
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly uniform Fe3O4 nanoparticle–rGO composites as anode materials for high performance lithium-ion batteries†
Shoupu Zhu,
Lei Fan and
Yingying Lu*
and
Yingying Lu*
State Key Laboratory of Chemical Engineering, Institute of Pharmaceutical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yingyinglu@zju.edu.cn; Tel: +86-0571-87953906
First published on 1st December 2017
Abstract
Current lithium-ion batteries (LIBs) based on carbonaceous anodes are close to their theoretical performance limits and can hardly meet the demand for high energy applications. Anode materials based on transition metal oxides are promising alternatives to graphite, stemming from their high lithium storage capacity. Among them, iron oxides have the advantages of rich raw materials, low prices, and high theoretical capacities. Herein, we report a facile strategy of improving the capacity and cycling stability of LIBs via the use of reduced graphene oxide-doped Fe3O4 nanoparticles (around 6.45 nm) as anode materials. Galvanostatic cycling measurements show that cells with Fe3O4/rGO nanocomposites deliver a reversible specific capacity of 1108 mA h g−1 at a current density of 0.5 A g−1 even after 400 cycles. The unique structure of Fe3O4/rGO nanocomposites is responsible for the high cycling performance. The rGO component enables high electrical conductivity while the homogeneous distribution of nano-sized Fe3O4 in rGO favors the diffusion and charge transfer of ions. The void space amongst the nanoparticles and the rGO nanosheets can accommodate volume expansion during cycling. This novel tactic can be used in the preparation of other transition metal oxides with ultra-small and uniform nanoparticles such as SnO2, Co2O3, TiO2 and RuO2 for high-energy LIBs.
Introduction
Since the great commercial success of the lithium-ion battery in 1991, there has been dramatic progress in order to satisfy the requirements of fast developing cellphones, electric vehicles, and large-scale energy storage systems. However, conventional lithium-ion batteries that are based on graphite anodes can hardly meet the increasing demand for high-energy, cost-effective, and reliable electrical energy storage. The lithiated graphite anode in currently marketed lithium-ion batteries has a gravimetric capacity of only 372 mA h g−1, and lithium-ion battery technology based on carbonaceous materials is approaching its theoretical performance limits. An urgent requirement for lithium-ion batteries is the exploration of new materials possessing high energy densities that could potentially serve as alternatives for lithium-ion batteries based on this design. New battery technologies such as batteries based on lithium metal or silicon have emerged, targeted on high energy. However, these batteries suffer from intense safety issues, stemming from uneven lithium electrodeposition/formation of lithium dendrites or electrode volume expansion. Lithium-ion batteries are designed for great reliability since lithium ions are hosted in a supporting material, preventing formation of lithium dendrites and reducing the huge volume expansion.Transition metal oxides as anodes for lithium-ion batteries have received much attention since 2000 when Poizot et al. first reported their high lithium storage capacity and special conversion mechanism.1 The challenges for transition metal oxides are often associated with the large voltage hysteresis, which limits the energy efficiency of electrochemical redox reactions.2–5 Although the mechanism is still not clear, it could be due to the poor electronic conductivity, the slow charge transfer kinetics and the structural instability.6–11 Most of the cells using transition metal oxides display voltage hysteresis from 0.7 to 1.0 V, among which cells with iron oxide/rGO show a lower overpotential of around 0.7 V. Studies show that three-dimensional electrode materials can effectively reduce the voltage hysteresis. Wang et al. fabricated cells with 3D mesostructured Ni scaffolded Fe2O3 electrodes, whose voltage hysteresis is 0.62 V at 100 mA g−1 and can be further reduced to 0.42 V at elevated temperature.3 In addition, iron oxides have the advantages of rich raw materials, low prices, and high theoretical capacities (1007 mA h g−1 for α-Fe2O3 and 926 mA h g−1 for Fe3O4). However, common iron oxides often undergo large volume changes (193% for Fe2O3, calculated from the densities of Fe2O3, Fe and Li2O) based on their electrochemical reactions in the charging–discharging process,12,13 which can cause crushing of the crystal and aggregation of particles and lead to almost constantly falling capacity decay during the initial few cycles. Fortunately, the electrochemical performance can be effectively improved by combining iron oxides with conducting materials14 such as reduced graphene oxide (rGO),15–18 carbon,19–22 carbon nanofibers (CNFs),23,24 carbon nanotubes,25,26 and conductive polymers.27,28 Coating carbon materials or conductive polymers can fix the locations of iron oxides in the composite electrode, buffer the volume expansion and shrinkage, and retain the integrity of the crystal structures. The highly conductive nature of the coating materials can also remedy the weak conductivity of iron oxides and improve the transport properties of lithium ions. On the other hand, the morphology of iron oxides is also critical to the electrochemical performance, and structural modifications on iron oxides such as three-dimensional α-Fe2O3,29 porous α-Fe2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 and α-Fe2O3 nanoflakes31 have been proven to stabilize the cycling behavior and increase the lithium storage capacity. Iron oxides with small particle size possess large specific surface areas, facilitating the contact between electrode and electrolyte, and shortening the diffusion path of lithium ions in the vicinity of the electrode. The void space amongst the nano-sized particles can accommodate the volume expansion and maintain the structural stability during cycling. The uniform particle size can generate uniform stress/strain over the entire electrode, preventing local cracking of the electrode during the lithiation and delithiation processes.32
30 and α-Fe2O3 nanoflakes31 have been proven to stabilize the cycling behavior and increase the lithium storage capacity. Iron oxides with small particle size possess large specific surface areas, facilitating the contact between electrode and electrolyte, and shortening the diffusion path of lithium ions in the vicinity of the electrode. The void space amongst the nano-sized particles can accommodate the volume expansion and maintain the structural stability during cycling. The uniform particle size can generate uniform stress/strain over the entire electrode, preventing local cracking of the electrode during the lithiation and delithiation processes.32
Owing to its excellent electrochemical properties such as high electrical conductivity and high lithium storage capacity,15 graphene has attracted great research interest for application in lithium-ion batteries. For the fabrication of graphene, oxygen-containing groups such as C–O–C, –OH and –COOH are often introduced onto the surface of graphene to yield graphene oxide (GO). This enables good solubility in water, generates an electrostatic force with the electropositive groups and forms assembled nanocomposites. Therefore, FexOy/rGO composites using GO as a precursor for rGO have been fabricated for lithium-ion batteries.17,18,33–36 However, the precursors used for the preparation, which are important for exploring the reaction mechanisms, have rarely been discussed in previous literature. Additionally, FexOy/rGO with nano-sized (typically less than 10 nm) iron oxides that uniformly anchor on the surface of the rGO nanosheet has rarely been reported. Herein, we report a facile strategy of improving the capacity and cycling stability of lithium-ion batteries via the use of rGO doped Fe3O4 nanoparticles as anode materials. Different precursors for making the iron oxides were extensively discussed and ultra-small iron oxides were successfully prepared, uniformly embedded in rGO nanosheets.
Experimental section
Synthesis of Fe3O4, rGO, Fe3O4/rGO-180, Fe3O4/rGO, Fe2O3/rGO and Fe(OH)3/GO
0.5 mL FeCl3 solution (2 mol L−1) was added to 20 mL boiling deionized (DI) water drop by drop, and the solution was kept boiling for about 3 minutes to yield the Fe(OH)3 sol. 80 mg graphene oxide was mixed with 20 mL DI water and the solution was then ultrasonically dispersed for 120 min to ensure the uniform distribution of graphene oxide nanosheets in DI water. The Fe(OH)3 sol and graphene oxide solution were mixed together by vigorous stirring. Then 1 mmol vitamin C and 1.25 mL hydrogen were successively added into the mixture. After stirring for 15 minutes, the mixed solution was transferred into a 50 mL Teflon-lined stainless steel autoclave and heated at 180 °C for 12 h in a drying oven. The precipitate was collected and washed with DI water until the supernatant liquid was clear. Then the Fe3O4/rGO-180 was obtained after freeze drying. The Fe2O3/rGO was prepared via the same process without adding vitamin C, and pure Fe3O4 and rGO were fabricated in the absence of GO and Fe(OH)3, respectively. Fe3O4/rGO-180 was further calcined at 500 °C for 2 h in an Ar atmosphere with a heating rate of 5 °C min−1, to yield Fe3O4/rGO. Fe(OH)3/GO nanocomposites were collected from centrifuging the mixed solution of the Fe(OH)3 sol and the GO solution.Materials characterization
TGA was performed in an air atmosphere from room temperature to 800 °C at a heating rate of 10 °C min−1 on a PE TGA7 thermal analyzer. Powder XRD measurements were carried out in a PANalytical X’Pert PRO X-ray diffractometer with Cu-Kα radiation at a scanning rate of 10° min−1. XPS measurements were carried out in a VG ESCALAB spectrometer with an Al-Kα (1486.8 eV) X-ray source to characterize the crystal phase of the iron oxide. SEM was performed with a Hitachi SU8010 scanning electron micro-analyzer with an accelerating voltage of 5 kV. TEM was conducted at 200 kV with a Philips Tecnai 12 field emission microscope. Raman spectra were recorded by a Horiba Jobin Yvon LabRAM HR Evolution Raman spectrometer with 532 nm wavelength. The Brunauer–Emmett–Teller (BET) surface areas of the samples were measured using a Micromeritics TriStar II porosity and surface area analyzer. The resistances of Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO were tested using a 4 Probes Tech ST-21 four-point probe square resistance tester.Electrochemical measurements
CR 2032 coin cells were used to measure the electrochemical performance of Fe3O4, rGO, Fe3O4/rGO-180 and Fe3O4/rGO. The working electrode consists of 80 wt% of active material (about 1–2 mg), 10 wt% of super P conductive carbon black and 10 wt% of PVDF. Cu foil was used as the current collector. The assembly was carried out in a high-purity argon-filled glovebox, using a lithium plate as the counter/reference electrode, Celgard 2400 polypropylene as the separator, and 1 M LiPF6 in a mixture of ethylene carbonate (EC), diethyl carbonate (DMC) and ethyl-methyl carbonate (EMC) (1:1:1 by volume) as the electrolyte. Galvanostatic charge and discharge cycling of the cells was performed using a Land CT2001A battery test system. Cyclic voltammetry (CV) measurements and electrochemical impedance spectroscopic (EIS) measurements were conducted on a Solartron Analytical 1400 CellTest System.
Results and discussion
The synthesis procedure is schematically depicted in Fig. 1. As the surface of the GO nanosheets has oxygen-containing groups, Fe(OH)3 nanoparticles can be uniformly adsorbed on the surface, forming Fe(OH)3/GO nanocomposites under electrostatic force,37 which was verified by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) analyses (Fig. S1a–c†). The homogeneously attached Fe(OH)3 nanoparticles were created for uniform Fe3O4 loading during the subsequent reactions. Additionally, hydrazine was added for the pre-reduction of Fe(OH)3/GO, and the GO in Fe(OH)3/GO was slightly reduced, which is demonstrated by the X-ray diffraction (XRD) patterns in Fig. S1d.† The XRD pattern for the Fe(OH)3/GO mixture shows a strong characteristic diffraction peak at about 11.0°, which disappeared after the addition of hydrazine. It indicates that hydrazine can partially reduce GO by simply stirring the mixed solution at room temperature. After vitamin C and hydrazine were added to the solution, the Fe(OH)3 nanoparticles were transformed into Fe3O4 nanoparticles and the partially reduced GO was fully transformed to rGO via the hydrothermal reaction. The product is named as Fe3O4/rGO-180. During the process, Fe3+ can be reduced to Fe2+ by the reductant VC (H2A) via reaction (1)38 and GO is understood to be reduced by hydrazine to rGO.33,34
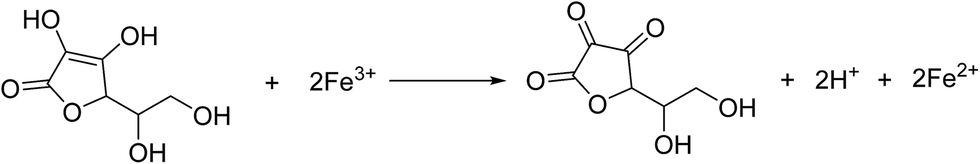 | (1) |
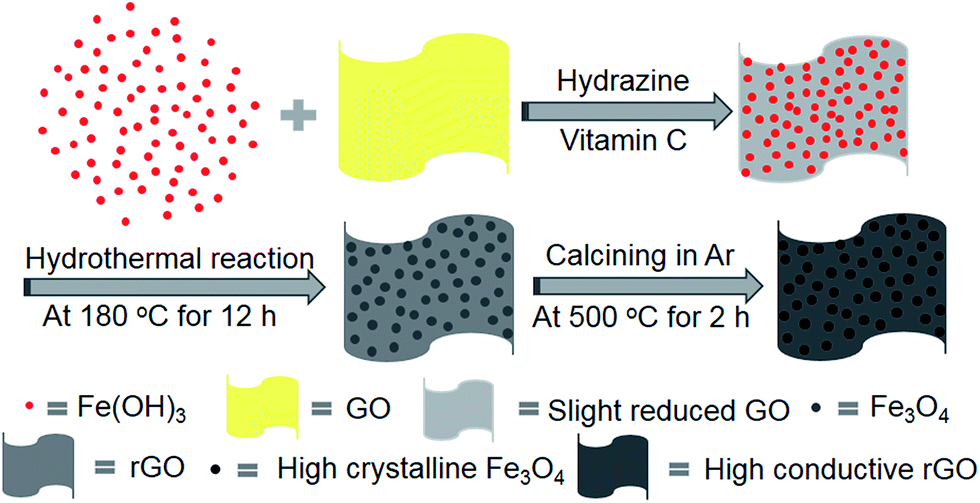 | ||
| Fig. 1 Schematic illustration of the preparation of the two-dimensional Fe3O4/rGO composite electrode. | ||
We also compared Fe2O3/rGO produced from a hydrothermal reaction with Fe(OH)3 and GO as precursors and hydrazine as a reductant (Fig. S2a and b†). This shows that employing VC as the reducing agent not only yields Fe3O4 but also enables the formation of iron oxides with smaller particle sizes. The reductive product formed using bare hydrazine is Fe2O3, which was verified by XRD analysis (Fig. S2c†). To further enhance the electrical conductivity of rGO and increase the crystallinity of Fe3O4, the Fe3O4/rGO-180 was calcined at 500 °C under argon gas protection. The as-prepared Fe3O4/rGO shows a uniform morphology consisting of Fe3O4 with increased crystallinity and rGO with enhanced conductivity. Brunauer–Emmett–Teller (BET) measurement was conducted to measure the surface areas of Fe3O4, rGO and Fe3O4/rGO (Fig. S3†). It showed that Fe3O4/rGO has a BET surface area of 114.7 m2 g−1 which is higher than that for Fe3O4 and rGO. The void space amongst the nano-sized Fe3O4 particles allows the diffusion and migration of ions and can also buffer the volume changes during cycling.
Further evidence of the successful synthesis of the Fe3O4/rGO composites is provided by XRD and X-ray photoelectron spectroscopy (XPS) analyses. Fig. 2a shows the XRD patterns of rGO, Fe3O4, Fe3O4/rGO and Fe3O4/rGO-180. The identified diffraction peaks of Fe3O4 are indexed to the standard card of magnetic Fe3O4 (JCPDS card no. 19-0629) and no additional peak is detected, implying the high purity of Fe3O4. The identifying peaks of rGO at about 26.4° and 44.4° are assigned to the (002) and (101) lattice planes of hexagonal graphite (JCPDS card no. 41-1487), respectively. Other than the peaks for Fe3O4, Fe3O4/rGO-180 has an extra peak at about 26.4° corresponding to the above-mentioned (002) lattice plane in graphite. This indicates that Fe3O4/rGO-180 contains both Fe3O4 and rGO. All of the diffraction peaks which emerge for Fe3O4/rGO-180 become stronger in the XRD pattern of Fe3O4/rGO, showing that the calcination of Fe3O4/rGO-180 increases the crystallinity of Fe3O4 and enhances the degree of reduction of rGO. The chemical composition of the as-prepared Fe3O4/rGO-180 was further determined by XPS analysis. The peak binding energies of 711.5 and 724.9 eV in Fig. 2b are associated with Fe 2p3/2 and Fe 2p1/2 in Fe3O4/rGO-180, respectively. This demonstrates the existence of Fe3O4 in Fe3O4/rGO-180, consistent with previous reports.15,19,39
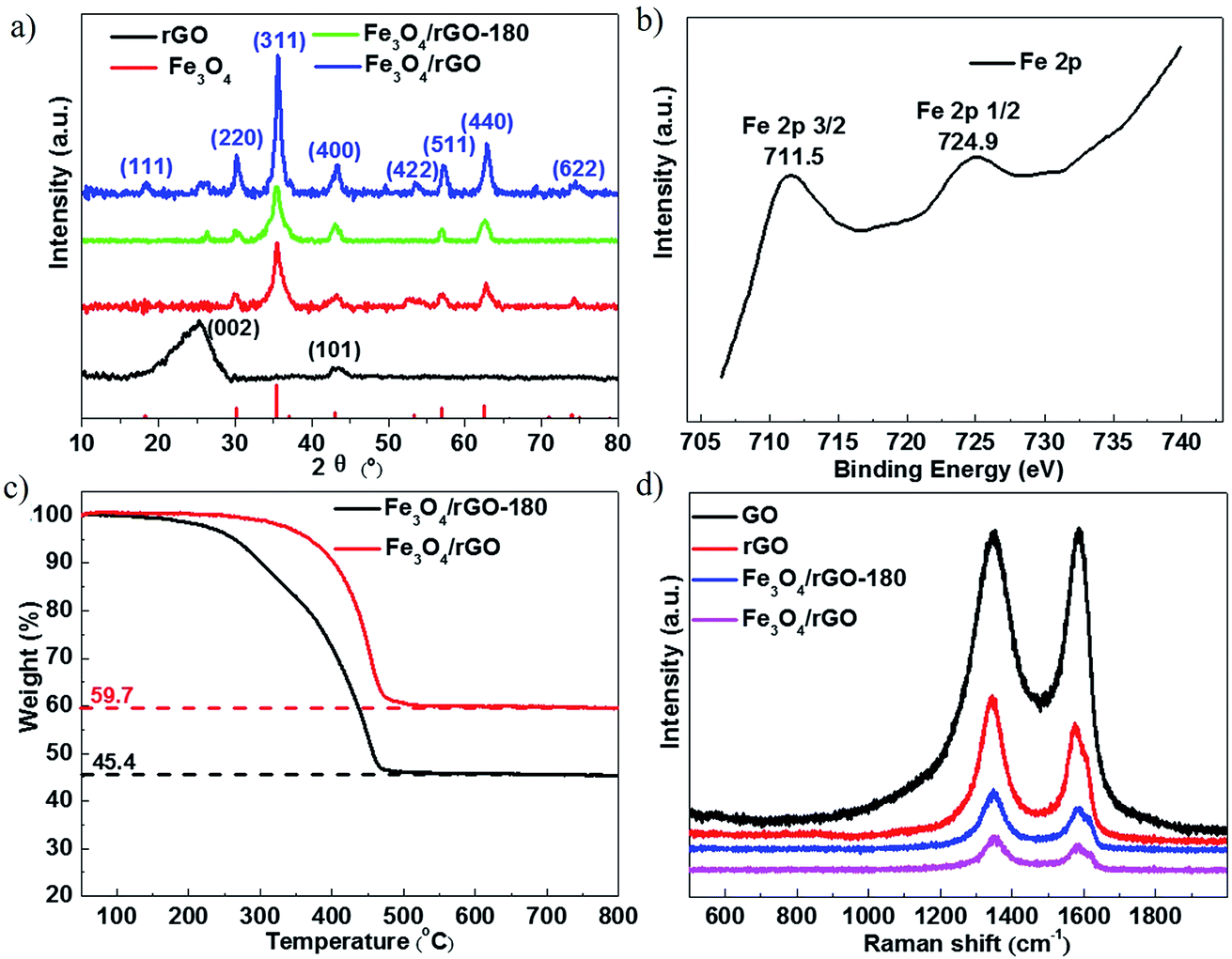 | ||
| Fig. 2 (a) XRD patterns of rGO, Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO. (b) The high resolution Fe 2p spectrum of Fe3O4/rGO-180. (c) TG curves of Fe3O4/rGO-180 and Fe3O4/rGO. (d) Raman spectra of GO, rGO, Fe3O4/rGO-180 and Fe3O4/rGO. | ||
The weight fractions of Fe3O4 and rGO in Fe3O4/rGO-180 and Fe3O4/rGO were characterized by thermogravimetric (TG) analysis in air. During the thermal process, the amount of Fe2O3 should be 1.035 times the amount of Fe3O4, calculated from the reaction of 4Fe3O4 + O2 = 6Fe2O3, and rGO would convert to CO2. Therefore, according to the ratio of the residual reddish brown Fe2O3 in the original Fe3O4/rGO-180 shown in Fig. 2c, the calculated proportions of Fe3O4 and rGO in Fe3O4/rGO-180 are 43.9 wt% and 56.1 wt%, respectively. Additionally, the proportions of Fe3O4 and rGO in Fe3O4/rGO were calculated to be 57.7 wt% and 42.3 wt%, respectively. The results indicate that the Fe3O4/rGO contains more Fe3O4 and thus more rGO with a higher level of reduction. Fig. 2d shows the Raman spectra of GO, rGO, Fe3O4/rGO-180 and Fe3O4/rGO. This allows us to assess the degree of disorder in the carbon materials. The spectra of these four materials show two pronounced peaks: a D peak at about 1350 cm−1 and a G peak at about 1590 cm−1. The D peak corresponds to the disordered structures with defects, while the graphitic structures with order are responsible for the G peak. The degree of disorder in the carbon materials is often determined from the ratio of the peak intensities (R), ID/IG. The values of R for GO and rGO are 1.00 and 1.26, respectively. It should be noted that Fe3O4 has a peak at about 1300 cm−1, thus the values of R for Fe3O4/rGO-180 and Fe3O4/rGO are difficult to calculate.33 The increased value of R in rGO reflects the presence of more disorder or carbon defects.40–42
Fig. 3a and c show the morphologies of Fe3O4/rGO-180 and Fe3O4/rGO. They demonstrate that Fe3O4 nanoparticles are uniformly embedded in rGO nanosheets. The morphology of the composite electrode created using Fe(OH)3 is more uniform than that using FeCl3·6H2O as a precursor.43 We also created pure Fe3O4 nanoparticles using hydrazine and VC at 180 °C and they display similar morphology to the nanoparticles in Fe3O4/rGO-180 (Fig. S4a and b†). Fig. 3b and d show the particle diameter distribution of Fe3O4 nanoparticles in Fe3O4/rGO-180 and Fe3O4/rGO. These show that the average size of Fe3O4 in Fe3O4/rGO-180 is 6.42 nm with a diameter range from 4.1 to 8.8 nm, and the average size of Fe3O4 in Fe3O4/rGO is 6.45 nm with a diameter range from 3.9 to 9.6 nm. The low magnification TEM image of Fe3O4/rGO-180 and the SEM images of Fe3O4/rGO further confirm the uniform distribution of Fe3O4 in rGO (Fig. S5a–c†).
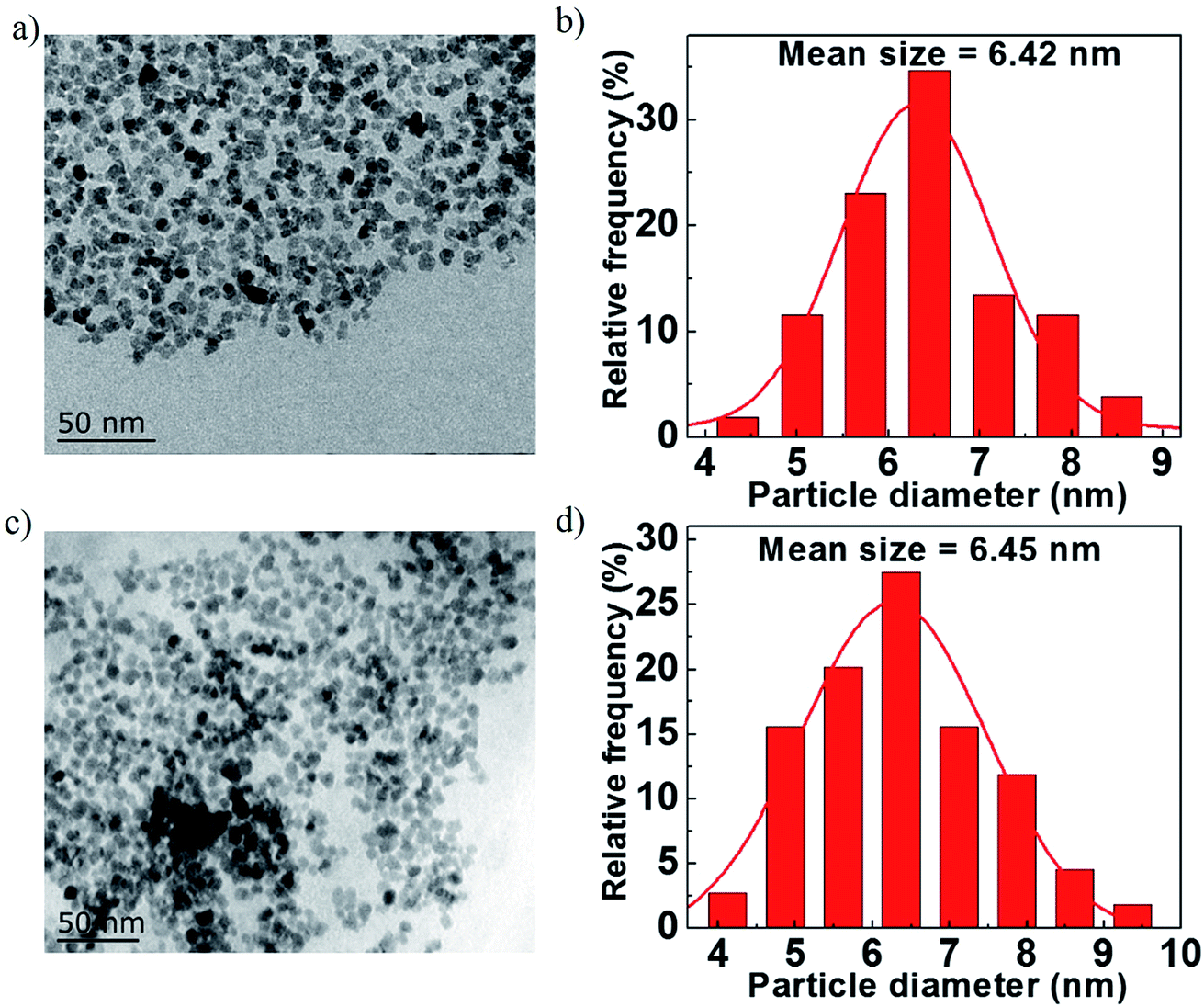 | ||
| Fig. 3 (a and c) TEM images of Fe3O4/rGO-180 and Fe3O4/rGO. (b and d) The diameter distribution of Fe3O4 nanoparticles in Fe3O4/rGO-180 and Fe3O4/rGO. | ||
Fig. 4 shows the electrochemical cycling performance of cells with rGO, Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO as the electrode. Fig. 4a compares the cycling performance of the first 100 cycles of the four electrodes. The cells with Fe3O4/rGO-180 or Fe3O4/rGO have much higher charge–discharge capacities than the cells with rGO or Fe3O4, indicating that the employment of rGO with Fe3O4 nanoparticles increases the capacity compared to bare Fe3O4. It can also be seen that the cells with Fe3O4/rGO display a higher capacity than the cells with Fe3O4/rGO-180, meaning that the calcination process used to make Fe3O4/rGO is responsible for the enhancement in cycling performance. This might be due to the increased crystallinity of Fe3O4 and the increased degree of graphitization of rGO. Fig. 4a also shows that the capacities of the cells with Fe3O4/rGO-180 or Fe3O4/rGO decrease dramatically in the first 10 cycles and gradually increase during the subsequent cycles. It should be noted that the capacity after the initial decrease can be higher than the theoretical specific capacity of Fe3O4/rGO (around 849.6 mA h g−1). The exceeded capacity is attributed to the reversible formation of a polymeric gel-like film,44–47 which grows continuously and delivers additional reversible capacity after the first few cycles. The charge–discharge voltage profiles for the four electrodes at the first cycle are provided in Fig. S6.† The long-term charge–discharge characteristics of cells with Fe3O4 or Fe3O4/rGO are shown in Fig. 4b. The discharge capacities of Fe3O4/rGO at the 100th, 150th, 250th and 400th cycles are 1126, 1143, 1193 and 1108 mA h g−1, respectively, while those of Fe3O4 at the 150th, 200th, 250th and 300th cycles are 128, 149, 173 and 192 mA h g−1, respectively. This indicates that Fe3O4/rGO delivers higher capacity than bare Fe3O4, and has good capacity retention for deep cycling. The coulombic efficiencies of these four electrodes are displayed in Fig. S7.† The rate capability of cells with Fe3O4/rGO is measured by galvanostatically cycling the cells at different current densities, as shown in Fig. 4c. Cells with Fe3O4/rGO render high specific discharge capacities of 1188, 1086, 1011, 903, 811, 718 and 593 mA h g−1 at current densities of 0.05, 0.1, 0.2, 0.5, 1.0, 2.0 and 5.0 A g−1, respectively. Additionally, when cycled back to a lower current density of 0.1 A g−1, the cell maintains the initial discharge capacity and delivers 1030 mA h g−1. Fig. 4d shows the corresponding specific charge–discharge curves of cells using Fe3O4/rGO at different current densities. The approximate symmetrical shapes of these curves indicate good reversibility of Li+ insertion/extraction.
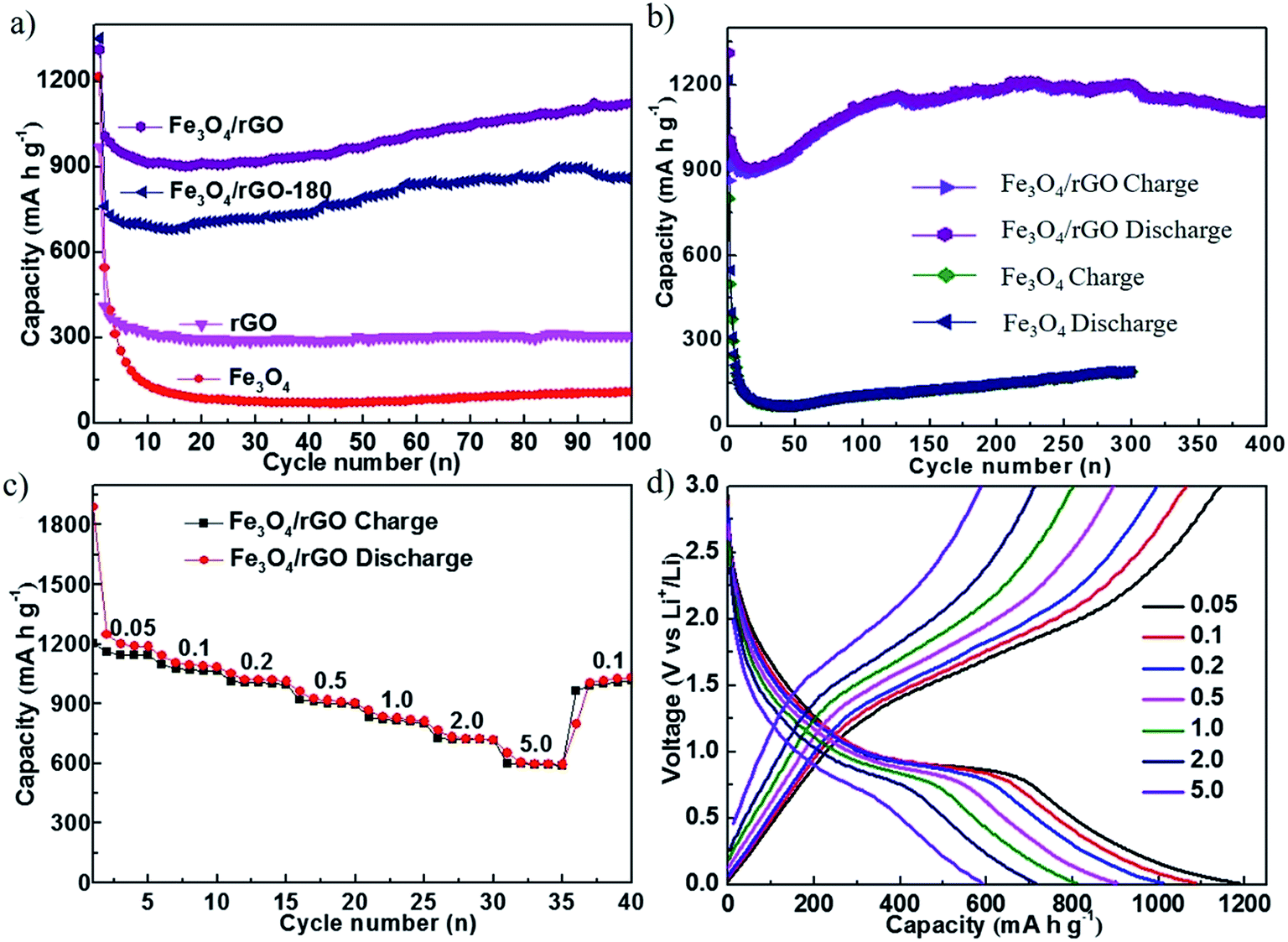 | ||
| Fig. 4 (a) The cycling performance of cells with rGO, Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO at a current density of 0.5 A g−1. (b) The long-term charge–discharge characteristics of cells using Fe3O4 and Fe3O4/rGO. (c) The rate capability of cells with the Fe3O4/rGO electrode. The galvanostatic cycling measurements were performed at various current densities from 0.05 A g−1 to 5 A g−1. (d) The corresponding charge–discharge curves of (c). | ||
Fig. 5a–c show the cyclic voltammograms (CVs) of cells with Fe3O4/rGO, Fe3O4 and rGO, respectively. The electrochemical measurements were carried out at a voltage of 0.01–3.00 V and at a scan rate of 0.1 mV s−1. During the first cycle, the cathodic peaks were observed at about 1.59 V (P1), 1.37 V (P2) and 1.00 V (P3), which are assigned to the reaction Fe3O4 + xLi+ + xe− → LixFe3O4,20–22 and the peak at about 0.67 V (P4) is attributed to the reaction LixFe3O4 + (8 − x)Li+ + (8 − x)e− → 3Fe0 + 4Li2O and the generation of a solid electrolyte interface (SEI) film.20–22 The broad anodic peak (P5) containing two connected peaks at about 1.63 V and 1.87 V is attributed to the oxidation reactions of Fe0 to Fe2+ and Fe2+ to Fe3+, respectively.26,47 During the subsequent cycles, the cathodic peak P4 shifts to 0.79 V, which is due to the diminished polarization effect.37 It can also be seen that there are no visibly pronounced changes in the peak position and current amplitude after the first cycle, indicating that Fe3O4/rGO has excellent cycling stability. Fig. 5b summarizes the CVs of cells with Fe3O4 for the first three cycles. The peak corresponding to the reaction of Fe3O4 to Fe is at about 0.70 V (P3) and the peaks for the reverse reaction appear at about 1.62 V and 1.82 V (P4) in the first cycle. These peak currents continuously decline in the 2nd and 3rd cycles, which could be caused by crushing of the crystal, volume expansion and shrinkage of Fe3O4 and the continuous breaking and formation of the SEI film.14 Fig. 5c shows the CVs for cells with rGO. The peak at about 0.19 V corresponds to the extraction of lithium ions from graphitized carbon, and the anodic peak at about 1.22 V corresponds to the irreversible reaction with the electrolyte.24
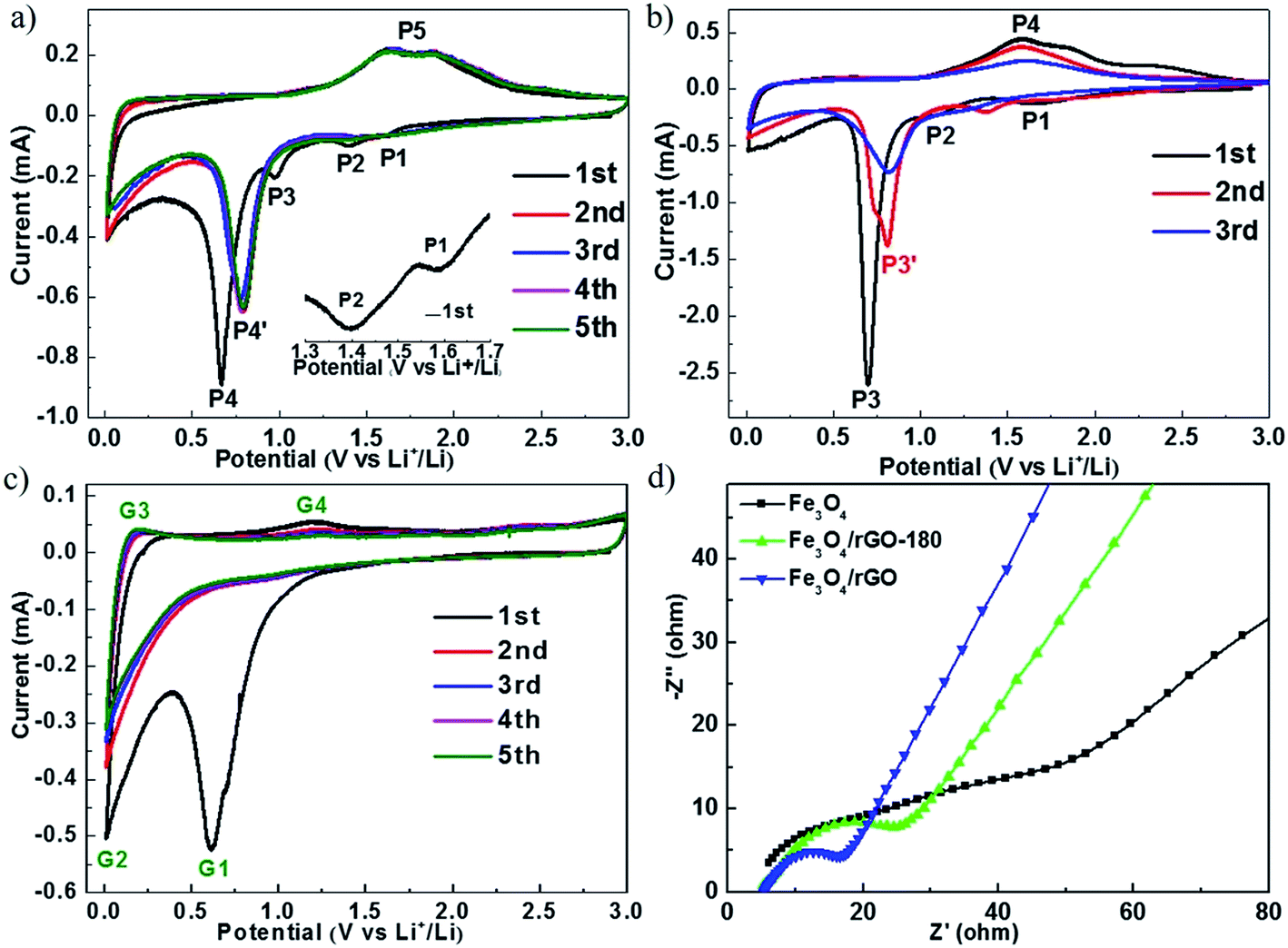 | ||
| Fig. 5 CV curves of (a) Fe3O4/rGO, (b) Fe3O4 and (c) rGO at a scan rate of 0.1 mV s−1 in a voltage range of 0.01–3.0 V. (d) Nyquist plots of Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO. | ||
The electrochemical resistances for cells with Fe3O4, Fe3O4/rGO-180 and Fe3O4/rGO were monitored after one charge–discharge cycle at 0.5 A g−1, as shown in Fig. 5d. The measurements were conducted at an open circuit potential with an AC voltage amplitude of 5.0 mV from 100 kHz to 0.01 Hz. The start of the semicircle is associated with the electrolyte resistance (Rs), the semicircle at high frequency corresponds to the charge transfer resistance (Rct) and the line near the end of the semicircle reveals the diffusion of lithium ions. The electrolyte resistances are almost the same for the three types of cell, and cells with Fe3O4/rGO-180 or Fe3O4/rGO have a much smaller Rct than cells with Fe3O4. This indicates that the combination of Fe3O4 with rGO promotes the charge transfer kinetics and the diffusion of lithium ions in the vicinity of the electrode. The equivalent circuit is used for fitting (Fig. S8†).
Conclusions
In summary, we fabricated a uniform Fe3O4/reduced graphene oxide nanocomposite electrode by a hydrothermal reaction using Fe(OH)3 and graphene oxide as precursors, and vitamin C and hydrazine as reductants. The oxygen containing groups in the graphene oxide nanosheets facilitated the uniform distributions of Fe(OH)3 nanoparticles on the surfaces of rGO via electrostatic forces. Fe3O4/rGO nanoparticles were further calcined to increase the crystallinity of Fe3O4 and to increase the degree of graphitization of rGO. From electrochemical measurements, it was found that cells using Fe3O4/rGO nanoparticles as the electrode deliver a reversible capacity of 1108 mA h g−1 after 400 cycles at 0.5 A g−1 and maintain a reversible capacity of 593 mA h g−1 at a high current density of 5.0 A g−1. The tiny particle size and uniform particle distribution of Fe3O4 in rGO are responsible for the excellent electrochemical performance. The void space amongst the Fe3O4 nanoparticles and rGO serves as a reservoir for the volume changes during cycling, and the graphitized nature of rGO remedies the weak conductivity of iron oxides. This novel synthesis method can potentially be applied to other transition metal oxides such as SnO2, Co2O3, NiO, TiO2 and RuO2 for high-energy lithium-ion batteries.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Y. Lu acknowledges the financial support from the National Key R&D Program of China (2016YFA0202900), the Natural Science Foundation of China (NSFC, grant number 21676242), and the State Key Laboratory of Chemical Engineering (No. SKL-ChE-17D01). Y. Lu appreciates the help from Yongjuan Xie and Jingjing Hua for the TEM and SEM characterization in the Test Center of Yangzhou University.References
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef PubMed.
- J. J. Wang, H. Zhou, J. Nanda and P. V. Braun, Chem. Mater., 2015, 27, 2803–2811 CrossRef CAS.
- M. V. Reddy, C. Yu, J. H. Fan, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 4361–4366 CAS.
- Y. Sharma, N. Sharma, G. V. S. Rao and B. V. R. Chowdari, J. Power Sources, 2007, 173, 495–501 CrossRef CAS.
- S. K. Martha, J. Nanda, H. Zhou, J. C. Idrobo, N. J. Dudney, S. Pannala, S. Dai, J. J. Wang and P. V. Braun, RSC Adv., 2014, 4, 6730–6737 RSC.
- X. P. Gao and H. X. Yang, Energy Environ. Sci., 2010, 3, 174–189 CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- R. E. Doe, K. A. Persson, Y. S. Meng and G. Ceder, Chem. Mater., 2008, 20, 5274–5283 CrossRef CAS.
- N. Yamakawa, M. Jiang, B. Key and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 10525–10536 CrossRef CAS PubMed.
- F. Wang, H. C. Yu, M. H. Chen, L. J. Wu, N. Pereira, K. Thornton, A. Van der Ven, Y. M. Zhu, G. G. Amatucci and J. Graetz, Nat. Commun., 2012, 3, 8 Search PubMed.
- J. Jiang, Y. Y. Li, J. P. Liu, X. T. Huang, C. Z. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- H. W. Zhang, L. Zhou, O. Noonan, D. J. Martin, A. K. Whittaker and C. Z. Yu, Adv. Funct. Mater., 2014, 24, 4337–4342 CrossRef CAS.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- L. Pan, X. D. Zhu, X. M. Xie and Y. T. Liu, Adv. Funct. Mater., 2015, 25, 3341–3350 CrossRef CAS.
- W. Wei, S. B. Yang, H. X. Zhou, I. Lieberwirth, X. L. Feng and K. Mullen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- H. L. Fei, Z. W. Peng, L. Li, Y. Yang, W. Lu, E. L. G. Samuel, X. J. Fan and J. M. Tour, Nano Res., 2014, 7, 502–510 CrossRef CAS.
- Y. R. Wang, L. Zhang, X. H. Gao, L. Y. Mao, Y. Hu and X. W. Lou, Small, 2014, 10, 2815–2819 CrossRef CAS PubMed.
- L. W. Su, Y. R. Zhong and Z. Zhou, J. Mater. Chem. A, 2013, 1, 15158–15166 CAS.
- C. N. He, S. Wu, N. Q. Zhao, C. S. Shi, E. Z. Liu and J. J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- T. Yoon, C. Chae, Y. K. Sun, X. Zhao, H. H. Kung and J. K. Lee, J. Mater. Chem., 2011, 21, 17325–17330 RSC.
- L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, J. Power Sources, 2008, 183, 717–723 CrossRef CAS.
- S. Zhu, M. Chen, J. Sun, J. Liu, T. Wu, H. Su, S. Qu, Y. Xie, S. Wang, X. Su and G. Diao, RSC Adv., 2016, 6, 58529–58540 RSC.
- G. M. Zhou, D. W. Wang, P. X. Hou, W. S. Li, N. Li, C. Liu, F. Li and H. M. Cheng, J. Mater. Chem., 2012, 22, 17942–17946 RSC.
- Y. He, L. Huang, J. S. Cai, X. M. Zheng and S. G. Sun, Electrochim. Acta, 2010, 55, 1140–1144 CrossRef CAS.
- Y. Shi, J. Zhang, A. M. Bruck, Y. M. Zhang, J. Li, E. A. Stach, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi and G. H. Yu, Adv. Mater., 2017, 29, 8 Search PubMed.
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, S. J. Chang, Y. K. Han, Y. B. Lee, H. U. Lee, S. Kwon, G. Lee, C. S. Lee and Y. S. Huh, Adv. Mater., 2013, 25, 6250–6255 CrossRef CAS PubMed.
- X. W. Li, L. Qiao, D. Li, X. H. Wang, W. H. Xie and D. Y. He, J. Mater. Chem. A, 2013, 1, 6400–6406 CAS.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- M. V. Reddy, T. Yu, C. H. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Chem.–Eur. J., 2011, 17, 661–667 CrossRef CAS PubMed.
- L. W. Ji, Z. K. Tan, T. R. Kuykendall, S. Aloni, S. D. Xun, E. Lin, V. Battaglia and Y. G. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 7170–7177 RSC.
- O. Gerber, S. Begin-Colin, B. P. Pichon, E. Barraud, S. Lemonnier, C. Pham-Huu, B. Daffos, P. Simon, J. Come and D. Begin, J. Energy Chem., 2016, 25, 272–277 CrossRef.
- F. Han, W. C. Li, D. Li and A. H. Lu, J. Energy Chem., 2013, 22, 329–335 CrossRef CAS.
- S. P. Zhu, M. Chen, W. J. Ren, J. R. Yang, S. S. Qu, Z. C. Li and G. W. Diao, New J. Chem., 2015, 39, 7923–7931 RSC.
- A. Babaei, M. Zendehdel, B. Khalilzadeh and A. Taheri, Colloids Surf., B, 2008, 66, 226–232 CrossRef CAS PubMed.
- J. Lu, X. L. Jiao, D. R. Chen and W. Li, J. Phys. Chem. C, 2009, 113, 4012–4017 CAS.
- Z. Li, Y. J. Chen, Y. K. Du, X. M. Wang, P. Yang and J. W. Zheng, Int. J. Hydrogen Energy, 2012, 37, 4880–4888 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- J. A. Zheng, C. A. Di, Y. Q. Liu, H. T. Liu, Y. L. Guo, C. Y. Du, T. Wu, G. Yu and D. B. Zhu, Chem. Commun., 2010, 46, 5728–5730 RSC.
- J. Su, M. H. Cao, L. Ren and C. W. Hu, J. Phys. Chem. C, 2011, 115, 14469–14477 CAS.
- L. Fan, W. Zhang, S. Zhu and Y. Lu, Ind. Eng. Chem. Res., 2017, 56, 2046–2053 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- Y. Chen, B. H. Song, X. S. Tang, L. Lu and J. M. Xue, Small, 2014, 10, 1536–1543 CrossRef CAS PubMed.
- Y. Suo, Q. Q. Zhao, J. K. Meng, J. Li, X. C. Zheng, X. X. Guan, Y. S. Liu and J. M. Zhang, Mater. Lett., 2016, 174, 36–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and SEM images, and XRD patterns of Fe(OH)3/GO with or without adding hydrazine (Fig. S1); TEM images of Fe3O4/rGO-180 and Fe2O3/rGO, and the XRD pattern of Fe2O3/rGO (Fig. S2); BET surface areas of Fe3O4, rGO and Fe3O4/rGO (Fig. S3); TEM images of Fe3O4 nanoparticles at different magnifications (Fig. S4); a TEM image of Fe3O4/rGO-180 and SEM images of Fe3O4/rGO at different magnifications (Fig. S5); the first cycle charge–discharge profiles of cells with Fe3O4, rGO, Fe3O4/rGO-180 or Fe3O4/rGO (Fig. S6); coulombic efficiencies of cells with rGO, Fe3O4, Fe3O4/rGO-180 or Fe3O4/rGO at a current density of 0.5 A g−1 and long-term charge–discharge coulombic efficiencies of cells with Fe3O4 or Fe3O4/rGO (Fig. S7); the equivalent circuit used for fitting cell resistances (Fig. S8). See DOI: 10.1039/c7ra11779e |
| This journal is © The Royal Society of Chemistry 2017 |