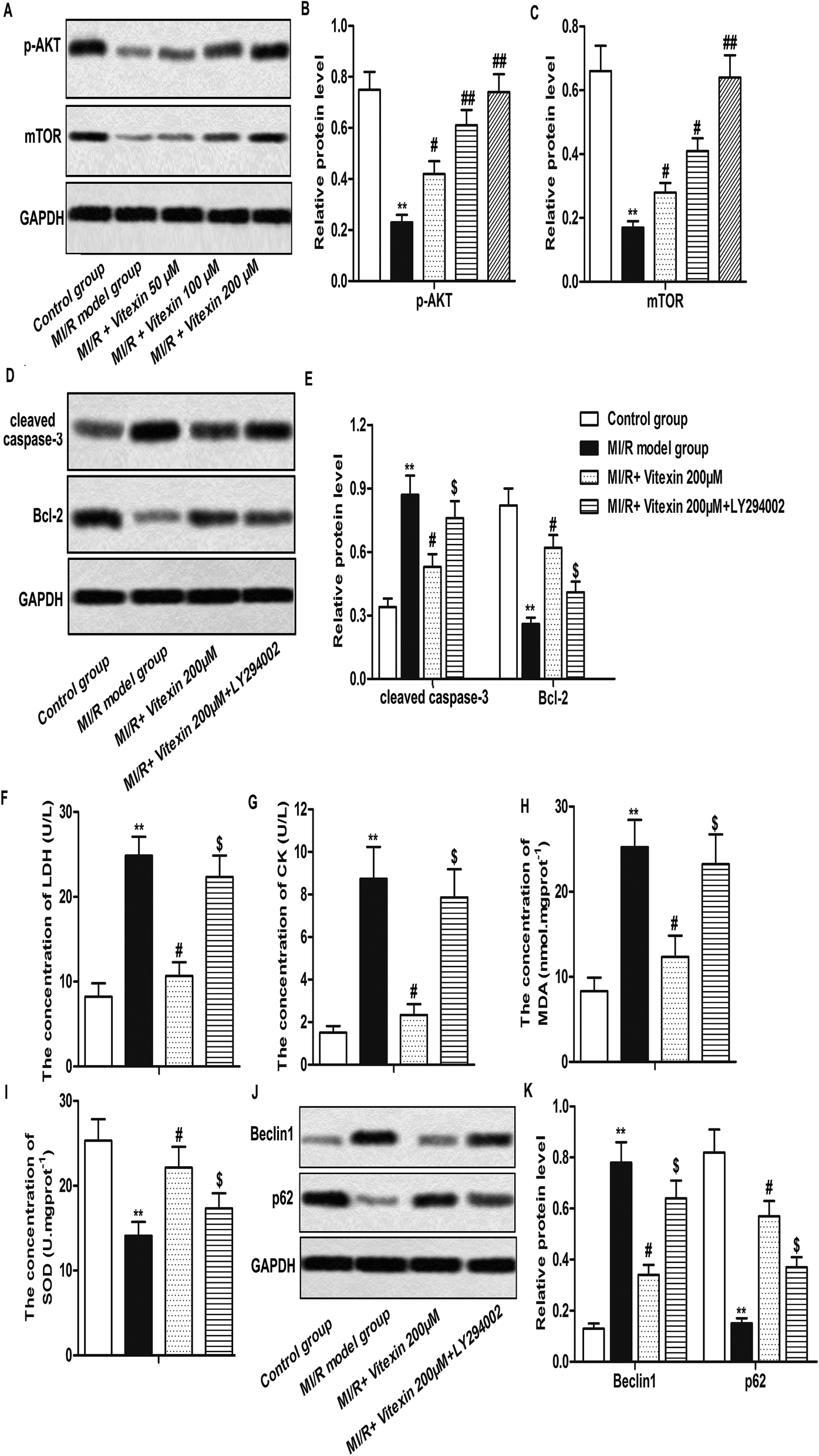 | ||
| Fig. 5 Vitexin mitigates MI/R injury through regulating of PI3K/Akt/mTOR signaling pathway. H9c2 cells were divided into control group, MI/R model, MI/R + vitexin treated group (50 μM, 100 μM and 200 μM respectively) and MI/R + vitemin 200 μM + LY294002 group. (A–C) Relative protein level of p-Akt and mTOR in different groups was measured by western blot. (D and E) Relative protein level of cleaved Caspase-3 and Bcl-2 in different groups was measured by western blot. (F–I) The concentrations of LDH, CK, SOD and MDA were measured by detection kits respectively. (J and K) Relative protein level of Beclin1 and p62 in different groups was measured by western blot. The bars showed means ± SD of three independent experiments. **P < 0.01 compared with control group, #P < 0.05 compared with MI/R model group, $P < 0.05 compared with MI/R + vitexin 200 μM group. | ||
In order to prove that vitexin ameliorated MI/R injury via activation of PI3K/Akt/mTOR signaling pathway, PI3K inhibitor LY294002 was used in our study. Our data showed that relative protein level of cleaved Caspase-3 was increased while relative protein level of Bcl-2 was decreased by LY294002 treatment compared with MI/R + vitexin 200 μM group (**P < 0.01, #P < 0.05, $P < 0.05, Fig. 5D and E). Moreover, the concentrations of LDH, CK and MDA were up-regulated while the concentration of SOD was down-regulated by LY294002 treatment compared with MI/R + vitexin 200 μM group (**P < 0.01, #P < 0.05, $P < 0.05, Fig. 5F–I). LY294002 treatment also increased the expression of Beclin1 and decreased the expression of p62 compared with MI/R + vitexin 200 μM group (**P < 0.01, #P < 0.05, $P < 0.05, Fig. 5J and K). The above results indicated that PI3K inhibitor LY294002 abolished the cardioprotective effects of vitexin, suggesting that vitexin suppressed apoptosis and autophagy of H9c2 cells through activating PI3K/Akt/mTOR signaling pathway in MI/R injury.
Cardioprotective effects of vitexin in vivo
Animal model of MI/R injury was constructed to investigate the effect of vitexin in vivo. Increased mean myocardial infarction area and decreased HR were detected in MI/R model group compared with Sham group. The concentration of SOD decreased while the concentration of MDA increased in MI/R model group compared with Sham group. HE stained showed that there were more pathological changes of apoptosis and necrosis in MI/R model group than Sham group. However, vitexin treatment obviously improved MI/R injury by decreasing mean myocardial infarction area and the concentration of MDA and increasing HR and the concentration of SOD compared with MI/R model group in a dose-dependent manner (**P < 0.01, #P < 0.05, $P < 0.05, Fig. 6A–E). Results from TUNEL assay showed that the high apoptosis index caused by MI/R injury was down-regulated by vitexin treatment significantly (Fig. 6F and G). Apart from that, the expression of Beclin1 and the rate of LC3 II/I was down-regulated while the expression of p-Akt and mTOR was up-regulated by vitexin treatment dose-dependently compared with MI/R group (**P < 0.01, #P < 0.05, ##P < 0.01, Fig. 6H–K). These results suggested that vitexin ameliorated MI/R injury by inhibiting apoptosis and autophagy through the activation of PI3K/Akt/mTOR signaling pathway in vivo.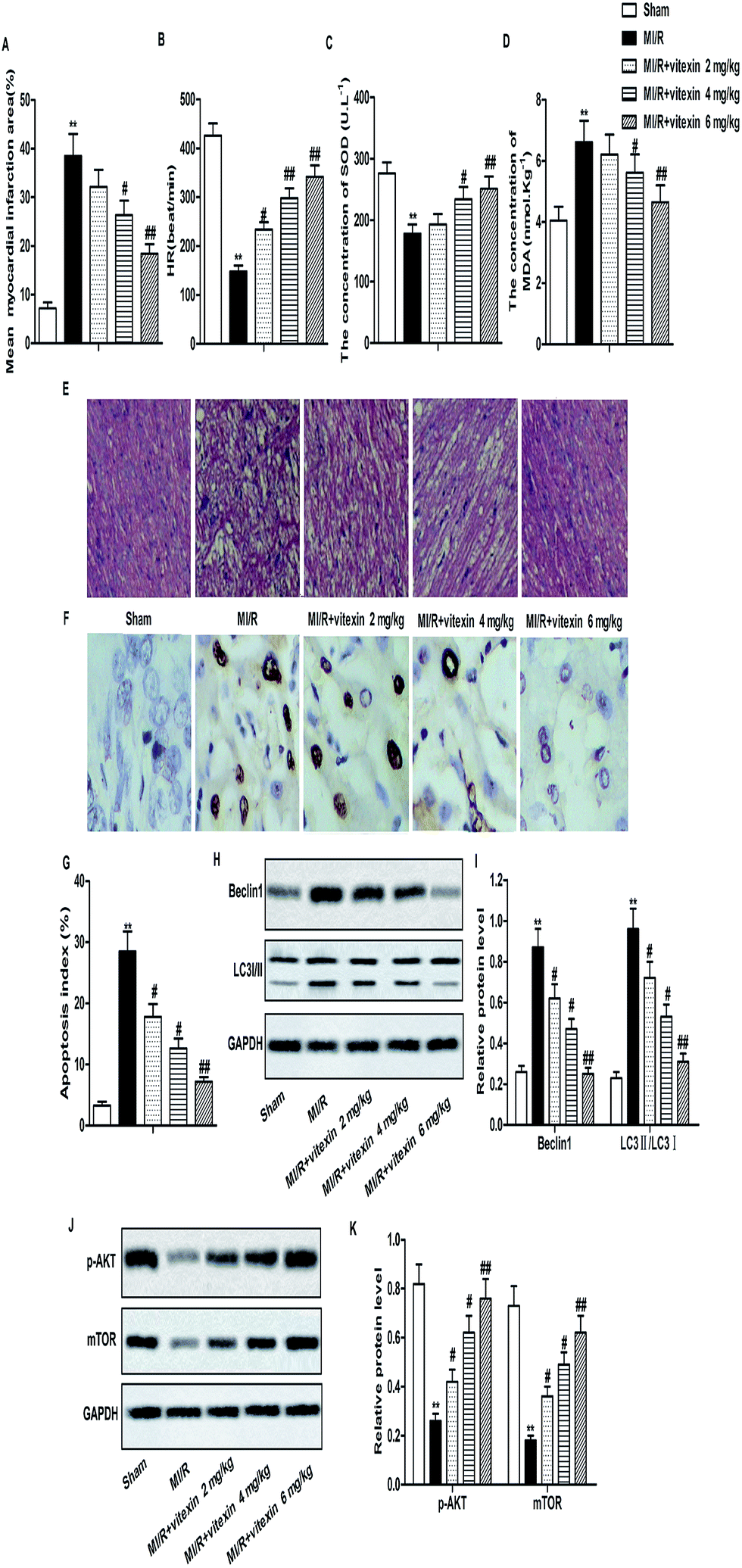 | ||
| Fig. 6 Cardioprotective effect of vitexin in vivo. C57BL/6 mice were divided into Sham group, MI/R group and MI/R + vitexin treated group (2, 4, 6 mg kg−1 respectively). (A) Mean myocardial infarct area was measured by Evans blue and tetrazoliumchloride (TTC) staining. (B) Heart rates were recorded by electrocardiogram. (C and D) The concentrations of SOD and MDA were measured by detection kits respectively. (E) Hematoxylin and eosin (H&E) staining was used to observe histopathologic changes in mice hearts. (F and G) Cell apoptosis rates in different groups were measured by TUNEL assay. (H and I) Relative protein level of Beclin1 and LC3I/II in different groups was measured by western blot. (J and K) Relative protein level of p-Akt and mTOR in different groups was measured by western blot. The bars showed means ± SD of three independent experiments. **P < 0.01 compared with Sham group, #P < 0.05, ##P < 0.01 compared with MI/R group. | ||
Discussion
Vitexin has been widely used for the treatment of asthma and coughs. It has also been reported that vitexin exerted anti-inflammation, anti-oxidation and anti-tumor effects.19,20 Here, in our present study, we showed that vitexin inhibited excessive autophagy to suppress apoptosis in rat cardiomyocytes H9c2 with stimulated MI/R injury and mitigated MI/R injury by inhibiting excessive autophagy in constructed MI/R injury animal model. Moreover, we also reported potential mechanism of vitexin's cardioprotective effects involving apoptosis, autophagy and PI3K/Akt/mTOR signaling pathway.Apoptosis is a self-suicide process which eliminates damaged, aged or mutant cells. Under some pathological conditions, insufficient or enhanced apoptosis can result in tissue damage and dysfunction.21,22 Apoptosis is often occurs in cardiomyocytes after MI/R injury. Song's study suggested that apoptosis played a vital role in the development of chronic MI/R injury.23 Therefore, inhibition of apoptosis can attenuate the irreversible injury induced by MI/R. Previous study indicated that apoptosis was regulated by numerous genes including Bcl-2 and Bax.24 Bcl-2 is an anti-apoptotic protein which binds to the mitochondrial membrane and blocks the release of cytochrome c to inhibit cellular death.25 Bax is considered as a pro-apoptotic molecular which remains in an inactive conformation in healthy cells. In response to apoptotic stimuli, Bax undergoes conformational activation and blocks the anti-apoptotic effect of Bcl-2.26,27 Caspase-3 is a class of cysteine proteases which is responsible for the proteolytic cleavage of many vital proteins related to apoptosis and plays an important role in apoptosis.27 In our study, we observed that vitexin suppressed apoptosis of rat cardiomyocytes H9c2 suffered from stimulated MI/R injury by decreasing the expression of cleaved Caspase-3 and Bax while increasing the expression of Bcl-2. Similarly, study by Che suggested that vitexin treatment suppressed apoptosis of rats' myocardial cells in MI/R injury.28 Another study from Dong reported that vitexin inhibited inflammatory response and apoptosis to protect MI/R injury in Langendorff-perfused rat.13 As we know, SOD is an important radical superoxide scavengers which protect cells from oxidative damage.29 MDA is a decomposition product of lipid hydroperoxides which is often used as an indicator of oxidative damage to cells and tissues.30 Previous studies also indicated that CK and LDH are both sensitive markers of myocardial damage.31,32 Thus we chose these markers to detect cardiomyocyte injury in our study. Moreover, Dong's another study also reported that vitexin exerted cardioprotective and anti-oxidative effects against MI/R injury through up-regulating activities of SOD and down-regulating activities of LDH, CK and the content of MDA.12 In agreement with his study, we also observed that the increased concentrations of LDH, CK and MDA in MI/R model group implied the deleterious effects to cardiocytes during ischemia/reperfusion. The concentration of SOD which protects cells from oxidative damage is decreased in MI/R injury. However, vitexin decreased the concentrations of LDH, CK and MDA and increased the concentrations of SOD, indicating that vitexin mitigates myocardial ischemia reperfusion-induced damage in H9c2 cells.
Several studies reported that MI/R injury induced various types of cell death of myocardial cells including apoptosis and autophagy.5 However, the crosstalk between autophagy and apoptosis is needed to be clear. Autophagy is an important intracellular self-degradation process which can be activated by MI/R injury. Autophagy is essential in cellular energy mobilization and homeostasis, but excessive autophagy leads to cardiac dysfunction and disorders during MI/R injury. A great number of proteins have been proved to participate in autophagy. Beclin1, which is increased during periods of cell stress and extinguished during the cell cycle, is also necessary for the induction of autophagy.33 P62 protein is reported to clear the autophagosome in order to keep hemostasis.34 The conversion of LC3I to LC3II is reported to contribute to the formation of autophagosome and LC3II is often considered as a marker of autophagy.35 He's research elucidated that vitexin suppressed autophagy in hepatocellular carcinoma.15 Whether vitexin suppressed autophagy in cardiomyocytes during MI/R was still remained unclear. In this present study, we found that vitexin treatment markedly decreased Beclin1, LC3I/II protein level and the number of GFP-LC3 dots while increased p62 protein level compared to MI/R model group, suggesting that vitexin may improve myocardial damage by inhibiting autophagy of cardiomyocytes in MI/R injury. In addition, we also found that activation of autophagy counteracted the inhibitory effect of vitexin on apoptosis and myocardial injury, indicating that vitexin inhibited apoptosis of H9c2 cells via suppressing excessive autophagy to reduce myocardial injury.
It was reported that PI3K/Akt/mTOR signaling pathway is essential for the regulation of autophagy and inhibition of PI3K/Akt/mTOR pathway activated autophagy.36 In the PI3K/Akt/mTOR signaling pathway, mTOR is in the downstream of the PI3K/Akt pathway and its activity is mainly regulated by the PI3K/Akt signaling pathway.37 Previous study indicated that autophagy is negatively regulated by the activation of mTOR.38 In this present study, vitexin treatment up-regulated the expression of p-Akt and mTOR compared with MI/R model group, suggesting that vitexin might suppress autophagy via activation of PI3K/Akt/mTOR signaling pathway. In order to verify our hypothesis, PI3K inhibitor LY294002 was administrated to H9c2 cells. The co-treatment with LY294002 increased cleaved Caspase-3 protein level and decreased Bcl-2 protein level compared with MI/R + vitexin 200 μM group. The down-regulation of Beclin1 protein level by vitexin was reversed by LY294002, as well as the up-regulation of p62. These results supported our conclusion that vitexin inhibited apoptosis and autophagy to ameliorate MI/R injury through activation of PI3K/Akt/mTOR signaling pathway.
Having known the cardioprotective effects of vitexin in vitro, in vivo experiments were further carried out to investigate the effects of vitexin in MI/R injury. Previous studies demonstrated that vitexin exerted cardioprotective effect on MI/R injury in rats.12,13 Similarly, in our study, we observed that vitexin treatment mitigated myocardial ischemia reperfusion-induced damage in the heart of mice in a dose-dependent manner. Vitexin also suppressed apoptosis and autophagy through activation of PI3K/Akt/mTOR pathway in vivo dose-dependently.
In summary, vitexin has a protective effect against MI/R injury both in vivo and in vitro. Vitexin mitigated MI/R injury through inhibiting excessive autophagy to suppress apoptosis via activation of PI3K/Akt/mTOR signaling pathway, laying a foundation for the mechanism research and treatment for MI/R injury.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| MI/R | Myocardial ischemia reperfusion |
| HR | Heart rate |
| ECG | Electrocardiograph |
| HE | Hematoxylin and eosin |
| TTC | Tetrazoliumchloride |
| SD | Standard deviation |
References
- N. J. Kassebaum, A. Bertozzi-Villa, M. S. Coggeshall, K. A. Shackelford, C. Steiner, K. R. Heuton, D. Gonzalez-Medina, R. Barber, C. Huynh and D. Dicker, et al., Global, regional, and national levels and causes of maternal mortality during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013, Lancet, 2014, 384(9947), 980–1004 CrossRef.
- A. Frank, M. Bonney, S. Bonney, L. Weitzel, M. Koeppen and T. Eckle, Myocardial ischemia reperfusion injury: from basic science to clinical bedside, Semin. Cardiothorac. Vasc. Anesth., 2012, 16(3), 123–132 CrossRef PubMed.
- Z. V. Schofield, T. M. Woodruff, R. Halai, M. C. Wu and M. A. Cooper, Neutrophils–a key component of ischemia-reperfusion injury, Shock, 2013, 40(6), 463–470 CrossRef CAS PubMed.
- D. M. Yellon and D. J. Hausenloy, Myocardial reperfusion injury, N. Engl. J. Med., 2007, 357(11), 1121–1135 CrossRef CAS PubMed.
- X. F. Tian, M. X. Cui, S. W. Yang, Y. J. Zhou and D. Y. Hu, Cell death, dysglycemia and myocardial infarction, Biomed. Rep., 2013, 1(3), 341–346 CrossRef CAS PubMed.
- T. J. Bunch, S. H. Hohnloser and B. J. Gersh, Mechanisms of sudden cardiac death in myocardial infarction survivors: insights from the randomized trials of implantable cardioverter-defibrillators, Circulation, 2007, 115(18), 2451–2457 CrossRef PubMed.
- V. Nikoletopoulou, M. Markaki, K. Palikaras and N. Tavernarakis, Crosstalk between apoptosis, necrosis and autophagy, Biochim. Biophys. Acta, 2013, 1833(12), 3448–3459 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Autophagy fights disease through cellular self-digestion, Nature, 2008, 451(7182), 1069–1075 CrossRef CAS PubMed.
- K. Wei, P. Wang and C. Y. Miao, A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury, CNS Neurosci. Ther., 2012, 18(11), 879–886 CrossRef PubMed.
- Y. Wang, Y. Zhen, X. Wu, Q. Jiang, X. Li, Z. Chen, G. Zhang and L. Dong, Vitexin protects brain against ischemia/reperfusion injury via modulating mitogen-activated protein kinase and apoptosis signaling in mice, Phytomedicine, 2015, 22(3), 379–384 CrossRef CAS PubMed.
- M. C. Prabhakar, H. Bano, I. Kumar, M. A. Shamsi and M. S. Khan, Pharmacological investigations on vitexin, Planta Med., 1981, 43(4), 396–403 CrossRef CAS PubMed.
- L. Y. Dong, S. Li, Y. L. Zhen, Y. N. Wang, X. Shao and Z. G. Luo, Cardioprotection of vitexin on myocardial ischemia/reperfusion injury in rat via regulating inflammatory cytokines and MAPK pathway, Am. J. Chin. Med., 2013, 41(6), 1251–1266 CrossRef CAS PubMed.
- L. Dong, Y. Fan, X. Shao and Z. Chen, Vitexin protects against myocardial ischemia/reperfusion injury in Langendorff-perfused rat hearts by attenuating inflammatory response and apoptosis, Food Chem. Toxicol., 2011, 49(12), 3211–3216 CrossRef CAS PubMed.
- S. Hu, S. Cao and J. Liu, Role of angiopoietin-2 in the cardioprotective effect of fibroblast growth factor 21 on ischemia/reperfusion-induced injury in H9c2 cardiomyocytes, Exp. Ther. Med., 2017, 14(1), 771–779 CrossRef PubMed.
- J. D. He, Z. Wang, S. P. Li, Y. J. Xu, Y. Yu, Y. J. Ding, W. L. Yu, R. X. Zhang, H. M. Zhang and H. Y. Du, Vitexin suppresses autophagy to induce apoptosis in hepatocellular carcinoma via activation of the JNK signaling pathway, Oncotarget, 2016, 7(51), 84520–84532 Search PubMed.
- C. Chen, W. Lu, G. Wu, L. Lv, W. Chen, L. Huang, X. Wu, N. Xu and Y. Wu, Cardioprotective effects of combined therapy with diltiazem and superoxide dismutase on myocardial ischemia-reperfusion injury in rats, Life Sci., 2017, 183, 50–59 CrossRef CAS PubMed.
- C. Chen, Z. Nong, M. Meng, Q. Wen, X. Lin, F. Qin, J. Huang and R. Huang, Toxicological evaluation of Yulangsan polysaccharide in Wistar rats: A 26-week oral gavage study, Environ. Toxicol. Pharmacol., 2016, 41, 1–7 CrossRef CAS PubMed.
- H. Guan and H. Piao, 2,5-Hexanedione induces autophagic death of VSC4.1 cells via a PI3K/Akt/mTOR pathway, 2017 Search PubMed.
- M. Bhardwaj, S. Paul, R. Jakhar and S. C. Kang, Potential role of vitexin in alleviating heat stress-induced cytotoxicity: Regulatory effect of Hsp90 on ER stress-mediated autophagy, Life Sci., 2015, 142, 36–48 CrossRef CAS PubMed.
- Y. Zhou, Y. E. Liu, J. Cao, G. Zeng, C. Shen, Y. Li, M. Zhou, Y. Chen, W. Pu and L. Potters, et al., Vitexins, nature-derived lignan compounds, induce apoptosis and suppress tumor growth, Clin. Cancer Res., 2009, 15(16), 5161–5169 CrossRef CAS PubMed.
- K. Chinda, J. Sanit, S. Chattipakorn and N. Chattipakorn, Dipeptidyl peptidase-4 inhibitor reduces infarct size and preserves cardiac function via mitochondrial protection in ischaemia-reperfusion rat heart, Diabetes Vasc. Dis. Res., 2014, 11(2), 75–83 CrossRef PubMed.
- W. Z. Sun, M. H. Li, M. Chu, L. L. Wei, M. Y. Bi, Y. He and L. B. Yu, Id1 knockdown induces the apoptosis and inhibits the proliferation and invasion of ovarian cancer cells, Eur. Rev. Med. Pharmacol. Sci., 2016, 20(13), 2812–2818 Search PubMed.
- J. Q. Song, X. Teng, Y. Cai, C. S. Tang and Y. F. Qi, Activation of Akt/GSK-3beta signaling pathway is involved in intermedin(1-53) protection against myocardial apoptosis induced by ischemia/reperfusion, Apoptosis, 2009, 14(11), 1299–1307 CrossRef CAS PubMed.
- Y. Wang, X. Li, X. Wang, W. Lau, Y. Wang, Y. Xing, X. Zhang, X. Ma and F. Gao, Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3beta signaling and inhibition of the mitochondria-dependent apoptotic pathway, PLoS One, 2013, 8(8), e70956 CAS.
- C. L. Xu, Q. Z. Wang, L. M. Sun, X. M. Li, J. M. Deng, L. F. Li, J. Zhang, R. Xu and S. P. Ma, Asiaticoside: attenuation of neurotoxicity induced by MPTP in a rat model of Parkinsonism via maintaining redox balance and up-regulating the ratio of Bcl-2/Bax, Pharmacol., Biochem. Behav., 2012, 100(3), 413–418 CrossRef CAS PubMed.
- A. Gross, J. M. McDonnell and S. J. Korsmeyer, BCL-2 family members and the mitochondria in apoptosis, Genes Dev., 1999, 13(15), 1899–1911 CrossRef CAS PubMed.
- R. S. Whelan, V. Kaplinskiy and R. N. Kitsis, Cell death in the pathogenesis of heart disease: mechanisms and significance, Annu. Rev. Physiol., 2010, 72, 19–44 CrossRef CAS PubMed.
- X. Che, X. Wang, J. Zhang, C. Peng, Y. Zhen, X. Shao, G. Zhang and L. Dong, Vitexin exerts cardioprotective effect on chronic myocardial ischemia/reperfusion injury in rats via inhibiting myocardial apoptosis and lipid peroxidation, Am. J. Transl. Res., 2016, 8(8), 3319–3328 Search PubMed.
- P. A. Olsvik, T. Kristensen, R. Waagbo, B. O. Rosseland, K. E. Tollefsen, G. Baeverfjord and M. H. Berntssen, mRNA expression of antioxidant enzymes (SOD, CAT and GSH-Px) and lipid peroxidative stress in liver of Atlantic salmon (Salmo salar) exposed to hyperoxic water during smoltification, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2005, 141(3), 314–323 CrossRef CAS PubMed.
- D. Bonnes-Taourel, M. C. Guerin and J. Torreilles, Is malonaldehyde a valuable indicator of lipid peroxidation?, Biochem. Pharmacol., 1992, 44(5), 985–988 CrossRef CAS PubMed.
- S. L. Badole, S. M. Chaudhari, G. B. Jangam, A. D. Kandhare and S. L. Bodhankar, Cardioprotective Activity of Pongamia pinnata in Streptozotocin-Nicotinamide Induced Diabetic Rats, BioMed Res. Int., 2015, 2015, 403291 Search PubMed.
- M. Gu, A. B. Zheng, J. Jin, Y. Cui, N. Zhang, Z. P. Che, Y. Wang, J. Zhan and W. J. Tu, Cardioprotective Effects of Genistin in Rat Myocardial Ischemia-Reperfusion Injury Studies by Regulation of P2X7/NF-kappaB Pathway, J. Evidence-Based Complementary Altern. Med., 2016, 2016, 5381290 Search PubMed.
- R. Kang, H. J. Zeh, M. T. Lotze and D. Tang, The Beclin 1 network regulates autophagy and apoptosis, Cell Death Differ., 2011, 18(4), 571–580 CrossRef CAS PubMed.
- S. Wang and C. Wang, N-Acetylcysteine Attenuates Diabetic Myocardial Ischemia Reperfusion Injury through Inhibiting Excessive Autophagy, 2017, 2017, 9257291 Search PubMed.
- P. Gimenez-Xavier, R. Francisco, A. F. Santidrian, J. Gil and S. Ambrosio, Effects of dopamine on LC3-II activation as a marker of autophagy in a neuroblastoma cell model, NeuroToxicology, 2009, 30(4), 658–665 CrossRef CAS PubMed.
- S. Saiki, Y. Sasazawa, Y. Imamichi, S. Kawajiri, T. Fujimaki, I. Tanida, H. Kobayashi, F. Sato, S. Sato and K. Ishikawa, et al., Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition, Autophagy, 2011, 7(2), 176–187 CrossRef CAS PubMed.
- T. Luo, G. Liu, H. Ma, B. Lu, H. Xu, Y. Wang, J. Wu, P. Ge and J. Liang, Inhibition of autophagy via activation of PI3K/Akt pathway contributes to the protection of ginsenoside Rb1 against neuronal death caused by ischemic insults, Int. J. Mol. Sci., 2014, 15(9), 15426–15442 CrossRef CAS PubMed.
- C. Wang, X. Zhang, Z. Teng, T. Zhang and Y. Li, Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice, Eur. J. Pharmacol., 2014, 740, 312–320 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |