
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High pressure effects on hydrate Cu-BTC investigated by vibrational spectroscopy and synchrotron X-ray diffraction
Zhaohui Donga,
Zhongying Mi *b,
Weiguang Shi*c,
Hui Jianga,
Yi Zhenga and
Ke Yanga
*b,
Weiguang Shi*c,
Hui Jianga,
Yi Zhenga and
Ke Yanga
aShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, CAS, Shanghai, 201204, P. R. China
bSchool of Earth and Space Sciences, Peking University, Beijing, 100871, P. R. China. E-mail: mornsunlight313@gmail.com; Tel: +86-021-33933188
cCollege of Chemistry & Chemical Engineering, Northeast Petroleum University, Daqing, 163318, P. R. China
First published on 7th December 2017
Abstract
The high pressure behaviors of hydrate Cu-BTC metal–organic framework (MOF) in terms of phase stability, compressibility and reversibility were investigated in situ by synchrotron X-ray powder diffraction as well as vibrational spectroscopy. Two phase transitions, caused by the interaction of water and the sample framework, were revealed by the vibrational spectroscopies. Compressibility of the hydrate Cu-BTC also displays soft and hard regimes, which is the same scenario as non-hydrate Cu-BTC with a pressure transmitting medium. It is further confirmed that the residual water molecules in hydrate Cu-BTC can serve as a pressure transmitting medium with small molecule size under high pressure. Our results not only prove the high stability of Cu-BTC but also provide spectroscopic evidence for the interactions taking place between the guest molecules and the sample framework. Such findings could provide further guidelines for improving Cu-BTC's absorption and storage abilities.
1. Introduction
Metal organic framework materials (MOFs) are porous materials based on the classical metal ions (such as Zn2+, Cu2+, and Al3+) with electron donors (such as carboxylic acid ester or amine).1,2 In contrast to aluminosilicate zeolite, MOFs have more tunable architectures and unique chemical properties due to the variable and flexible organic chains in their structures. MOFs, as an intriguing class of porous functional materials, have been extensively studied for decades owing to their promising applications and excellent performance in gas storage,3–8 hydrocarbon separation,9,10 catalysis11–15 and drug delivery.16–19Normally, the porosity of MOFs, i.e., pore size and geometry, is controlled by synthetic routes, for example, by adjusting the size of the organic linkers.16–19 Such modifications are associated with changes in the functional groups. In addition to traditional synthetic routes, high pressure can provide a clean outer force to further tune pore structures without changing the functional groups. The pore structures are very sensitive to pressure, and many meaningful changes have occurred in a relatively low pressure range (<10 GPa); some of them even occur below 2 GPa.1,8,20–26 Previous studies have revealed that pressure could induce diverse changes in the pore geometry of MOFs,24,27 which consequently affect the absorption capacity, absorption selectivity, and access to binding sites. In addition to the pore geometry, how the pore traps or interacts with guest molecules under high pressure also affects MOFs' absorption abilities.23 Thus, exploring the pressure dependence of MOFs' functionality is crucial to develop application-specific products.
One of the common phenomena observed in MOFs is pressure induced amorphization (PIA). For example, in ZIF-8 (ref. 24) and MOF-5,27 PIA was observed at 0.34 GPa and 0.35 GPa, respectively. A similar scenario was also found in single crystal ZIF-4 but at a higher pressure, between 5.1 GPa and 6.5 GPa.1 In addition to PIA, pressure induced lattice rearrangement was also found in MOFs, normally below 1 GPa. For instance, ZIF-4 underwent a pressure induced phase transformation from orthorhombic to monoclinic phase between 0.13 GPa and 0.56 GPa.1 Spencer et al. observed a crystalline to crystalline phase transition occurring bin the range of 0.5–0.8 GPa using neutral zinc imidazolate (Zn(Im)2).26 It is widely recognized that the stability and mechanical properties of MOFs make them an advanced research hot spot owing to their rigid and soft regions in a group of materials which flex and squeeze upon compression. Previous high pressure studies have revealed that MOFs have high compressibility comparable to molecular crystals. Bulk moduli of most MOFs fall in the range of 5–30 GPa, such as ZIF-8 (6.5 GPa),24 ZIF-4 (7.8 GPa),1 NH2-MIL-53(In) (10.9 GPa),28 ZAG-4 (11.7 GPa),29, [tmenH2][Er(HCOO)4]2 (13.8 GPa),30 Zn(Im)2 (14 GPa),21 LiB(IM)4 (16.6 GPa),21 and MOF-5 (16.5 GPa).27
Cu based MOF is a commercially available highly porous material named copper benzene-1,3,5-tricarboxylate (Cu-BTC). It has been proposed as a suitable compound for CO2 capture and gas separation purposes.10,31 However, few high pressure experiments have been carried out on Cu-BTC. Moreover, few studies of Cu-BTC showed excellent results in comparison with other MOFs mentioned above. Chapman et al. documented a change in the equation of state in Cu-BTC upon compression by introducing different pressure transmitting media into the sample.23,25 Moreover, Cu-BTC exhibited an intrinsic property of high resistance to pressure when it was continually compressed up to 8 GPa.25 However, no high pressure study was carried out beyond 8 GPa, leaving the stability and high pressure behaviors of Cu-BTC beyond 8 GPa still unclear.
In this study, we extended the high pressure region of Cu-BTC up to 20 GPa. High pressure behaviors of Cu-BTC were investigated by in situ Raman spectroscopy, IR spectroscopy and X-ray diffraction with synchrotron radiation. Structural stability, compressibility, and phase transition reversibility were examined. By employing vibrational spectroscopy in addition to XRD, we expect to gain a better understanding of the extraordinary properties of Cu-BTC porous materials under high pressure, particularly the interactions taking place between guest molecules and sample frameworks.
2. Experiments
Copper benzene-1,3,5-tricarboxylate (Cu-BTC) was purchased from Sigma Aldrich. The powder Cu-BTC sample was used without further treatment. Morphologies of Cu-BTC before and after compression were examined by scanning electronic microscopy (SEM, FEI 1540XB FIB/SEM). In situ structural characterization was performed using vibrational spectroscopy and synchrotron X-ray diffraction (XRD).In situ high pressure Raman and XRD measurements were carried out using a symmetric diamond anvil cell (DAC) with a pair of type I diamonds. The diamond anvils have culet size of 500 μm. A stainless steel gasket with a hole of 180 μm drilled in the center was used as the sample chamber. For the in situ infrared studies, a DAC equipped with a pair of 400 μm type II diamonds was used. Similar to the Raman and XRD measurements, a 180 μm hole drilled in a stainless steel gasket was used as the sample chamber. For all experiments, the pressure was determined by the well-established ruby fluorescence method,32 and no pressure transmitting medium was used.
The Raman spectra were collected using a Renishaw inVia Raman microscopy system with a 633 nm line excited by a He–Ne laser. The infrared measurements were carried out on a Bruker VERTEX 70v FTIR spectrometer and a custom IR microscope at the Center for High Pressure Science and Technology Advanced Research, Beijing.
The angle dispersive XRD synchrotron experiments were accomplished at the 12.2.2 beamline at the Advanced Light Source (ALS) at Lawrence Berkeley Laboratory (LBL), as well as the BL15U beamline at Shanghai Synchrotron Facility (SSRF). High pressure XRD experiments at ALS were conducted using a monochromatic beam with a wavelength of 0.4132 nm (30 keV) and a focused beam size of 10 × 10 μm2. A MAR CCD detector was used to collect the 2D Debye–Scherrer patterns. The diffraction parameters were calibrated using LaB6 standard pattern. XRD measurements at SSRF were conducted using a 0.6188 nm monochromatic beam. CeO2 was used as the calibrant for experimental parameters. The beam size was focused down to 5 × 4 μm2 by a K-B mirror. The 2D Debye–Scherrer diffraction patterns were integrated using Fit2D program for further analysis.
3. Results and discussions
3.1 Characterization of Cu-BTC at ambient pressure
The porous framework of Cu-BTC with cubic structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) is depicted in Fig. 1. Cu(II) ions form bridges with the carboxylate groups on discrete 1,3,5-benzenetricarboxylate (BTC) ligands. Two adjacent Cu(II) ions constitute Cu(II) dimers. Such Cu(II) dimers link via the trigonal BTC molecules, and form extended three dimensional (3D) networks. The 3D network consists of three types of cages with different sizes, among which the largest has a diameter of 10 Å.33 The SEM result shows a particle size of tens of microns (Fig. 2).
m) is depicted in Fig. 1. Cu(II) ions form bridges with the carboxylate groups on discrete 1,3,5-benzenetricarboxylate (BTC) ligands. Two adjacent Cu(II) ions constitute Cu(II) dimers. Such Cu(II) dimers link via the trigonal BTC molecules, and form extended three dimensional (3D) networks. The 3D network consists of three types of cages with different sizes, among which the largest has a diameter of 10 Å.33 The SEM result shows a particle size of tens of microns (Fig. 2).
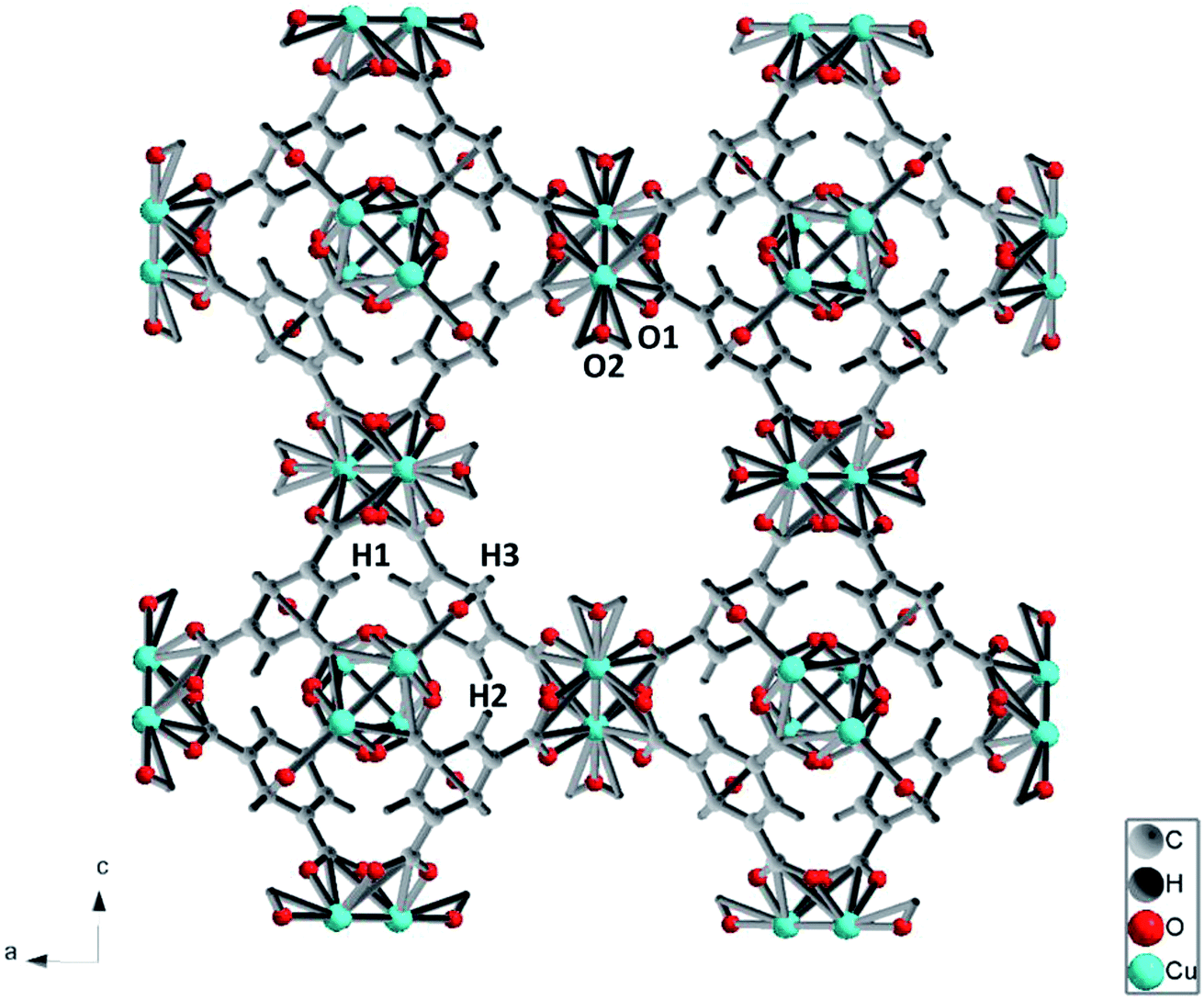 | ||
| Fig. 1 A presentation of the structure of Cu-MOF frame work. Color code: Cu atoms, green; O atoms, red; H atoms, dark grey; C atoms, grey. O1 and O2 denote oxygen atoms attached to Cu atom at equatorial and axial position, respectively. | ||
 | ||
| Fig. 2 SEM images of Cu-BTC taken before (a) and after (b) compression. | ||
The vibrational spectra of Cu-BTC at ambient conditions are shown in Fig. 3. In the Raman spectrum (Fig. 3a), regions of 1200–1400 cm−1 and >1800 cm−1 are excluded due to the intense Raman peaks of diamond at 1335 cm−1 and 2400 cm−1. Raman peaks located at 1618 cm−1 and 1006 cm−1 are associated with the benzene ring stretch modes, labeled as νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ar) in the figure. The peaks at 743 cm−1 and 830 cm−1 are assigned to out-of-plane ring bending (δCC(Ar)) and out-of-plane C–H bending of the benzene ring (δC–H(Ar)). The double peaks at 1465 cm−1 and 1548 cm−1 are ascribed to the symmetric and asymmetric stretching modes of the carboxylate groups, which are labeled as νsym COO− and νasym COO−, respectively. In the low frequency region (100–600 cm−1), two peaks are observed at 191 cm−1 and 501 cm−1, which are associated with the Cu–O stretch. As shown in Fig. 1, there are two similar Cu–O bonds in the frame network. The Cu–O1 lies on the equatorial position, while the Cu–O2 bond lies on the axial position. The bond lengths for Cu–O1 and Cu–O2 are obtained as 1.941 Å and 2.277 Å, respectively. Referring to the Hooke's law, the Cu–O1 stretch should be obtained at a higher frequency. Therefore, the two Raman modes observed at 501 cm−1 and 191 cm−1 are relevant to the equatorial and axial Cu–O, respectively, which is consistent with a previous study.34
C(Ar) in the figure. The peaks at 743 cm−1 and 830 cm−1 are assigned to out-of-plane ring bending (δCC(Ar)) and out-of-plane C–H bending of the benzene ring (δC–H(Ar)). The double peaks at 1465 cm−1 and 1548 cm−1 are ascribed to the symmetric and asymmetric stretching modes of the carboxylate groups, which are labeled as νsym COO− and νasym COO−, respectively. In the low frequency region (100–600 cm−1), two peaks are observed at 191 cm−1 and 501 cm−1, which are associated with the Cu–O stretch. As shown in Fig. 1, there are two similar Cu–O bonds in the frame network. The Cu–O1 lies on the equatorial position, while the Cu–O2 bond lies on the axial position. The bond lengths for Cu–O1 and Cu–O2 are obtained as 1.941 Å and 2.277 Å, respectively. Referring to the Hooke's law, the Cu–O1 stretch should be obtained at a higher frequency. Therefore, the two Raman modes observed at 501 cm−1 and 191 cm−1 are relevant to the equatorial and axial Cu–O, respectively, which is consistent with a previous study.34
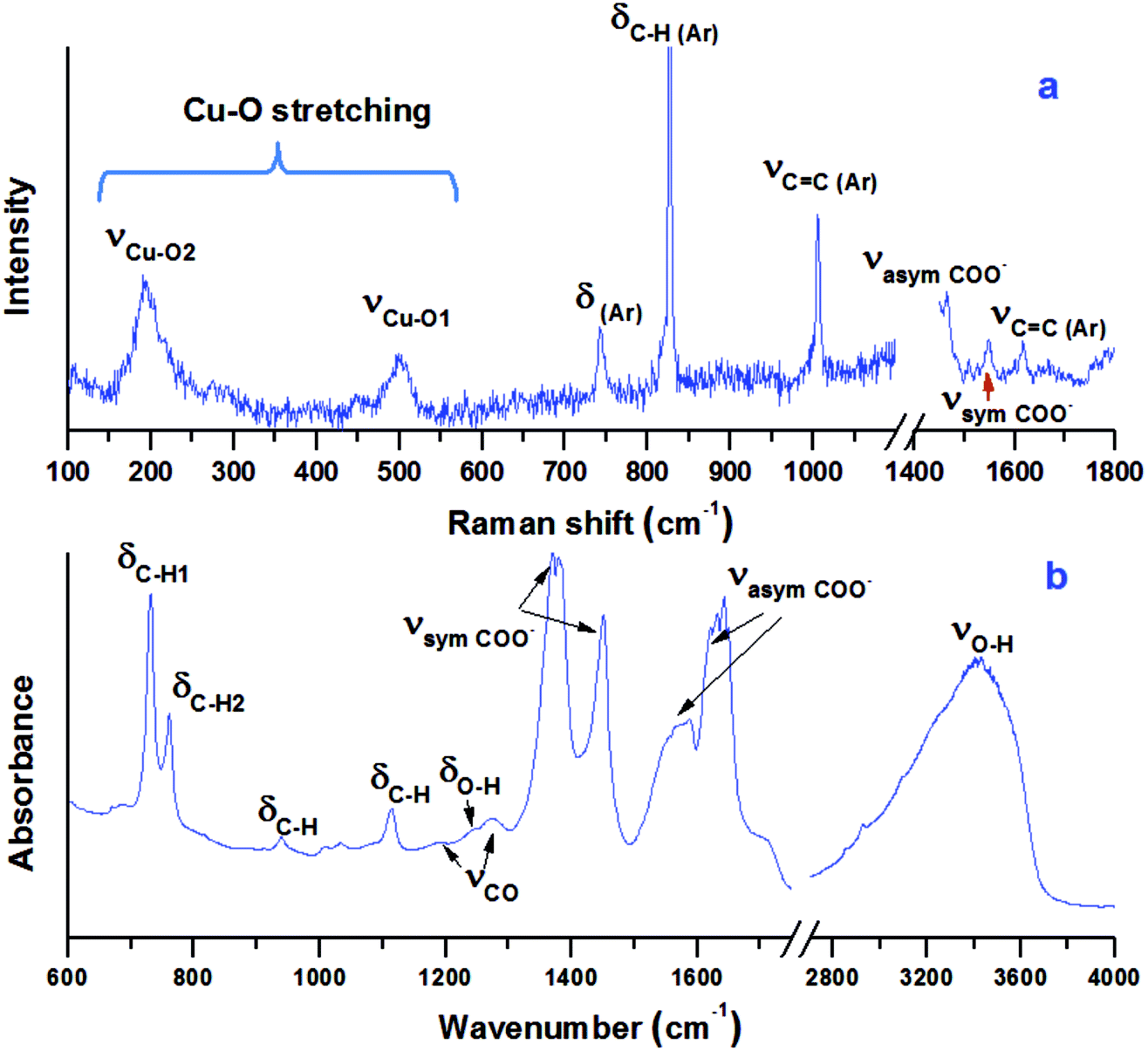 | ||
| Fig. 3 Raman (a) and IR (b) spectra of Cu-BTC obtained at ambient condition in a DAC. Both Raman and IR peaks are assigned and labeled. δ stands for bending, while ν denotes stretching. Ar stands for the benzene ring. | ||
The IR spectrum of the sample is shown in Fig. 3b. The spectrum can be divided into three regions. The first is the region below 1150 cm−1, in which most vibrational modes of the BTC ligand are detected. The two bands at 730 cm−1 and 758 cm−1 are the out-of-plane C–H bending modes, while the band at 1113 cm−1 represents the in-plane C–H bending mode, all of which are associated with the aromatic ring of the BTC.35,36 The second region is from 1150 cm−1 to 1700 cm−1, in which the characteristic bands associated with the carboxylate groups of the BTC ligand are detected. The weak peaks at 1191 cm−1 and 1274 cm−1 are attributed to the C–O stretching modes. The intense peaks at 1373/1449 cm−1 are assigned to the asymmetric stretching of the carboxylate groups, while the peaks at 1571/1630 cm−1 belong to their symmetric stretching. More precisely, the two peaks at 1449 cm−1 and 1571 cm−1 are attributed to the C–C stretching associated with the aromatic rings. The third region is above 2800 cm−1, in which the broad O–H stretch mode of H2O is observed. In addition, the O–H bending mode is detected at 1241 cm−1.37 The existence of O–H bending and stretching modes confirms that the starting sample contained a large amount of water. The IR modes associated with Cu lie below 600 cm−1,36 which are out of the detectable range of the IR system.
3.2 Raman results of Cu-BTC upon compression and decompression
A Cu-BTC sample was studied under high pressure upon compression and decompression by Raman spectroscopy. As shown in Fig. 4, the Raman profile remains unchanged up to 9 GPa, and then a shoulder peak is found at 830 cm−1. Peaks below 600 cm−1, associated with Cu–O vibration, completely vanish and a new broad peak at 280 cm−1 appears. This new peak could be assigned to the absent Cu–Cu stretching of Cu(II) dimers in the framework, which could be enhanced and become visible due to the cell shrinkage at high pressure. Moreover, two new peaks are observed at 675 cm−1 and 804 cm−1, and they become distinguishable beyond 9 GPa. Based on assignment, the Raman region of 600 cm−1 to 1200 cm−1 belongs to vibrations associated with benzene rings such as ring bending and ring related C–H stretching.3 The changes occuring in this region under high pressure strongly suggest that the benzene ring could interact with its surrounding atoms or guest molecules. In conclusion, the apparent changes in the Raman profile imply a possible phase transition occurring at 9 GPa. Upon compression, no more changes are observed up to 15 GPa.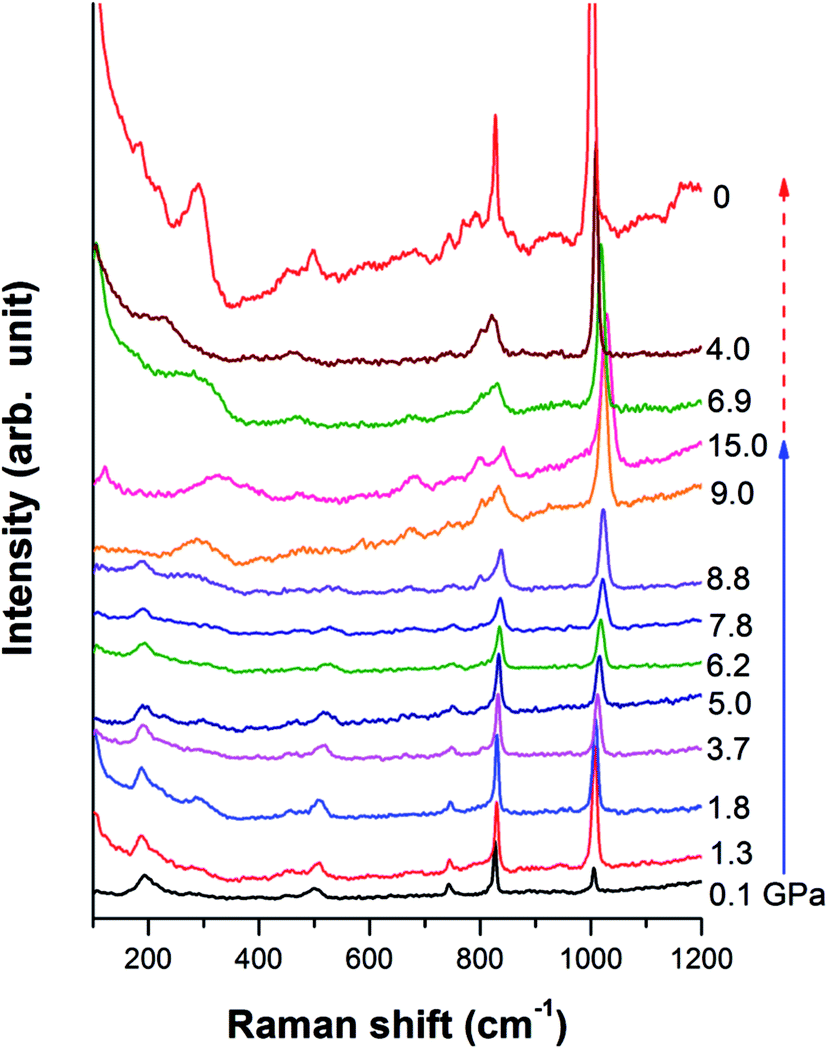 | ||
| Fig. 4 Selected Raman spectra of Cu-BTC upon compression and decompression. Pressure collected for each spectrum is labeled. The solid and dashed lines indicate the compression and decompression sequence, respectively. All the spectra are offset for clarity. | ||
Examination of the pressure effects on the crystal reversibility of Cu-BTC were also conducted upon decompression to gain important information on the transformation mechanism. In general, the intensity of all the Raman peaks increases gradually as pressure decreases, and all the Raman modes shift to lower frequencies. The three new peaks, which appear at high pressure, gradually disappear as pressure releases. When the pressure is completely removed, all Raman peaks observed before compression are recovered, indicating that the phase transition is reversible.
3.3 FTIR results of Cu-BTC upon compression and decompression
In addition to Raman measurements, the sample was also studied by in situ FTIR to investigate asymmetric vibrations. Fig. 5a presents behaviours of out-of-plane C–H bending modes δC–H upon compression. The two peaks remain steady up to 16 GPa and then merge upon further compression. As shown in Fig. 5b, the δCH band at 938 cm−1 split into two observed peaks at 930 cm−1 and 958 cm−1 starting at 4.1 GPa, which could be attributed to the interaction between water and the framework. At the same pressure, the νCO modes as well as the O–H bending mode are completely suppressed. The new peak at 930 cm−1 remains steady up to 19.2 GPa, the highest pressure achieved, while the peak at 958 cm−1 disappears at 17.4 GPa due to pressure suppression. Moreover, the weak νC–H(Ar) mode of the aromatic ring disappears at 4.1 GPa. In addition to the peak splitting of δCH and suppression of νCO, these observations suggest a possible on-site phase transition. The δCH mode found at 1113 cm−1 can still be clearly discerned at 19.2 GPa. The symmetric and asymmetric stretching modes of carboxylate groups (in Fig. 5c) are very stable up to 19.2 GPa with slightly decreased intensity and broadened profile. Upon compression, the νO–H mode exhibits a red shift, indicating bond softening of the hydrogen bond. It is suggested that H2O interacts with its environment (e.g., the Cu-MOF framework) upon compression owing to weakening of hydrogen bonds. At 19.2 GPa, the highest pressure achieved in this study, the characteristic IR peaks are still clearly discernable, suggesting that no complete PIA occurred in Cu-BTC, which is further confirmed by the subsequent XRD results.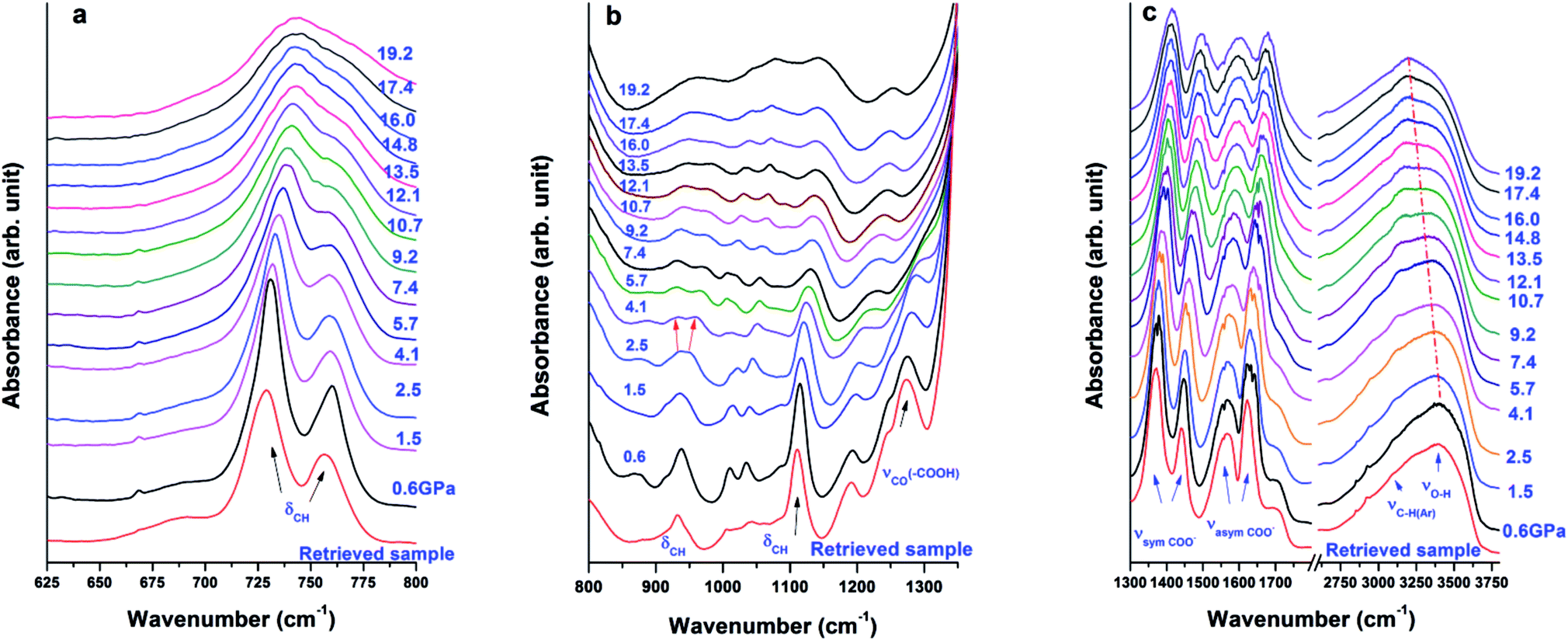 | ||
| Fig. 5 IR spectra of Cu-BTC collected in different regions upon compression, where (a), (b), (c) denote ranges below 800 cm−1, 800–1350 cm−1, and 1300–3800 cm−1, respectively. Pressure for each spectrum is labelled. The dot dashed line in (c) is drawn for eye guidance. The IR spectra at the bottom of each figure are of the retrieved sample after compression. All the spectra are offset for clarity. | ||
Upon decompression, the IR spectrum of the retrieved sample shows an almost identical profile as the one recorded before compression, indicating that the phase transition is completely reversible, which is consistent with the Raman results.
3.4 XRD results of Cu-BTC upon compression and decompression
Fig. 6 shows the structural evolution of Cu-BTC under high pressure. The XRD results confirm that Cu-BTC originally has a cubic structure with cell parameters of a = 26.45 Å at ambient pressure, which is consistent with reference value.34 Upon compression, no significant change is observed in the XRD pattern, except the suppression in intensity of all reflections. Such observation suggests that no structural change occurs in Cu-BTC upon compression. As the pressure is increased to 14.6 GPa, reflections below 6° are still clearly observed, indicating the sample is still in a crystalline phase.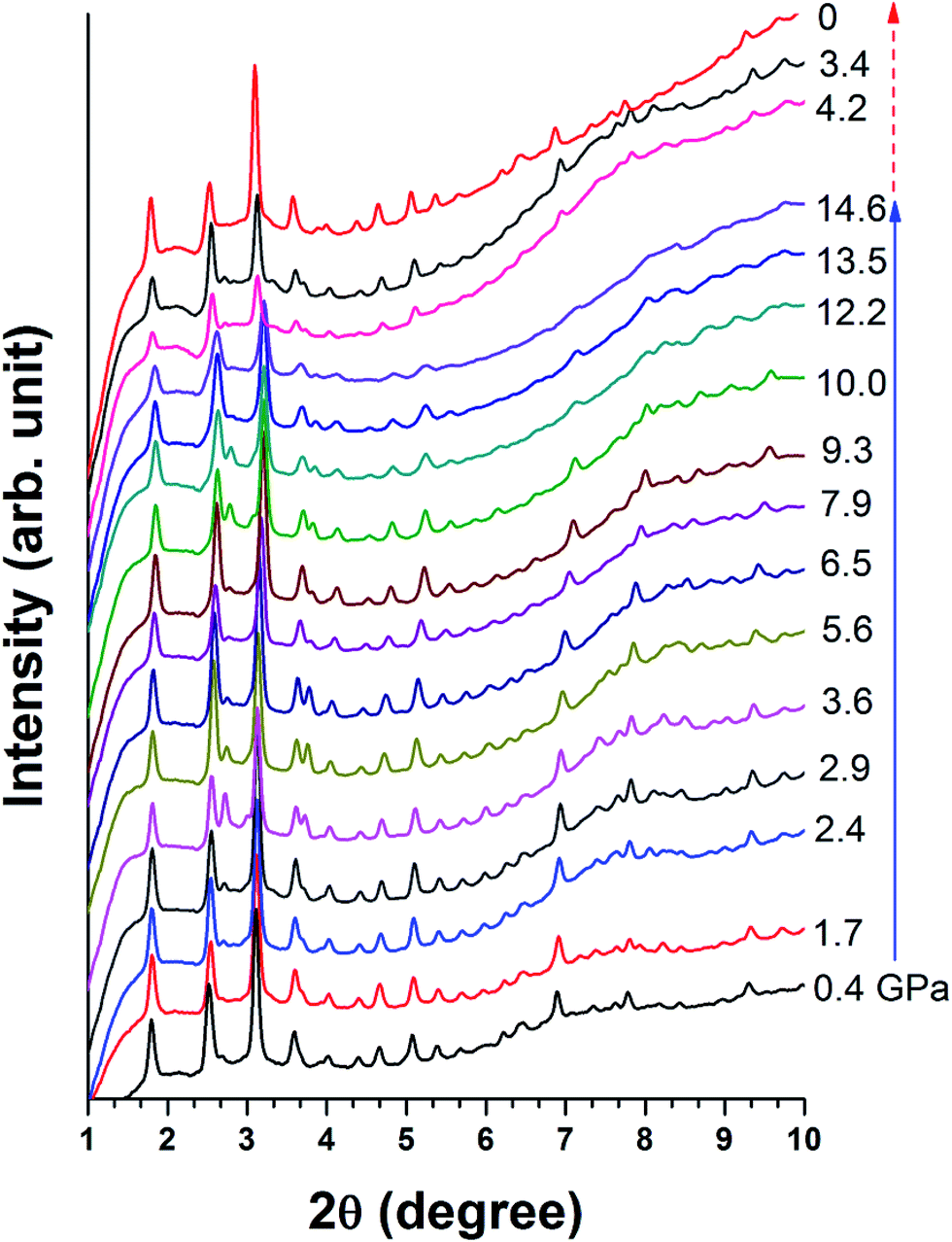 | ||
| Fig. 6 Collected XRD patterns for Cu-BTC upon compression and decompression. The experimental pressure is labeled beside each pattern in GPa. The solid and dashed arrows indicate the compression and decompression sequences, respectively. | ||
Upon decompression, reflections of high 2θ (>6°) that vanished at high pressure reappear. The XRD pattern of the recovered sample has the same profile as the starting material, which confirms that the recovered sample regains its original structure. However, in comparison with the XRD pattern taken at 1.1 GPa, the intensity of each reflection is weaker and slightly broadened. In addition, the diffraction background is also higher. Morphology of the recovered sample was also examined by SEM as shown in Fig. 2b, in which the particle size of Cu-BTC is significantly reduced. Such reduction in particle size and pressure induced sample disorder are attributed to the diffraction profile broadening.
3.5 Discussion
The pressure dependences for all Raman modes are shown in Fig. 7. With a linear fitting, the shifting rate (Δν/Δp) for each Raman mode was obtained and labeled. Upon compression, almost all Raman modes associated with organic linkers shift to higher frequencies at a similar rate of ∼1 cm−1 GPa−1 except the two Cu–O stretching modes (νCu–O1 and νCu–O2). The νCu–O1 mode shifts at a rate of 3.7 cm−1 GPa−1, which is two times more sensitive than the rest. In strong contrast, the νCu–O2 mode is insensitive to pressure, and even has a negative pressure dependence. Such striking difference is ascribed to the different locations of the two bonds in the framework of Cu-BTC. As shown in Fig. 1, the Cu–O1 and Cu–O2 bonds lie in the equatorial and axial positions, respectively. In contrast to Cu–O1, the axial position gives the Cu–O2 bond more room to respond to pressure.5 Accordingly, the Cu–O2 is least sensitive to compression. Moreover, compared to their original values, bond shortening and bond lengthening are respectively observed for Cu–O1 (1.700 Å vs. 1.941 Å) and Cu–O2 (2.410 Å vs. 2.277 Å) upon compression, which directly attribute to the red and blue shift of the Cu–O1 and Cu–O2, respectively, under high pressure. It is well known that chemical bonds shrink under high pressure. However, it is observed that the Cu–O1 bond softens under pressure, probably owing to the interactions between the Cu–O bond and surrounding atoms. In this study, the starting Cu-BTC is found to contain a large amount of residual water. Furthermore, the changes observed in Raman results are primarily related to the Cu–O1 bonds. It is reasonable to speculate that during compression the residual water was squeezed into cages and interacted with the Cu–O bond, which will be discussed in subsequent sections.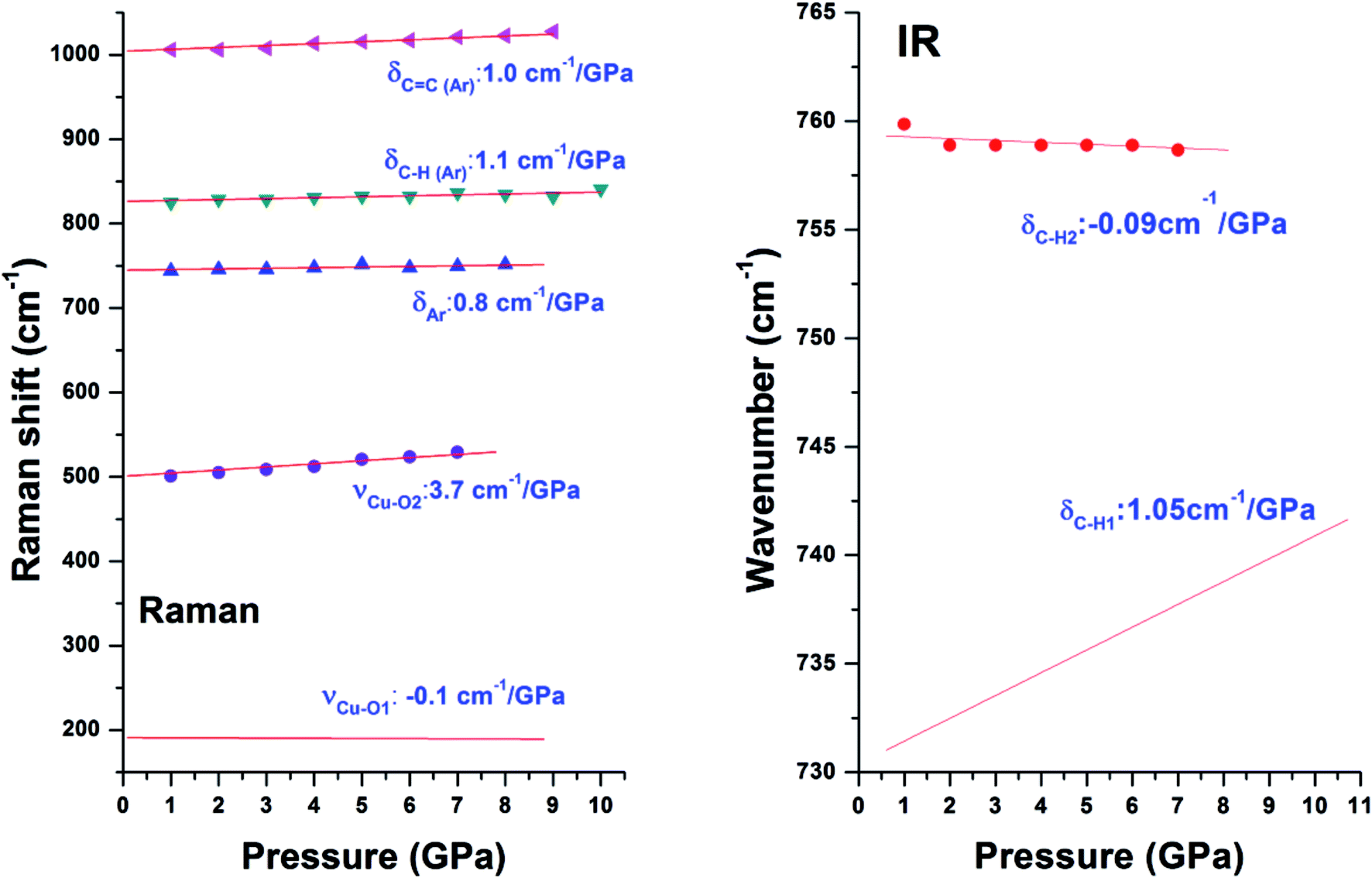 | ||
| Fig. 7 Pressure dependences of Raman (left) and IR (right) modes in Cu-BTC. Different symbols denote different Raman/IR modes which are labeled. Shift rate of each mode is also labeled. | ||
Similar to Raman modes, IR modes of Cu-BTC also gradually shift to higher wavenumbers upon compression except the doublet peak δC–H1 and δC–H2. For comparison purposes, pressure dependences of δC–H1 and δC–H2 modes are plotted in Fig. 7. It is clearly shown that the δC–H1 is more sensitive to pressure than δC–H2 mode (1.05 cm−1 GPa−1 vs. −0.09 cm−1 GPa−1). As shown in Fig. 1, benzene rings with carbonyl groups form the cage structures. Hydrogen atoms located in three different positions point to different cages, where H1 and H2 have the same chemical atmosphere. Thus, δC–H1 and δC–H2 are assigned to the bending of C–H1/H2 and C–H3 bonds respectively. Moreover, the area integral ratio of AδC–H1/AδC–H2 is 2.11, which further confirms the amount of C–H1 is twice that of C–H2. Therefore, we can ascribe the different pressure sensitivities of δC–H to their different environments, where the C–H3 has more space to collide or interact with its surroundings upon compression.
In this study, we observed that the phase transition results are inconsistent among the different probe techniques. In the results obtained from vibrational spectroscopy, one phase transition is observed for both Raman and IR measurements, but at different pressures. In Raman results, the phase transition is found at ∼9 GPa, which was deduced from peak altering in the Cu–O and aromatic ring vibrational regions. In contrast, the phase transition in IR results occurred between 4.1 and 5.7 GPa, which was suggested by a series of IR mode changes associated with the framework. However, in strong contrast to both Raman and IR, no phase transition was found from the XRD patterns. The absence of the two phase transitions in XRD results strongly suggests that the phase transitions observed in Raman and IR only involve chemical environment changes without any crystal structural alteration. Thus, no crystal phase transition is observed in Cu-BTC using either vibrational spectroscopy or XRD. The inconsistency between Raman and IR is probably due to the differences between Raman and IR techniques. It is well known that IR has a higher sensitivity to asymmetric vibrational changes due to the weaker scattering effect of Raman technique. Therefore, the lower phase transition observed in IR could not be revealed by Raman because of the weak interactions.
However, as shown in Fig. 8, the pressure dependence of the cell parameters clearly reveals two transitions at ∼5.8 GPa (Pt1) and 10 GPa (Pt2). Such transitions are known as compressibility changes that are not accompanied by any changes in the crystal structure. The Pt1 compressibility transition is the same as that reported in a previous high-pressure study of Cu-BTC.25 Chapman et al. have studied non-hydrate Cu-BTC with different pressure transmitting media, and concluded that this compressibility transition only occurred in Cu-BTC with small molecule alcohol-based pressure transmitting media (e.g. water, MEW and isopropanol).25 No apparent transition was observed in Cu-BTC without PTM or in large molecule Fluorinert fluid. In addition, it has been proposed that residual water in a hydrate sample could act as a pressure transmitting medium.23 In this study, the starting Cu-BTC was confirmed to contain a large amount of water. Compared with results obtained by Chapman et al., our results indicate the same scenario as that in Cu-BTC with water. Therefore, our findings further confirm that the residual water acted as the guest molecules upon compression. Furthermore, as shown in Fig. 8, the compressibility transition pressure varies as the PTM changes. The Cu-BTC with water demonstrates the highest pressure compressibility transition pressure compared with other hydrostatic pressure media including MEW, isopropyl alcohol and Fluorinert. The compressibility transition pressure increases in the order of no-PTM → Fluorinert → isopropyl alcohol → MEW → water as the molecular size decreases, which strongly suggests that the compressibility transition pressure is dependent on the molecular size of the hydrostatic fluid used.
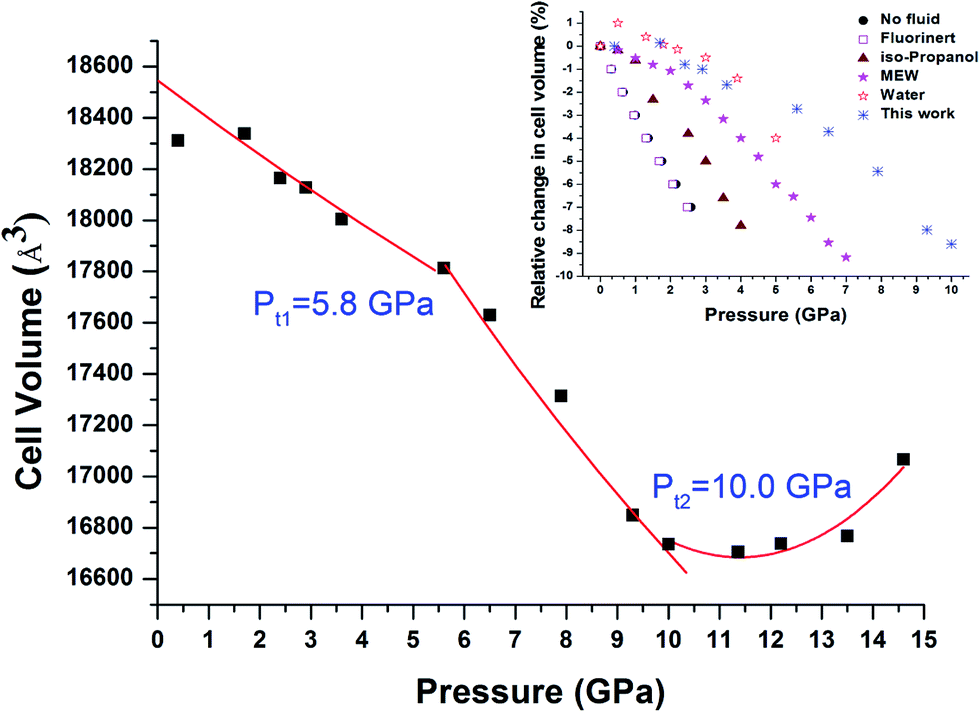 | ||
| Fig. 8 Pressure dependence of the unit cell volume for Cu-BTC. Square solid symbols are the experimental data obtained upon compression, respectively, from this study. Experimental data points in the regions 0–Pt1 and Pt1–Pt2 are fitted with the third Birch Murnaghan equation of state. The solid line in the region of >Pt2 is only for eye guidance. The inset figure is relative change of cell volume of Cu-BTC with different pressure transmitting media adopted from references. The data of Cu-BTC with water are from ref. 23. The rest are all adopted from ref. 25. | ||
For comparison, bulk moduli for the first two regions, 0–Pt1 and Pt1–Pt2, were obtained by fitting the experimental data points using the third Birch Murnaghan equation of state with B′0 fixed at 4. The obtained bulk moduli are 122.6 GPa and 38.2 GPa for regions 0 GPa–Pt1 and Pt1–Pt2, respectively, which are considered hard and soft regimes. As shown in Table 1, the values obtained are slightly higher, but still reasonable in comparison with values reported by Chapman et al. considering the sample and experimental difference.25 Such results support the theory that the hard regimes are present due to the up-taking of guest molecules, which helps to sustain the framework under high pressure.25 The bulk moduli in the soft regime are all similar to the bulk modulus of pure Cu-BTC without pressure transmitting medium, indicating that compressibility in the soft regime can be attributed to intrinsic properties of the framework itself, independent from the type of pressure transmitting medium used.25
Moreover, it is interesting that Pt1 and Pt2 are respectively coincident with phase transition pressures observed in IR and Raman results, taking experimental error into consideration. As mentioned earlier, below Pt1, the sample is in the process of up-taking water into its cages. High pressure study of H2O insertion in AlPO4-54·xH2O has revealed that H2O molecules tended to concentrate near the pore walls once entering the pores.5 In this study, the first phase transition Pt1 observed in IR is deduced by vibrational changes linked to the BTC, which constitutes the main part of the framework wall. Therefore, it is reasonable to speculate that during the 0–Pt1 pressure period, water molecules could also accumulate in the pore particularly along the framework wall. Below Pt1, the interaction is too weak to be detected by Raman spectroscopy. At Pt2, pressure induced sample shrinking becomes substantial enough that interactions and Cu–Cu stretching of the Cu(II) dimer can be detected by Raman. However, the Cu associated IR modes lying below 600 cm−1 could not studied by IR spectroscopy due to the equipment edge limitation. Thus, the phase transition occurring at Pt2 is missing in our IR results.
A pressure induced volume expansion is observed at pressure >Pt2, which is reported for the first time. The phenomenon of negative thermal expansion is speculated to be ascribed to the translation, vibration and twisting vibration of aromatic rings and Cu(II) dimers, which increases distortion of the neighboring two units and further induces negative thermal expansion. Future studies are necessary to verify such speculation.
4. Conclusion
In conclusion, hydrate Cu-BTC was studied under high pressure up to 20 GPa without any pressure transmitting medium by vibrational spectroscopy and XRD with synchrotron radiation. The hydrate Cu-BTC shows strong stability up to 20 GPa with no structural change. Vibrational spectroscopy successfully revealed two non-crystal structural phase transitions (∼5.8 and 9 GPa) caused by interaction changes between water and sample framework. These two phase transition pressures are consistent with the compressibility transition observed in XRD results, which together confirm that the sample underwent a water up-take process and then a pressure induced compression. Moreover, our finding also proves the compressibility transition pressure is dependent on the molecular size of the pressure transmitting medium, while the compressibility was an intrinsic property of the Cu-BTC framework, independent from the experimental conditions. This work not only helps to study the mechanical stability of Cu-BTC, but also provides spectroscopic evidence to understand the interaction between the framework and guest molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Natural Science Foundation of China (#11404362), and the Shanghai Sailing Plan (14YF1407400). We acknowledge Dr Dongxu Liang, Dr Shangming He for technique support at BL15U1 at Shanghai Synchrotron Facility. We would also thank Dr Martin Kunz and Dr Jinyuan Yan at beamline 12.2.2 at ALS for their kindly technique support. We appreciate Dr Haiyan Zheng's help with Infrared spectroscopy measurements.References
- T. D. Bennett, P. Simoncic, S. A. Moggach, F. Gozzo, P. Macchi, D. A. Keen, J.-C. Tan and A. K. Cheetham, Chem. Commun., 2011, 47, 7983–7985 RSC.
- K. W. Chapman, P. J. Chupas and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 15630–15636 CrossRef CAS PubMed.
- P. B. Somayajulu Rallapalli, M. C. Raj, D. V. Patil, K. P. Prasanth, R. S. Somani and H. C. Bajaj, Int. J. Energy Res., 2013, 37, 746–753 CrossRef CAS.
- E. Deniz, F. Karadas, H. A. Patel, S. Aparicio, C. T. Yavuz and M. Atilhan, Microporous Mesoporous Mater., 2013, 175, 34–42 CrossRef CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- R. Krishna and J. M. van Baten, Sep. Purif. Technol., 2012, 87, 120–126 CrossRef CAS.
- W. Wong-Ng, I. Levin, J. A. Kaduk, L. Espinal and H. Wu, J. Alloys Compd., 2016, 656, 200–205 CrossRef CAS.
- Y. Hu, B. Lin, P. He, Y. Y. Li, Y. N. Huang and Y. Song, Chem.–Eur. J., 2015, 21, 18739–18748 CrossRef CAS PubMed.
- V. F. D. Martins, A. M. Ribeiro, A. Ferreira, U. H. Lee, Y. K. Hwang, J.-S. Chang, J. M. Loureiro and A. E. Rodrigues, Sep. Purif. Technol., 2015, 149, 445–456 CrossRef CAS.
- G. Autie-Castro, M. A. Autie, E. Rodriguez-Castellon, C. Aguirre and E. Reguera, Colloids Surf., A, 2015, 481, 351–357 CrossRef CAS.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- J. Kim, H.-Y. Cho and W.-S. Ahn, Catal. Surv. Asia, 2012, 16, 106–119 CrossRef CAS.
- R.-Q. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542–2546 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- J. Gordon, H. Kazemian and S. Rohani, Mater. Sci. Eng., C, 2015, 47, 172–179 CrossRef CAS PubMed.
- V. André, S. Quaresma, J. L. F. da Silva and M. T. Duarte, Beilstein J. Org. Chem., 2017, 13, 2416–2427 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- Y. Hu, H. Kazemian, S. Rohani, Y. Huang and Y. Song, Chem. Commun., 2011, 47, 12694–12696 RSC.
- T. D. Bennett, J.-C. Tan, S. A. Moggach, R. Galvelis, C. Mellot-Draznieks, B. A. Reisner, A. Thirumurugan, D. R. Allan and A. K. Cheetham, Chem.–Eur. J., 2010, 16, 10684–10690 CrossRef CAS PubMed.
- A. Clearfield, Dalton Trans., 2016, 45, 4100–4112 RSC.
- A. J. Graham, J. C. Tan, D. R. Allan and S. A. Moggach, Chem. Commun., 2012, 48, 1535–1537 RSC.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546–17547 CrossRef CAS PubMed.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2008, 130, 10524–10526 CrossRef CAS PubMed.
- E. C. Spencer, R. J. Angel, N. L. Ross, B. E. Hanson and J. A. K. Howard, J. Am. Chem. Soc., 2009, 131, 4022–4026 CrossRef CAS PubMed.
- A. J. Graham, D. R. Allan, A. Muszkiewicz, C. A. Morrison and S. A. Moggach, Angew. Chem., Int. Ed., 2011, 50, 11138–11141 CrossRef CAS PubMed.
- P. Serra-Crespo, E. Stavitski, F. Kapteijn and J. Gascon, RSC Adv., 2012, 2, 5051–5053 RSC.
- K. J. Gagnon, C. M. Beavers and A. Clearfield, J. Am. Chem. Soc., 2013, 135, 1252–1255 CrossRef CAS PubMed.
- E. C. Spencer, M. Kiran, W. Li, U. Ramamurty, N. L. Ross and A. K. Cheetham, Angew. Chem., Int. Ed., 2014, 53, 5583–5586 CrossRef CAS PubMed.
- H. Wang, Z. G. Qu, W. Zhang, Q. N. Yu and Y. L. He, Int. J. Heat Mass Transfer, 2016, 92, 859–863 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., [Solid Earth Planets], 1986, 91, 4673–4676 CrossRef CAS.
- A. I. Skoulidas, J. Am. Chem. Soc., 2004, 126, 1356–1357 CrossRef CAS PubMed.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- S. Maiti, A. Pramanik, U. Manju and S. Mahanty, Microporous Mesoporous Mater., 2016, 226, 353–359 CrossRef CAS.
- J. Ethiraj, F. Bonino, C. Lamberti and S. Bordiga, Microporous Mesoporous Mater., 2015, 207, 90–94 CrossRef CAS.
- Y.-K. Seo, G. Hundal, I. T. Jang, Y. K. Hwang, C.-H. Jun and J.-S. Chang, Microporous Mesoporous Mater., 2009, 119, 331–337 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |