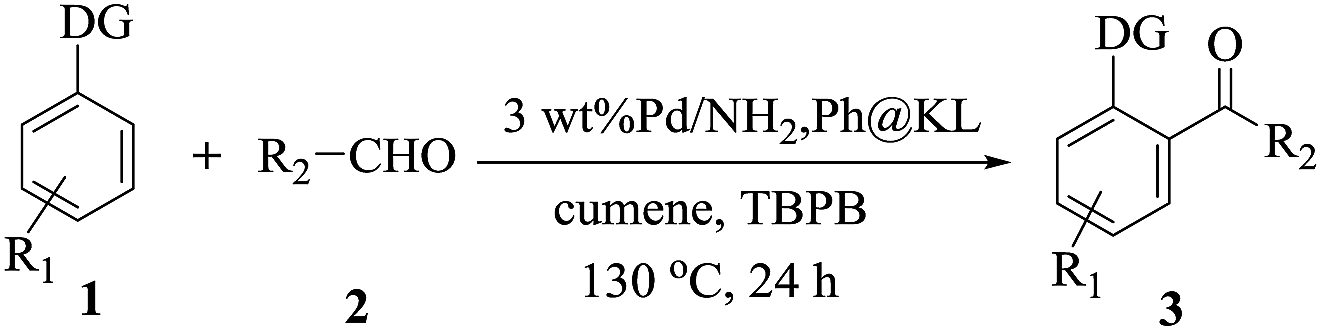
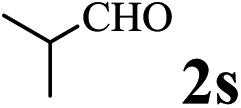DOI:
10.1039/C7RA11800G
(Paper)
RSC Adv., 2017,
7, 53878-53886
Palladium nanoparticles supported on organofunctionalized kaolin as an efficient heterogeneous catalyst for directed C–H functionalization of arylpyrazoles†
Received
26th October 2017
, Accepted 17th November 2017
First published on 23rd November 2017
Abstract
A heterogeneous catalyst system based on the immobilization of Pd0 nanoparticles onto organofunctionalized kaolin is reported with a view to introducing new synthetic routes of directed C–H functionalization of arylpyrazoles. Various characterization techniques revealed that the functional groups, 3-aminopropyltriethoxysilane (APTES) and phenyltrimethoxysilane, become strongly attached to the kaolin surface through Si–O–Si bonds and 3–6 nm sized Pd0 nanoparticles are uniformly decorated and stabilized through the organic amine moieties. The nano-palladium catalyst is durable, undergoing five times reuse with moderate catalytic activity. The XPS analysis of the catalyst before and after reaction suggested that the reaction might be performed via a catalytic cycle that begins with Pd0.
1. Introduction
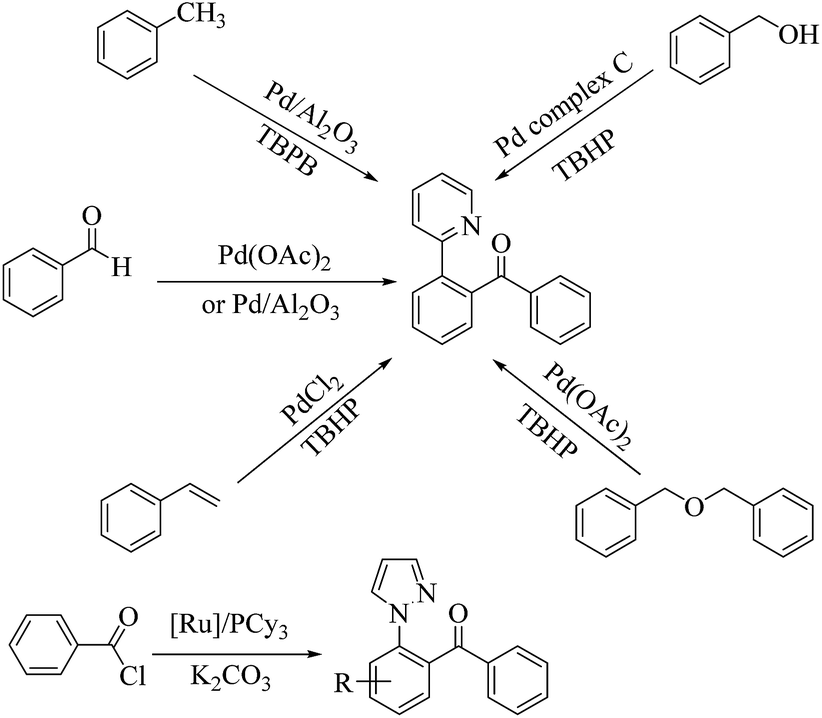
The transition-metal-catalyzed direct arylation of arenes bearing different directing groups (such as arylpyridines, benzamides, acetophenone oximes and anilides) represents a direct and promising approach to access ketones.1–11 In this context, pyrazole serves as an effective directing group for C–H functionalization methods involving cyclometalation. But, at present, methods for directed C–H acylation of 1-arylpyrazole12 are relatively scarce while there are numerous reports on directed C–H acylation of 2-phenyl pyridine (see Scheme 1).13–20 And though these homogeneous transition-metal catalysts offer high selectivity and yields under relatively mild operating conditions, their industrial applicability is limited by the inherent problem of catalyst separation from the product and its recycling.21,22 Considering the easy separation and reusability of a heterogeneous system, it would be desirable to develop highly efficient heterogeneous catalysts for these transformations.
 |
| | Scheme 1 Previous reported directed C–H acylation of 2-phenyl pyridine and 1-arylpyrazole. | |
In recent years, supported palladium nanoparticles (PdNPs) have attracted the broad interest of chemists due to their high selectivity and efficiency as heterogeneous catalysts for various reactions, such as hydrogenation of conjugated dienes, enantioselective allylic alkylation, carbon–carbon coupling, asymmetric allylic substitution and electrocatalytic formic acid oxidation.23–27 A wide range of inorganic and organic supports have been utilised for the preparation of supported PdNPs catalysts, such as carbon,28–30 alumina,31 magnetically recoverable Fe3O4,32,33 MgO,34 composite oxide,35 modified silicas,36,37 carbon nanotube,38 polymers39,40 so on. Even though various approaches to PdNPs immobilization have been considered, progress is still required to further improve supported PdNPs catalytic systems for use in industries regarding the green chemistry context. Clay minerals are naturally abundant minerals of hydrous aluminum phyllosilicates and can be obtained from certain regions in high purity. By the organofunctionalization, clay can support various metal nanoparticles including Au, Ag, Pd, Pt, etc.41–46 General organic modifiers of clay include quaternary ammonium salts47–50 and silane coupling agents.51,52 Among the different types of clay minerals, kaolin seems to be more studied and used in environmental engineering, cosmetics and medicine because of its physical and chemical properties, crystal structure, and surface chemistry.53 However, there is rare report on kaolin supported nano-metal catalysts,54 while there are numerous literatures55–58 on the montmorillonite clay supported nano-metal catalysts.
Kaolin, Al2Si2O5(OH)4, is a dioctahedral-layered hydrated aluminosilicate clay of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 type with two distinct interlayer surfaces: a gibbsite-like with aluminum atoms coordinated octahedrally to corner oxygen atoms and hydroxyl groups, and a silica-like structure, where the silicon atoms are coordinated tetrahedrally to oxygen atoms.59–61 The alternating adjacent layers are linked by hydrogen bonds involving aluminol (Al–OH) and siloxane (Si–O) groups. As a consequence of this structure, the silica/oxygen and alumina/hydroxyl sheets are exposed and interact with different molecules. So kaolin provides the advantage of their relatively easy functionalization with organic groups by host–guest interactions. In order to achieve higher efficiency in most environmental engineering applications using kaolin as catalyst support, the kaolin mineral has to be modified to ensure a proper anchor for the Pd catalyst during catalyst precursor incorporation stage.
:1 type with two distinct interlayer surfaces: a gibbsite-like with aluminum atoms coordinated octahedrally to corner oxygen atoms and hydroxyl groups, and a silica-like structure, where the silicon atoms are coordinated tetrahedrally to oxygen atoms.59–61 The alternating adjacent layers are linked by hydrogen bonds involving aluminol (Al–OH) and siloxane (Si–O) groups. As a consequence of this structure, the silica/oxygen and alumina/hydroxyl sheets are exposed and interact with different molecules. So kaolin provides the advantage of their relatively easy functionalization with organic groups by host–guest interactions. In order to achieve higher efficiency in most environmental engineering applications using kaolin as catalyst support, the kaolin mineral has to be modified to ensure a proper anchor for the Pd catalyst during catalyst precursor incorporation stage.
Based on our research on the supported PdNPs catalyzed C–H activation reaction,19,20 here we verify that the organofunctionalized kaolin supported PdNPs can be used to drive the ortho-directed C–H activation reaction of arylpyrazoles. To our knowledge, this transformation represents the first example of heterogeneous transition-metal catalyzed directed C–H acylation of 1-arylpyrazole. We reported on a simple method to decorate kaolin particles with palladium nanoparticles using 3-aminopropyltriethoxysilane (APTES) as the linkage, where the silane group of APTES formed Si–O–Si bonds with kaolin and the –NH2 group bound to the palladium nanoparticle covalently through a Pd–N bond.62 Meantime, aim to increase the adsorption of organic reactants and the hydrophobicity of the catalysts, along with amine group, the surface of kaolin also was functionalized with phenyl group. In a series of kaolin supported PdNPs, the 3 wt% Pd/NH2,Ph@KL catalyst with a PdNP mean diameter of 4.12 nm exhibited the best catalytic performance and it could be used five times without significant loss in catalytic activity. A possible mechanism was proposed based on the experimental results and relative literature reports.
2. Experimental section
2.1. Catalyst preparation
2.1.1. Synthesis of organofunctionalized kaolin [NH2,Ph@KL]. A 1 g sample of kaolin (KL), which is exploited from Inner Mongolia Autonomous Region of China, was added to a mixture of 1.2 mmol of 3-aminopropyltriethoxysilane (APTES) and 1.2 mmol of phenyltrimethoxysilane solution in n-heptane (30 mL). The suspension was stirred at room temperature for 6 h. Then the solvent was removed by centrifugation and the functionalized kaolin was washed with n-heptane and dried under vacuum for 12 h. The sample is assigned as NH2,Ph@KL. In a similar procedure, NH2@KL and Ph@KL were prepared adding 1.2 mmol of 3-aminopropyltrimethoxysilane or 1.2 mmol of phenyltrimethoxysilane to the KL, respectively.
2.1.2. Synthesis of Pd0 loaded organofunctionalized kaolin [Pd/NH2,Ph@KL]. Pd0 loaded organofunctionalized kaolin was prepared by an impregnation-reduction method using the preparation method of Pd/γ-Al2O3 as our previous reports.19,20 For example, 3 wt% Pd NH2,Ph@KL catalyst was prepared by the following procedure: 0.97 g of organofunctionalized kaolin NH2,Ph@KL was dispersed in tetrahydrofuran (20 mL) solvent with constant stirring for 1 h at room temperature. Then the two kinds of aqueous solutions of PdCl2 (0.01 M, 28.2 mL) and L-Lysine (0.03 M, 1 mL) were added to the mixture consecutively under vigorous stirring for 20 min. Subsequently, 0.1 M NaOH aqueous solution was added into the mixture to adjust the pH to 7. An aqueous solution of NaBH4 (0.35 M, 4.5 mL) was added gradually in about 10 min to the suspension. Finally, the mixture was left to stand for 24 h and the solid was separated by centrifugation and washed with distilled water (4 times) and ethanol (once) followed by air drying overnight. The dried solid residue on grinding furnished a black powder, denoted as 3 wt% Pd/NH2,Ph@KL. With the use of a similar procedure, 3 wt% Pd/NH2@KL and 3 wt% Pd/Ph@KL were prepared using NH2@KL and Ph@KL as the supports, respectively.
2.1.3. Synthesis of Pd0 loaded kaolin [Pd@KL]. For comparison, 3 wt% Pd/KL was prepared according to the above steps presented in this section using neat KL as the support instead of NH2,Ph@KL.
2.1.4. Synthesis of Pd0 loaded CTAB functionalized kaolin [Pd/CTAB@KL]. For comparison, 3 wt% Pd/CTAB@KL was prepared by a modified impregnation-reduction method. (see ESI†)
2.2. Characterization techniques
The TEM study on the samples was recorded with a JEM-2100 transmission electron microscope with an accelerating voltage of 200 kV. The samples were fine powders deposited on a copper microgrid coated with a holey carbon film. X-ray photoelectron spectroscopy (XPS) analysis was performed on an ESCALAB 250Xi X-ray photoelectron spectrometer of ThermoFisher Scientific and Al Kα radiation was used as the X-ray source. All binding energies were referenced to the C1s hydrocarbon peak at 284.80 eV. X-ray diffraction (XRD) was carried out using a Rigaku Ultimal IV X-ray diffractometer with Cu-Kα radiation (λ = 1.5406 Å) from 2θ = 10° to 80°, at a scan rate of 8° min−1, operating at 40 kV and 40 mA. Elemental analysis was performed by vario EL cube Elementar analyzer of Elementar Analysensysteme GmbH. The specific surface areas of all samples were taken with an ASAP-2020 accelerated surface area and porosity analyzer of Micromeritics company, calculated by the BET method from the data in a P/P0 range between 0.06 and 0.30. The Pd loadings in the catalysts were measured on TAS-990 flame atomic absorption spectrophotometer (AAS) of the Beijing Purkinje General Instrument Company. The Pd sensitive wavelength is 244.8 nm. The FTIR (Fourier transform infrared) spectra of the samples were recorded using a Thermo Scientific Nicolet 6700 infrared spectrometric analyzer in KBr matrix in the range 4000–650 cm−1. The H2-TPR (temperature programmed reduction) of the samples was carried out in a Chem BET TPR/TPD Chemisorption Analyzer. An about 0.05 g sample was taken inside a quartz U tube and degassed at 120 °C for 0.5 h with argon gas flow. The sample was then cooled to 30 °C and at this temperature the gas flow was changed to 5% H2 in argon. It was then heated at a heating rate of 10 °C min−1 up to 600 °C and the spectra were recorded. The solid-state NMR experiments were performed on a Bruker Avance III 400 WB spectrometer equipped with a 9.39 T magnet at 297 K. The 1H–13C cross-polarization magic angle spinning (CP/MAS) NMR spectra were recorded using a Bruker 4 mm standard bore MAS probe head with the ZrO2 rotors spinning at 5.0 kHz rate with a Larmor frequency of 100.62 MHz. The spinning sidebands were suppressed by total sideband suppression (TOSS) technique. The 29Si {1H} CP MAS spectra were recorded on a Bruker AVANCE III 400 WB spectrometer equipped with a 4 mm standard bore CP MAS probe head whose X channel was tuned to 79.50 MHz for 29Si and the other channel was tuned to 400.18 MHz for broad band 1H decoupling, using a magnetic field of 9.39 T at 297 K. The dried and finely powdered samples were packed in the ZrO2 rotor closed with Kel-F cap which were spun at 8 kHz rate. A total of 4000 scans were recorded with 6 s recycle delay for each sample. All 29Si CP MAS chemical shifts are referenced to the resonances of 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) standard (d = 0.0). The 27Al MAS spectra were recorded on a Bruker AVANCE III 400 WB spectrometer equipped with a 4 mm standard bore CP MAS probe head whose X channel was tuned to 104.27 MHz for 27Al, using a magnetic field of 9.39 T at 297 K. The dried and finely powdered samples were packed in the ZrO2 rotor closed with Kel-F cap which were spun at 5 kHz rate, with a π/12 pulse. A total of 300 scans were recorded with 2 s recycle delay for each sample. All 27Al MAS chemical shifts are referenced to the resonances of [Al(H2O)6]3+ standard (d = 0.00). Thin layer chromatography (TLC) was performed on pre-coated silica gel GF254 plates. The 1H NMR and 13C NMR spectra were obtained on a 500 MHz Bruker Avance III nuclear magnetic resonance spectrometer with CDCl3 as the solvent. All chemical shifts are reported in ppm using TMS the internal standard. The structures of known compounds were further corroborated by comparing their 1H NMR data with those of literature.
2.3. Activity test
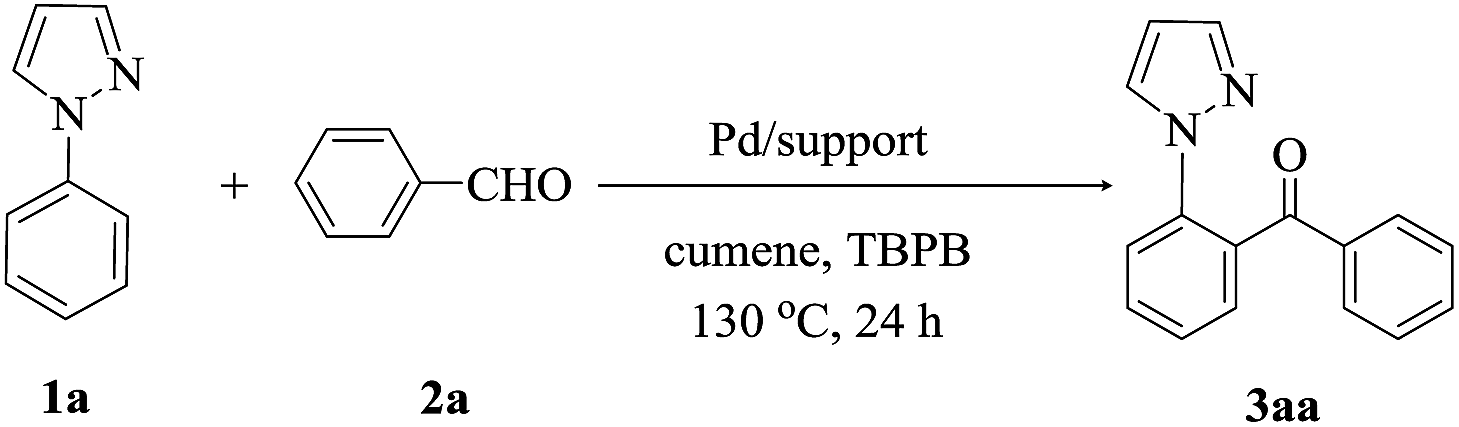
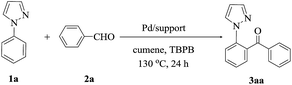
1-phenylpyrazole 1a and benzaldehyde 2a were used as the model reaction. In a typical reaction, 1-phenylpyrazole (27 mg, 0.20 mmol), benzaldehyde (65 mg, 0.60 mmol), tert-butyl peroxybenzoate (TBPB, 156 mg, 0.80 mmol), catalyst (45 mg), and the solvent of cumene (2.0 mL) were charged in a 25 mL oven-dried reaction tube. Reaction was carried out 130 °C for 24 h in an oil bath under air condition. After being cooled to room temperature, the reaction solution was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethylacetate/petroleum ether = 1:3−1:5 as an eluent) to afford the desired product 3. All the products were also confirmed by comparing the 1H NMR and 13C NMR data with authentic samples.
2.4. Catalysts recycle experiment
After each reaction cycle, the substrate, solvent, and products were removed by centrifugal separation; the separated catalyst was washed drastically with 0.1 M NaOH ethanol solution (twice), distilled water (4 times), and then washed twice with ethanol followed by centrifugation and drying at 80 °C for 12 h. The recovered catalyst was used for the next cycle.
3. Results and discussion
3.1. Characterization of catalysts and support
3.1.1. SEM. The SEM image of kaolin, which is exploited from Inner Mongolia Autonomous Region of China, indicated that the kaolin possess smooth surface and flake-like morphology (see Fig. 1). Yang's work proves that the smaller Pd nanoparticles were more easily formed and more highly dispersed on the flake-like kaolinite than rod-like and tube-like kaolinite.54
 |
| | Fig. 1 SEM of kaolin (Inner Mongolia Autonomous Region of China). | |
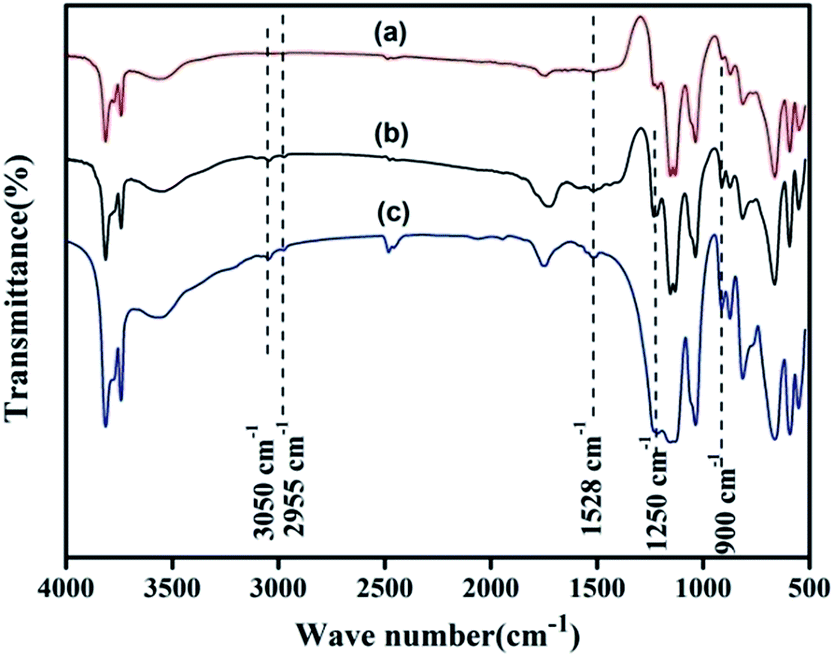
3.1.2. FTIR. The FTIR spectra of KL, NH2,Ph@KL and 3 wt% Pd/NH2,Ph@KL are presented in Fig. 2. The grafting of the amine moiety onto the kaolin surface can be observed from the investigation of the CH2 peaks and the symmetric stretching vibrations of –CH2 groups from grafted APTES can be seen at 2955 cm−1. Again, the presence of a weak N–H bending vibration at 1528 cm−1, suggests the successful grafting of the organosilane on the kaolin surface. Three other distinct peaks around 900 cm−1 and 1250 cm−1 for δ (C–H) and 3050 cm−1 for ν (C–H) of the benzene ring suggest the grafting of phenyl groups on the kaolin surface. After Pd0 loading the basic structure of the functionalized kaolin remains unaltered.
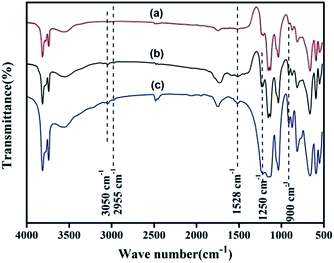 |
| | Fig. 2 FTIR spectra of (a) KL, (b) NH2,Ph@KL and (c) 3 wt% Pd NH2,Ph@KL. | |
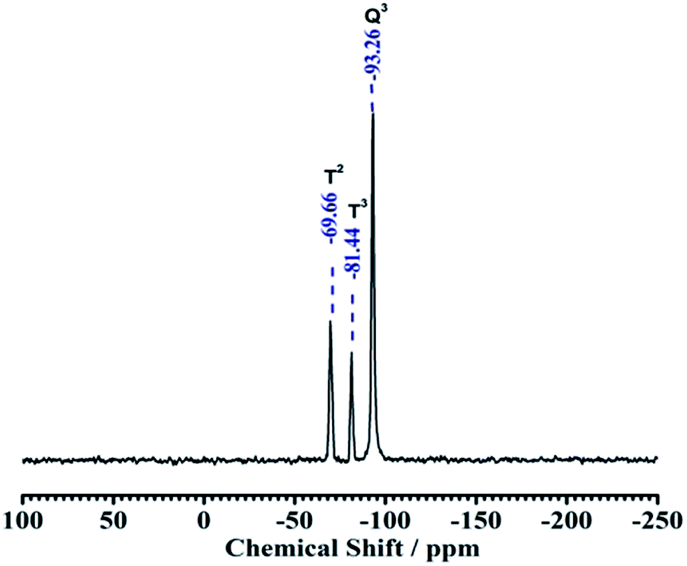
3.1.3. Solid state NMR. The grafting of the binary functional groups into the kaolin framework could also be evidenced by solid state NMR spectra. Basically, the solid state 29Si CP MAS NMR spectra of NH2,Ph@KL shows 29Si NMR shifts assigned as Q3(0Al) units of silica, and δ = −93.26 ppm in natural kaolinite spectra corresponds to the signal of Si nuclei in the Q3 polymerization state, in which three Si atoms bonded to a SiO4 tetrahedron. On functionalization, apart from the Q3 peaks, another two downfield peaks T3 and T2 were observed at −81.44 and −69.61 ppm, respectively, where Tn = [RSi(OEt)n(OSi)3−n], in the case of NH2,Ph@KL, confirming the grafting of the organic silane moieties onto the kaolin surface (see Fig. 3).
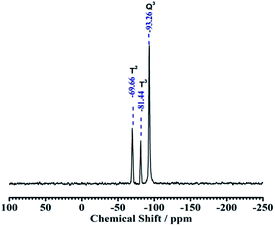 |
| | Fig. 3 29Si CP MAS NMR spectra of NH2,Ph@KL. | |
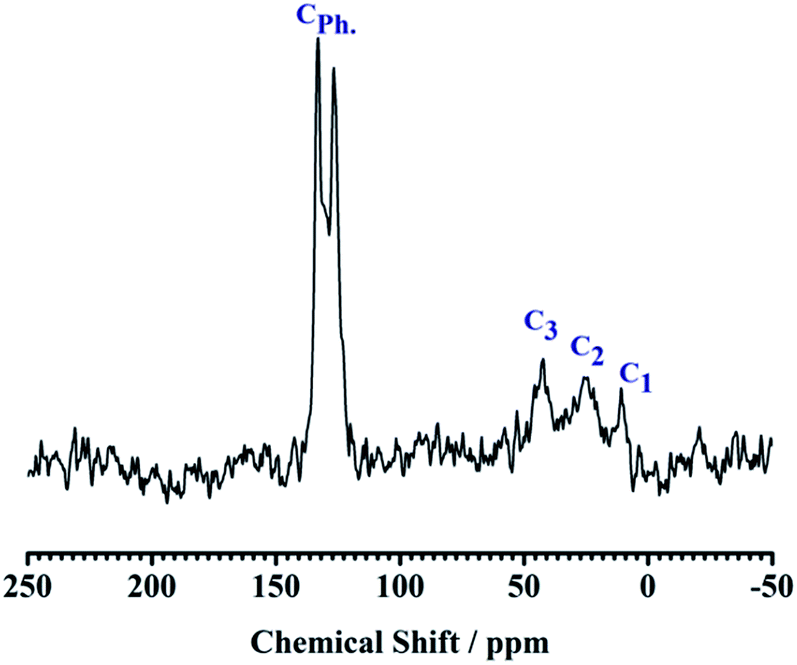
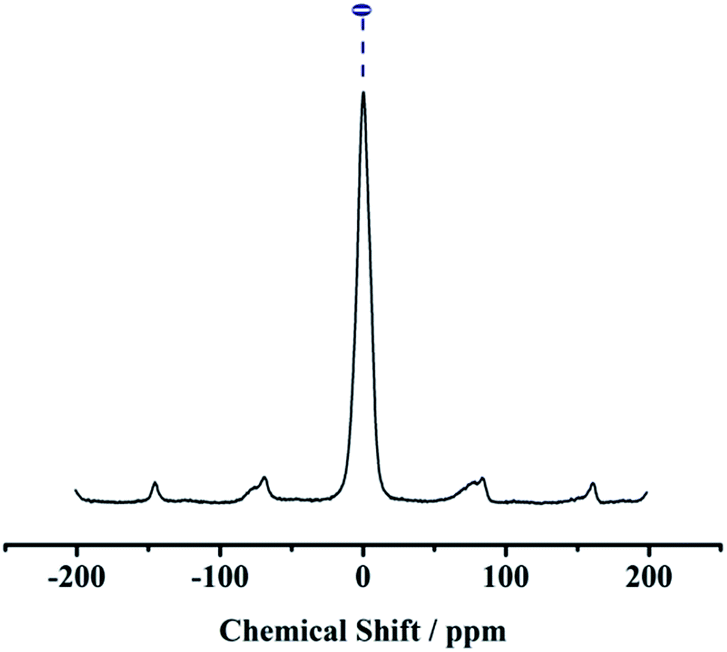
The 13C CP MAS NMR spectra have three distinct resonances at 10.9, 22.2, and 42.8 ppm, which correspond to the carbon atoms in the >Si–CH2–CH2–CH2–NH2 group in the sequence from left to right, proving the existence of APTES. In the spectra, another two peaks at 133.0 and 126.7 ppm were observed indicating the presence of carbon atoms of the Ph group (see Fig. 4). In 27Al CP MAS NMR spectra, kaolin gives 27Al line at ∼0 ppm corresponding to the hexa-coordinated Al (see Fig. 5).
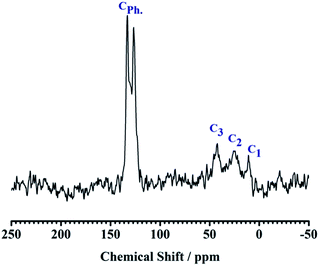 |
| | Fig. 4 13C CP MAS NMR spectra of NH2,Ph@KL. | |
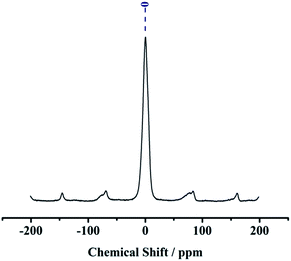 |
| | Fig. 5 27Al CP MAS NMR spectra of NH2,Ph@KL. | |
3.1.4. Elemental analysis and AAS. The results of elemental analysis of the prepared materials are shown in Table 1. Table 1 suggests that the C or N content increased correspondingly along with the organofunctionalization of kaolin. C and N were grafted onto the kaolin at 1.36 and 1.04 mmol g−1, respectively, for 3 wt% Pd/NH2,Ph@KL. The amounts of Pd loaded in the samples were determined by an AAS, and the Pd content of the 3 wt% Pd/NH2,Ph@KL catalyst is approximately 3 wt%. But the Pd content of 3 wt% Pd/KL is only 1.43%. This result indicated that without organofunctionalization, the binding palladium capacity of kaolin is poor.
Table 1 The Elemental Analysis and AAS Results
| Sample |
Chemical composition |
Pd loading (wt%) |
| C (mmol g−1) |
N (mmol g−1) |
| KL |
0.32 |
— |
— |
| NH2@KL |
0.34 |
1.08 |
— |
| Ph@KL |
1.47 |
— |
— |
| NH2,Ph@KL |
1.44 |
0.96 |
— |
| 3 wt% Pd/NH2@KL |
0.40 |
0.98 |
3.1 |
| 3 wt% Pd/Ph@KL |
1.41 |
— |
2.97 |
| 1 wt% Pd/NH2,Ph@KL |
1.47 |
1.01 |
0.88 |
| 3 wt% Pd/NH2,Ph@KL |
1.36 |
1.04 |
3.08 |
| 5 wt% Pd/NH2,Ph@KL |
1.44 |
1.03 |
5.06 |
| Used 3 wt% Pd NH2,Ph@KL |
1.38 |
0.78 |
2.03 |
| 3 wt% Pd/KL |
0.35 |
— |
1.43 |
In addition, we did note a slight decrease in the Pd content after being cycled 5 times (2.03%, Table 1), which can decrease the catalytic activity on the basis of available Pd on the support surface.
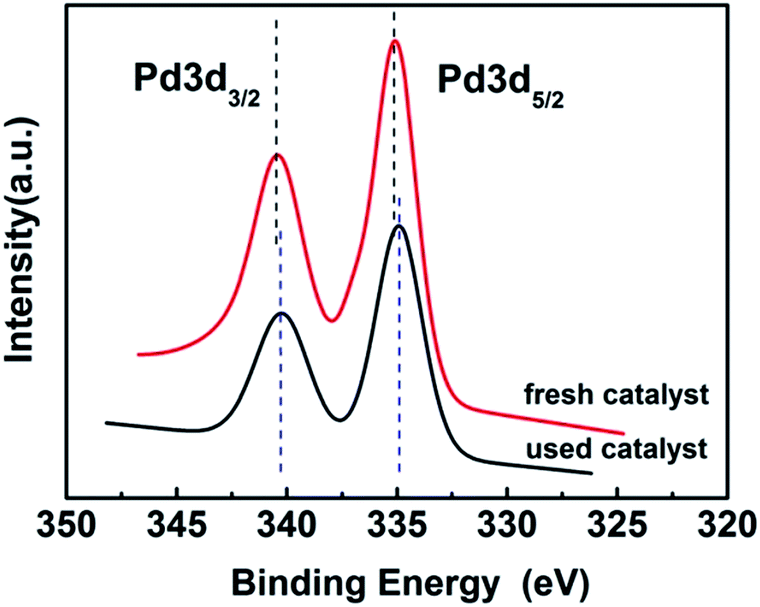
3.1.5. XPS. In order to investigate the electronic state of the Pd, the X-ray photoelectron spectroscopy (XPS) of the fresh and used (after 5th recycle) 3 wt% Pd/NH2,Ph@KL catalysts was investigated (see Fig. 6). It is shown that PdNPs on the support exist in the metallic state, corresponding to the binding energies of 334.83 and 340.23 eV over fresh catalyst and 335.13 and 340.43 eV over used catalyst, which are characteristic of the 3d5/2 and 3d3/2 peaks of Pd0.63 The unchanged valence of Pd confirmed that PdNPs-catalyzed directed C–H functionalization of arylpyrazoles was performed via a catalytic cycle that began with Pd0.
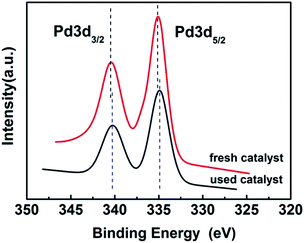 |
| | Fig. 6 The XPS spectra of fresh and used 3 wt% Pd/NH2,Ph@KL. | |
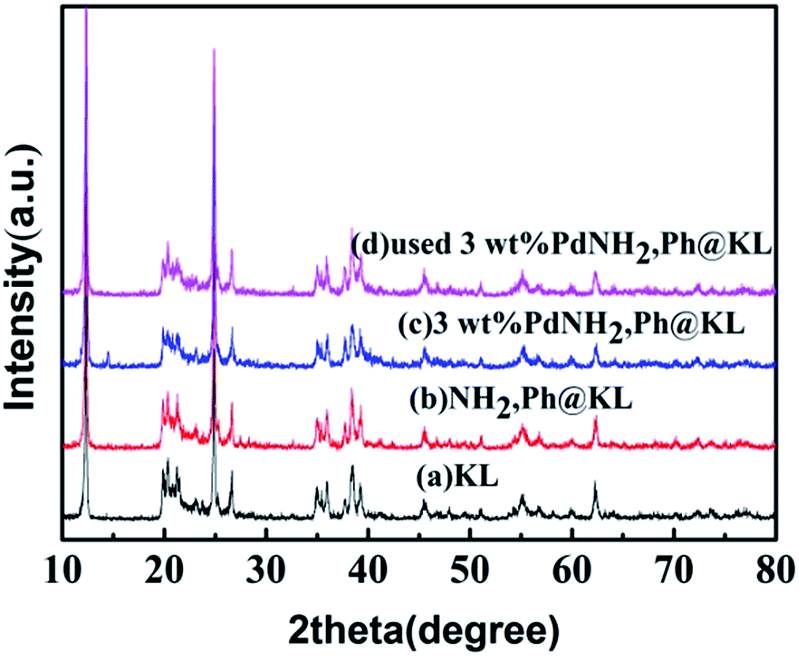
3.1.6. XRD. The XRD patterns of the fresh and used 3 wt% Pd/NH2,Ph@KL catalysts are shown in Fig. 7. The broad angle XRD patterns of NH2,Ph@KL indicate no structural change in the kaolin after functionalization with the organic groups. Three diffraction peaks originated at 12.32°, 20.22° and 24.86° correspond to the kaolin diffraction peaks respectively. 2θ = 20.81°, 26.59° is characteristic diffraction peaks of silicon dioxide. No evident peaks from palladium crystals were observed compared with kaolin sample. It is obvious that the spacing of the kaolin barely changed after surface modification and introduction of the reduced Pd nanoparticles, which indicated that the structure of kaolin was maintained and Pd nanoparticles were not intercalated in kaolin but present at the exterior of the kaolin layer.
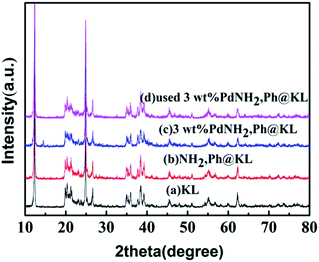 |
| | Fig. 7 XRD patterns of (a) KL, (b) NH2,Ph@KL, (c) 3 wt% Pd/NH2,Ph@KL and (d) used 3 wt% Pd/NH2,Ph@KL. | |
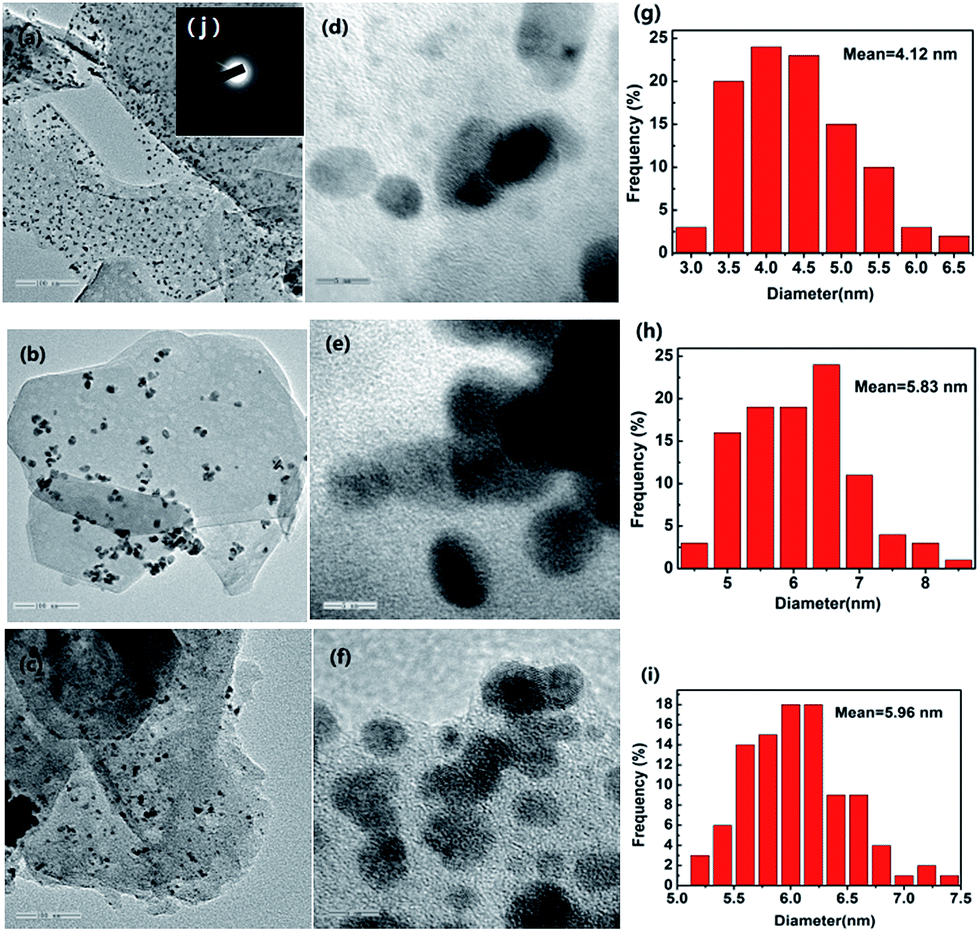
3.1.7. TEM. The morphology and distribution of grown Pd nanoparticles on NH2,Ph@KL and KL were characterized by TEM. As shown in Fig. 8, the PdNPs of fresh and used (after 5th recycle) 3 wt% Pd/NH2,Ph@KL catalysts were both well dispersed and had narrow size distributions, which is quite consistent with the data obtained from the XRD analysis. The HRTEM images depicted clearly visible lattice fringes that evince the formation of crystalline Pd nanoparticles (Fig. 8d). The spacing between adjacent lattice fringes is approximately 0.224 nm, which coincided with the (111) d-spacing of the Pd crystal. The Pd particle size was measured and found to be 4.12 and 5.83 nm for fresh and used catalysts, respectively (Fig. 8g and h). The used catalyst had a slightly larger Pd particle size, but PdNPs still distributed evenly on the support. The TEM image of 3 wt% Pd/CTAB@KL is shown in Fig. S1 (see ESI†).
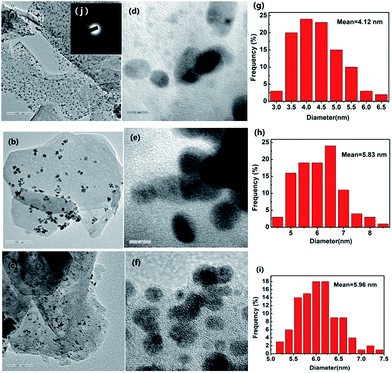 |
| | Fig. 8 (a–c) 100 nm TEM images of fresh 3 wt% Pd/NH2,Ph@KL, used 3 wt% Pd/NH2,Ph@KL and 3 wt% Pd/KL, respectively; (d–f) 5 nm HRTEM images of fresh 3 wt% Pd/NH2,Ph@KL, used 3 wt% Pd/NH2,Ph@KL and 3 wt% Pd/KL, respectively; (g–i) PdNP size distributions of fresh 3 wt% Pd/NH2,Ph@KL, used 3 wt% Pd/NH2,Ph@KL and 3 wt% Pd/KL, respectively; (j) the SAED of TEM images of the fresh 3 wt% Pd/NH2,Ph@KL catalyst. | |
In contrast, apparent agglomeration phenomenon happened in the case of 3 wt% Pd/KL (Fig. 8c and f). Therefore, the presence of the organic amine moiety on the kaolin surface facilitated dispersion of the PdNPs on the KL surface through coordination and acts as a stabilizing agent.
Fig. 8j showed the selected area electron diffraction (SAED) of TEM images of the fresh 3 wt% Pd/NH2,Ph@KL catalyst. It exhibited four diffused rings due to (111), (200), (220), and (311) reflections of face centered cubic (fcc) Pd and indicated the crystalline nature of nanoparticles.64
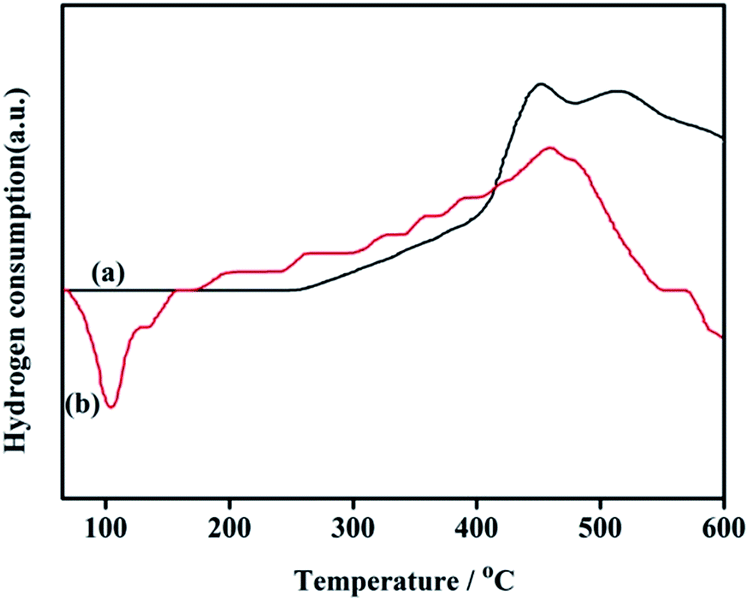
3.1.8. TPR. The reducibility of the supported Pd catalyst plays an important role in influencing its catalytic property. The H2-TPR of NH2,Ph@KL and 3 wt% Pd/NH2,Ph@KL are reported in Fig. 9. The organofunctionalized kaolin (NH2,Ph@KL) does not show any reduction peak below 110 °C. Meanwhile, the TPR curve of 3% Pd/NH2,Ph@KL displayed one strong negative peak around 104 °C, which is characteristic of the decomposition of the β-hydride PdH2.
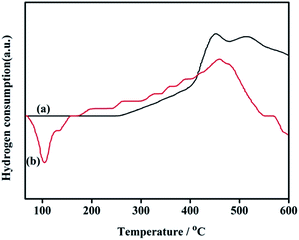 |
| | Fig. 9 H2-TPR spectra of (a) NH2,Ph@KL and (b) 3 wt% Pd/NH2,Ph@KL. | |
3.2. The catalytic performance for directed C–H functionalization of arylpyrazoles
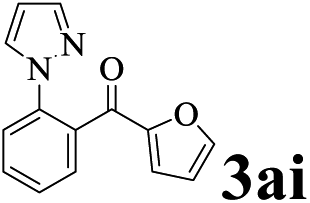
Initially, to examine the efficiency of different PdNPs catalysts prepared by us in the ortho-directed C–H functionalization reaction, we used 1-phenylpyrazole 1a with benzaldehyde 2a as the model substrates, tert-butyl peroxybenzoate (TBPB) as oxidant and cumene as solvent. As can be observed form Tables 2 and 3 wt% Pd/NH2,Ph@KL showed higher activity than other catalysts including Pd/C, Pd/KL and Pd/CTAB@KL, affording (2-(1H-pyrazol-1-yl)phenyl)(phenyl) methanone 3aa in excellent yield (entries 1–4). Compared with Pd/NH2,Ph@KL, Pd/NH2@KL and Pd/Ph@KL exhibited lower catalytic activity (entries 5 and 6). These results indicated that the synergetic promoting effects from Ph and NH2 functionalizations increased the catalytic performance of supported PdNPs. The NH2 functionalization helps in the uniform distribution of the active species PdNPs on the kaolin surface. The Ph functionalization increased the surface hydrophobicity, which facilitated the adsorption of organic molecules. We also examined the effect of different Pd loadings on the reaction. It was found that the catalytic efficiency was significantly influenced by palladium loading and that the catalyst with 3 wt% Pd exhibited the best performance (entries 7 and 8). The yield of product was decreased along with the reduction of oxidant's amount (entry 9). A control experiment was carried out in the absence of TBPB and it failed to give the expected product 3aa, suggesting the importance of the oxidant (entry 10).
Table 2 Activity of various catalyst toward ortho-directed C–H activation reaction of 1-phenylpyrazolea
|
| Entry |
Catalyst |
Oxidant |
Yieldb (%) |
TONc |
| Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (45 mg), TBPB (0.8 mmol), cumene (2 mL), 130 °C, 24 h, under air. Isolated yield. The TON values were calculated on the basis of the amount of Pd metal. Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (45 mg), TBPB (0.2 mmol), cumene (2 mL), 130 °C, 24 h, under air. |
| 1 |
3 wt%Pd/NH2,Ph@KL |
TBPB |
81 |
13 |
| 2 |
5 wt% Pd/C |
TBPB |
63 |
10 |
| 3 |
3 wt% Pd/KL |
TBPB |
52 |
8 |
| 4 |
3 wt% Pd/CTAB@KL |
TBPB |
65 |
10 |
| 5 |
3 wt% Pd/NH2@KL |
TBPB |
67 |
11 |
| 6 |
3 wt% Pd/Ph@KL |
TBPB |
59 |
9 |
| 7 |
1 wt%Pd/NH2,Ph@KL |
TBPB |
69 |
11 |
| 8 |
5 wt%Pd/NH2,Ph@KL |
TBPB |
70 |
11 |
| 9d |
3 wt%Pd/NH2,Ph@KL |
TBPB |
26 |
4 |
| 10 |
3 wt%Pd/NH2,Ph@KL |
— |
— |
— |
Table 3 Substrate scope of ortho-directed C–H activation of arylpyrazolesa
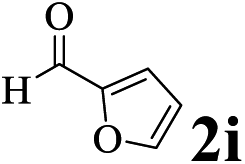
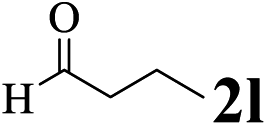
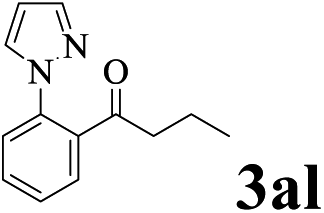
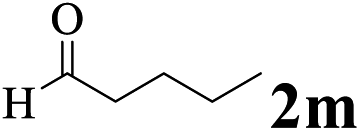
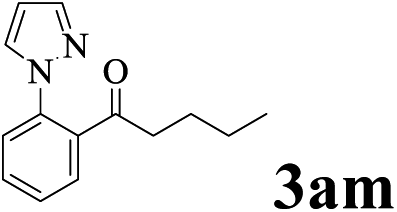
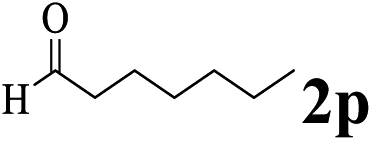
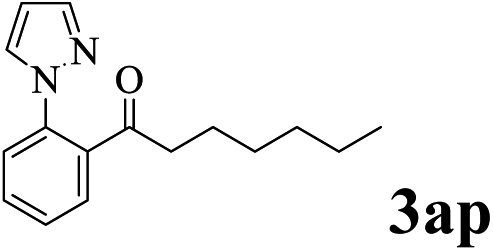
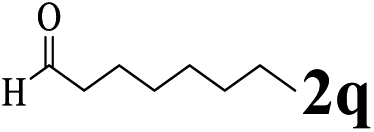
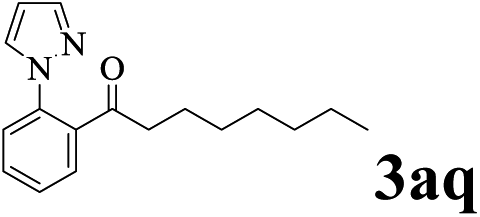
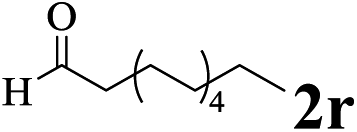
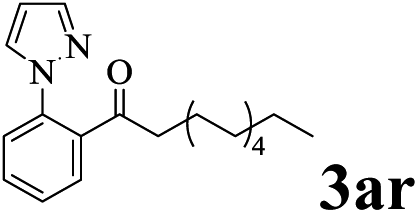
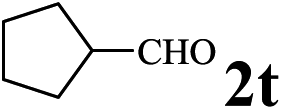
The directed acylation of 1-phenylpyrazole 1a with a variety of aldehydes was performed under the optimized reaction conditions, and the results are presented in Table 3. Various functional groups including methyl, methoxyl, chloro, bromo and cyano were compatible and the desired products were achieved in moderate to good yields (entries 1–6). Compared to its para isomers, o-chlorobenzaldehyde 2e delivered a lower yield, which probably resulted from the steric hindrance effect (entries 3 and 4). Naphthaldehyde 2h as well as benzaldehyde derivatives performed the acylation reaction expediently to give the corresponding product 3ah in 73% yield (entry 7). Using furan-2-carbaldehyde 2i as a heteroaromatic aldehyde resulted desired product (2-(1H-pyrazol-1-yl)phenyl)(furan-2-yl) methanone 3ai in comparatively lower yield (entry 8). Encouragingly, apart from aromatic aldehydes, the methodology was also successful in converting various aliphatic aldehydes with different chain lengths to the corresponding products: all of three to twelve carbons fatty aldehydes 2k–r react with 1-phenylpyrazole 1a smoothly to give the corresponding ketones (entries 9–17), in which five carbons n-pentanal 2m showed the best activity affording 1-(2-(1H-pyrazol-1-yl)phenyl)pentan-1-one 3am in 90% yield (entry 12). Interestingly, when α-substituted aliphatic aldehydes 2s and 2t were used as substrates, no product formation was observed (entries 18 and 19). This indicated that the steric hindrance effect of aliphatic aldehydes is a determining factor in the reaction. In addition, the optimized reaction conditions were implemented in the coupling reactions between other phenyl-N-heteroarene 1 and benzaldehyde 2a (entries 20 and 21). When 2-phenylpyridine 1b and benzo[h]-quinoline 1c, six membered nitrogen containing heterocycle served as the directing group, were employed instead of 1-phenylpyrazole 1a, the acylation reaction also proceed expediently to afford phenyl(2 (pyridin-2-yl)phenyl)methanone 3ba in 77% yield and benzo[h]quinolin-10-yl(phenyl) methanone 3ca in 65% yield, respectively.
The recyclability of the catalyst was examined in the reaction of 1-phenylpyrazole 1a and benzaldehyde 2a as provided in the Experimental section. As shown in Fig. 10, the catalyst can be reused for five cycles and the yield about 3aa held above 49% even after recycling five times. Therefore, the kaolin supported PdNPs-catalyzed C–H activation reaction is more aligned with green catalysis than the homogeneous palladium catalysis is.
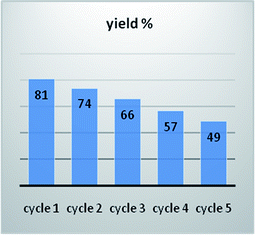 |
| | Fig. 10 Recyclability of catalysts. | |
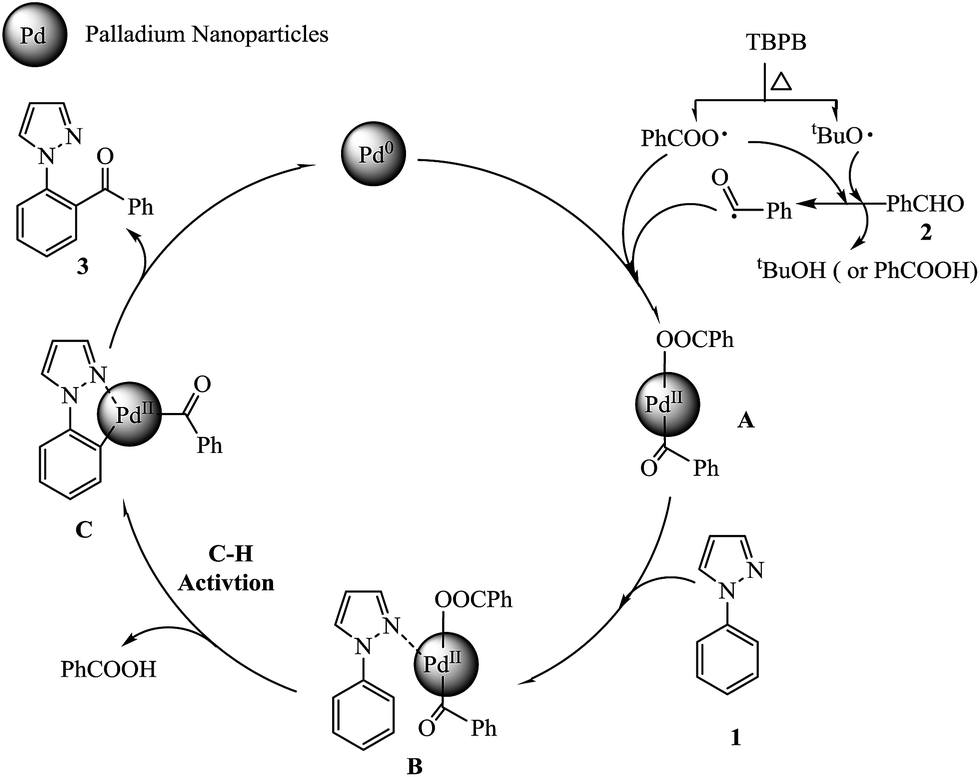
The radical trapping experiment was carried out to have a good understanding of the reaction mechanism. When the 1-phenylpyrazole 1a with benzaldehyde 2a reaction was carried out in the presence of 0.4 mmol TEMPO [(2,2,6,6-tetramethyl piperidin-1-yl)oxy], a radical scavenger, only a 30% yield of product was observed. And when 0.8 mmol TEMPO was added in model reaction, no product was detected, suggesting a possible radical approach. The comparison experiments of model reaction under air, oxygen and argon atmosphere situations were performed. Under argon atmosphere, the desired product 3aa was isolated in 71% yield. But surprisingly, under oxygen atmosphere, no product was detected. These results indicate that Pd0 is not oxidized to PdII by air (oxygen) and excess oxygen may hinder the catalytic cycle. Based on the previous reports65–69 and our own results, a tentative mechanism is illustrated in Scheme 2. First, the aldehyde 2 was oxidized by TBPB to give a benzoyl radical and releasing the benzoic acid or tert-butyl alcohol. Then, Pd0 was oxidized by benzoyl radical and benzoate radical to form a PdII complex A. Second, complex A coordinated with the nitrogen atom of the pyrazole group of 1-phenylpyrazole 1 to form a complex B. Subsequently, the ortho C–H bond is activated by PdII to generate five-membered palladacycle intermediate C by releasing benzoic acid. Finally, the final product 3 is generated through the reductive elimination of intermediate C and releases a Pd0 to continue catalytic cycle.
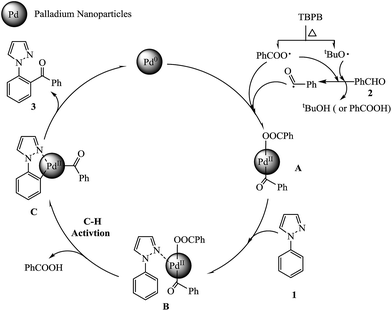 |
| | Scheme 2 Possible reaction mechanism. | |
4. Conclusions
In summary, we demonstrate a novel, highly efficient, and reusable heterogeneous nano-palladium catalyst supported on organofunctionalized kaolin for ortho-directed C–H activation of arylpyrazoles. Under the synergetic promoting effects from Ph and NH2 functionalizations, 3–6 nm size PdNPs were well-dispersed on kaolin surface and durable for five times reuse with moderate catalytic activity. Using supported palladium nanoparticles as the catalyst, a broad scope of aldehydes, including aryl aldehydes, heteroaromatic aldehydes and aliphatic aldehydes, react with arylpyrazoles to synthesize the corresponding aryl ketones in yield up to 90%. Compared with aromatic aldehydes, the steric hindrance effect of aliphatic aldehydes is a determining factor in the reaction. Further studies are in progress to develop kaolin supported palladium nanoparticles catalyst with higher activity and expand further the scope of heterogeneous palladium catalyzed C–H activation reaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by National Science Foundation of China (21462031), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-17-A22) and Inner Mongolia Science Foundation (2015MS0225).
Notes and references
- X. F. Jia, S. H. Zhang, W. H. Luo, F. Wang and J. Cheng, Org. Lett., 2009, 11, 3120–3123 CrossRef CAS PubMed.
- J. Park, E. Park, A. Kim, Y. Lee, K.-W. Chi, J. H. Kwak, Y. H. Jung and I. S. Kim, Org. Lett., 2011, 13, 4390–4393 CrossRef CAS PubMed.
- S. Sharma, E. Park, J. Park and I. S. Kim, Org. Lett., 2012, 14, 906–909 CrossRef CAS PubMed.
- S. Sharma, J. Park, E. Park, A. Kim, M. Kim, J. H. Kwak, Y. H. Jung and I. S. Kim, Adv. Synth. Catal., 2013, 355, 332–336 CAS.
- C.-W. Chan, Z. Zhou, A. S. C. Chan and W.-Y. Yu, Org. Lett., 2010, 12, 3926–3929 CrossRef CAS PubMed.
- Y. Yang, B. Zhou and Y. Li, Adv. Synth. Catal., 2012, 354, 2916–2920 CrossRef CAS.
- S. Sharma, A. Kim, J. Park, M. Kim, J. H. Kwak, Y. H. Jung, J. S. Park and I. S. Kim, Org. Biomol. Chem., 2013, 11, 7869–7876 CAS.
- J. Yao, R. Feng, Z. Wu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2013, 355, 1517–1522 CrossRef CAS.
- J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Organometallics, 2015, 34, 953–966 CrossRef CAS.
- C.-W. Chan, Z. Y. Zhou and W.-Y. Yu, Adv. Synth. Catal., 2011, 353, 2999–3006 CrossRef CAS.
- F. Szabó, J. Daru, D. Simkó, T. Zs Nagy, A. Stirling and Z. Novak, Adv. Synth. Catal., 2013, 355, 685–691 CrossRef.
- P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862–5865 CrossRef CAS PubMed.
- C. P. Perumgani, S. P. Parvathaneni, B. Kodicherla, S. Keesara and M. R. Mandapati, Inorg. Chim. Acta, 2017, 455, 105–111 CrossRef CAS.
- Y. Liang, X. Wang, C. Tang, T. Shen, J. Liu and N. Jiao, Chem. Commun., 2016, 52, 1416–1419 RSC.
- S. Han, S. Sharma, J. Park and M. Kim, J. Org. Chem., 2014, 79, 275–284 CrossRef CAS PubMed.
- A. B. Khemnar and B. M. Bhanage, Eur. J. Org. Chem., 2014, 6746–6752 CrossRef CAS.
- X. Jia, S. Zhang, W. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120–3123 CrossRef CAS PubMed.
- B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659–13663 CrossRef CAS PubMed.
- Y. S. Bao, D. Zhang, M. Jia and B. Zhaorigetu, Green Chem., 2016, 18, 2072–2077 RSC.
- D. Zhang, B. Zhaorigetu and Y. S. Bao, J. Phys. Chem. C, 2015, 119, 20426–20432 CAS.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS.
- H.-U. Blaser, A. Indolese, F. Naud, U. Nettekoven and A. Schnyder, Adv. Synth. Catal., 2004, 346, 1583 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed.
- L. X. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- A. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450 CrossRef CAS PubMed.
- D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809 CrossRef CAS PubMed.
- K. D. Collins, R. Honeker, S. Vásquez-Céspedes and D.-T. D. Tang, Chem. Sci., 2015, 6, 1816 RSC.
- S. Vásquez-Céspedes, A. Ferry, L. Candish and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 5772 CrossRef PubMed.
- J. Lee, J. Chung, S. M. Byun, B. M. Kim and C. Lee, Tetrahedron, 2013, 69, 5660 CrossRef CAS.
- L. Zhang, P. Li, C. Liu, J. Yang, M. Wang and L. Wang, Catal. Sci. Technol., 2014, 4, 1979 CAS.
- P. V. Reddy, M. Annapurna, P. Srinivas, P. R. Likhar and M. L. Kantam, New J. Chem., 2015, 39, 3399 RSC.
- R. Kishore, J. Yadav, B. Venu, A. Venugopal and M. L. Kantam, New J. Chem., 2015, 39, 5259 RSC.
- L. Wang, W.-B. Yi and C. Cai, Chem. Commun., 2011, 47, 806 RSC.
- Y. M. A. Yamada, Y. Yuyama, T. Sato, S. Fujikawa and Y. Uozumi, Angew. Chem., Int. Ed., 2014, 53, 127 CrossRef CAS PubMed.
- S. Korwar, K. Brinkley, A. R. B. Siamaki, F. Gupton and K. C. Ellis, Org. Lett., 2015, 17, 1782 CrossRef CAS PubMed.
- V. A. Zinovyeva, M. A. Vorotyntsev and I. Bezverkhyy, Adv. Funct. Mater., 2011, 21, 1064 CrossRef CAS.
- S. Keshipour and A. Shaabani, Appl. Organomet. Chem., 2014, 28, 116 CrossRef CAS.
- H. A. Patel, H. C. Bajaj and R. V. Jasra, J. Nanosci. Nanotechnol., 2009, 9, 5946–5952 CrossRef CAS PubMed.
- H. Wang, S.-X. Deng, Z.-R. Shen, J.-G. Wang, D.-T. Ding and T.-H. Chen, Green Chem., 2009, 11, 1499 RSC.
- D. Manikandan, D. Divakar, A. V. Rupa, S. Revathi, M. E. L. Preethi and T. Sivakumar, Appl. Clay Sci., 2007, 37, 193–200 CrossRef CAS.
- D. Dutta and D. K. Dutta, Appl. Catal., A, 2014, 487, 158–164 CrossRef CAS.
- Y. Masui, J. Wang, K. Teramura, T. Kogure, T. Tanaka and M. Onaka, Microporous Mesoporous Mater., 2014, 198, 129–138 CrossRef CAS.
- Z. Ren, F. Zhang, L. Yue, X. Li, Y. Tao, G. Zhang, K. Wu, C. Wang and B. Li, RSC Adv., 2015, 5, 52658–52666 RSC.
- S.-M. Paek, J.-U. Jang, S.-J. Hwang and J.-H. Choy, J. Phys. Chem. Solids, 2006, 67, 1020–1023 CrossRef CAS.
- V. Belova, H. Möhwald and D. G. Shchukin, Langmuir, 2008, 24, 9747–9753 CrossRef CAS PubMed.
- A. A. Teixeira-Neto, B. Faceto, L. S. Siebra and E. Teixeira-Neto, Appl. Clay Sci., 2015, 116–117, 175–181 CrossRef CAS.
- H. A. Patel, H. C. Bajaj and R. V. J. Jasra, J. Nanosci. Nanotechnol., 2009, 9, 5946–5952 CrossRef CAS PubMed.
- R. Zhang, M. Hummelgård and H. Olin, Langmuir, 2010, 26, 5823–5828 CrossRef CAS PubMed.
- G. B. B. Varadwaj, S. Rana and K. Parida, J. Phys. Chem. C, 2014, 118, 1640–1651 CAS.
- K. G. Bhattacharyya and S. S. Gupta, Adv. Colloid Interface Sci., 2008, 140, 114–131 CrossRef CAS PubMed.
- X. Li, J. Ouyang, Y. Zhou and H. Yang, Sci. Rep., 2015, 5, 13763 CrossRef PubMed.
- W. Xu, H. M. Sun, B. Yu, G. F. Zhang, W. Q. Zhang and Z. W. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 20261–20268 CAS.
- W. J. Zhang, M. K. S. Li, R. J. Wang, P.-L. Yue and P. Gao, Langmuir, 2009, 25, 8226–8234 CrossRef CAS PubMed.
- M. R. Xia, W. Ding, K. Xiong, L. Li, X. Q. Qi, S. G. Chen, B. S. Hu and Z. D. Wei, J. Phys. Chem. C, 2013, 117, 10581–10588 CAS.
- D. Varade and K. Haraguchi, Langmuir, 2013, 29, 1977–1984 CrossRef CAS PubMed.
- Y. Gomez, L. Fernandez, C. Borras, J. Mostany and B. Scharifker, Electroanalysis, 2009, 21, 1354–1362 CrossRef CAS.
- A. K. Pandaa, B. G. Mishraa, D. K. Mishrac and R. K. Singh, Colloids Surf., A, 2010, 363, 98–104 CrossRef.
- Z. H. Wang, L. Guo, D. M. Han and F. B. Gu, Sens. Actuators, B, 2015, 212, 264–272 CrossRef CAS.
- R. Y. Zhang, M. Hummelgård and H. Olin, Langmuir, 2009, 26, 5823–5828 CrossRef PubMed.
- X. Yang, Q. Li, H. Wang, J. Huang, L. Lin, W. Wang, D. Sun, Y. Su, J. B. Opiyo, L. Hong, Y. Wang, N. He and L. Jia, J. Nanopart. Res., 2010, 12, 1589–1598 CrossRef CAS.
- L. Adak, S. Bhadra and B. C. Ranu, Tetrahedron Lett., 2010, 51, 3811–3814 CrossRef CAS.
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- D. J. Cárdenas, B. Martin-Matute and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 5033–5040 CrossRef PubMed.
- K. M. Gericke, D. I. Chai, N. Bieler and M. Lautens, Angew. Chem., 2009, 121, 1475–1479 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis and characterization of Pd/CTAB@KL, characterization data for the products, 1H NMR and 13C NMR spectra of the products. See DOI: 10.1039/c7ra11800g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*