
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A series of planar tetranuclear lanthanide complexes: axial ligand modulated magnetic dynamics in Dy4 species†
Ya-Xin Zhanga,
Meng Lia,
Bi-Ying Liua,
Zhi-Lei Wu *a,
Hai-Ying Wei*a and
Wen-Min Wang*b
*a,
Hai-Ying Wei*a and
Wen-Min Wang*b
aCollege of Chemistry and Environmental Science, Hebei University, Baoding, 071002, P. R. China. E-mail: wuzhilei03@163.com; weihy@hbu.edu.cn
bDepartment of Chemistry, Tianjin University, Tianjin, 300072, China. E-mail: wangwenmin0506@126.com
First published on 7th December 2017
Abstract
Ten new lanthanide compounds were successfully obtained based on an 8-hydroxyquinoline Schiff base derivative and β-diketonates with different substituent groups, namely, [Ln4(μ3-OH)2L6(acac)4]·2.5CH3CN (Ln = Gd (1), Tb (2), Dy (3), Ho (4) and Yb (5); HL = 5-(4-methoxybenzylidene)-8-hydroxylquinoline; Hacac = acetylacetone), [Ln4(μ3-OH)2L6(tmhd)4]·xCH3CN·yCH3CH2OH (Ln = Eu (6), Gd (7), and Tb (8), x = 1, y = 1; Ln = Dy (9), x = 2, y = 0; Htmhd = 2,2,6,6-tetramethyl-3,5-heptanedione) and [Dy4(μ3-OH)2L6(beac)4]·CH3CN (10) (Hbeac = 1-benzoylacetone). Single crystal X-ray diffraction revealed that compounds 1–10 all contain planar tetranuclear lanthanide centers with a similar butterfly-shape arrangement, and each lanthanide ion is located in a distorted bicapped trigonal prism coordination sphere. 1·Gd and 7·Gd exhibit magnetic refrigeration behavior with the maximum values of −ΔSm being 19.39(2) and 16.21(3) J K−1 kg−1, respectively. Additionally, the effects of steric-hindrance on the magnetic anisotropy of the Dy3+ centers in 3·Dy, 9·Dy and 10·Dy were observed through comparative studies on their magnetic behaviors, which showed different SMM behaviors with energy barriers of 99.29(3) K for 3·Dy, 64.56(4) K for 9·Dy and 61.05(0) K for 10·Dy.
Introduction
Single molecule magnets (SMMs) have become more and more attractive in the fields of chemistry, physics and materials science in that they show potential optical applications and quantum phenomenena.1–3 A number of SMMs have been reported in recent years,4–7 especially for lanthanide-based magnetic compounds, many of which display intriguing magnetic behaviors due to the significant intrinsic magnetic anisotropy arising from their large and unquenched orbital angular momentum. However, intentional design and regulation of structural and magnetic changes is still a challenge. The investigations reveal that the magnetic properties can be deliberately tuned by altering the coordination geometry and ligand field around the metal centers. It should be noted that the regulation of magnetic transformations has been realized by changing the solvent molecules,8 doping with other elements,9 modifying ligands,10 etc. Impressively, Tong's group synthesized two similar dysprosium complexes coordinated by different peripheral anions (Cl− and Br−), displaying distinct magnetic dynamics with effective energy barriers of 708 K and 1025 K, respectively.11 In addition, two dinuclear dysprosium compounds with different magnetic properties were assembled by Tang et al. through employing two ligands with different coordination pockets.12 Hence, it can be seen that the SMMs behaviors were sensitive to the coordination geometry and ligand fields around the lanthanide sites, and even a slight change in the coordination environment of Ln(III) ion center might result in significant divergence of the magnetic behaviors.In our previous work, we employed two Schiff base ligands with different substituents to construct SMMs, and investigated the electronic effect on the magnetic behaviors, obtaining two dysprosium complexes with anisotropic barriers (ΔE/kBT) 67.6 K and 34.5 K, respectively.13 The different SMMs behaviors mainly originated from the different electron-donating effect, which to some extent influenced the coordination environment of center Dy3+ ions. Inspired by this case, we expect to prepare different series of lanthanide complexes based on Schiff base ligand HL (HL = 5-(4-methoxybenzylidene)-8-hydroxylquinoline; synthesized by condensation of 5-amino-8-hydroxyquinoline and 4-methoxybenzaldehyde) and different β-diketonates (Scheme 1 and 2) with objective to study the effect of auxiliary ligands on both structures and magnetic properties. Herein, as a continuation of our work, we report three series of tetranuclear lanthanide complexes with similar butterfly topology based on HL and β-diketonates, [Ln4(μ3-OH)2L6(acac)4]·2.5CH3CN (Ln = Gd (1), Tb (2), Dy (3), Ho (4) and Yb (5); Hacac = acetylacetone), [Ln4(μ3-OH)2L6(tmhd)4]·xCH3CN·yCH3CH2OH (Ln = Eu (6), Gd (7), and Tb (8), x = 1, y = 1; Ln = Dy (9), x = 2, y = 0; Htmhd = 2,2,6,6-tetramethyl-3,5-heptanedione) and [Dy4(μ3-OH)2L6(beac)4]·CH3CN (10) (Hbeac = 1-benzoylacetone). Thereinto, complexes 3·Dy, 9·Dy and 10·Dy display different magnetic relaxation behaviors with the energy barrier of 99.29(3) K for 3·Dy, 64.56(4) K for 9·Dy and 61.05(0) K for 10·Dy. As expected, their magnetic properties show a substituent-dependent phenomenon, which mainly originate from the different ligand field effects on the local Dy3+ sites. Additionally, complexes 1·Gd and 7·Gd exhibit magnetic entropy change (−ΔSm) of 19.39(2) J K−1 kg−1 and 16.21(3) J K−1 kg−1, respectively.
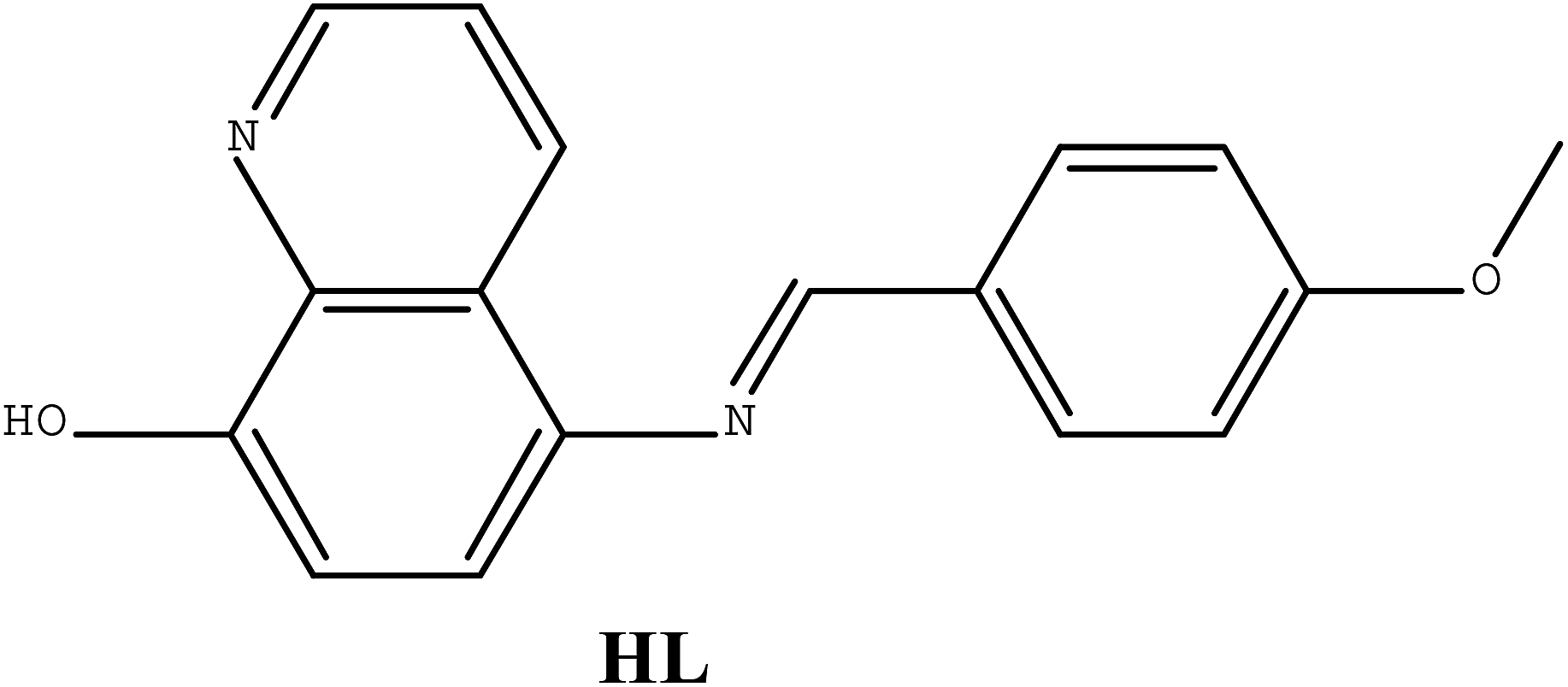 | ||
| Scheme 1 Structure of ligand HL. | ||
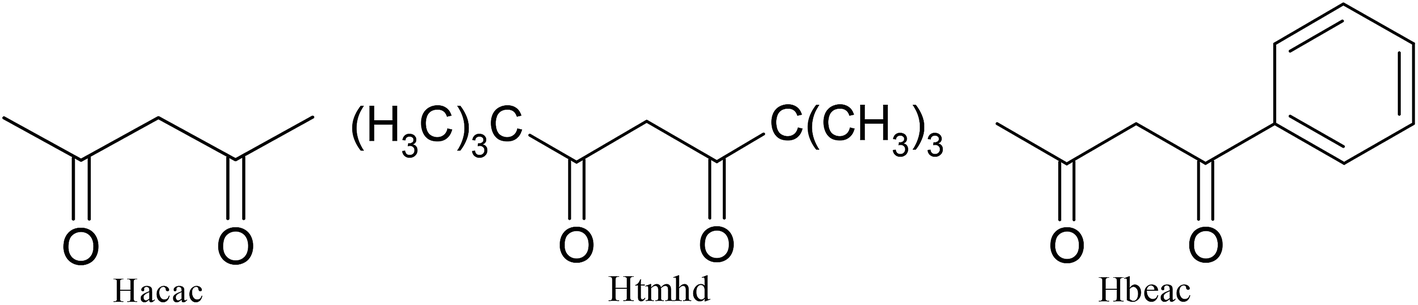 | ||
| Scheme 2 Structures of three different β-diketones. | ||
Experiment section
Materials and general methods
All the chemicals and solvents used for the syntheses were reagent grade without further purification. The Ln(acac)3·2H2O (Ln = Gd, Tb, Dy, Ho, Yb), Ln(tmhd)3·2H2O (Ln = Eu, Gd, Tb, Dy) and Dy(heac)3·2H2O were obtained according to the literature previously.14 The Schiff base ligand HL was synthesized by condensation of 5-amino-8-hydroxyquinoline and 4-methoxybenzaldehyde. 1H NMR spectra was performed on JEOL Eclipse β 400 MHz spectrometer. Elemental analyses for C, H and N were measured by a PerkinElmer 240 CHN elemental analyzer. IR spectra were recorded on a Bruker Tensor 27 spectrophotometer using KBr disks. Powder X-ray diffraction (PXRD) data were collected on a D/Max-2500 X-ray diffractometer using Cu Kα radiation (Fig. S3 in ESI†). Thermogravimetric analysis (TGA) was performed on a Netzsch TG 209 Setaram apparatus from room temperature to 800 °C with a heating rate of 10 °C min−1 (Fig. S4 in ESI†). Magnetic measurements were carried out with Quantum Design MPMS-XL7 and PPMS-9 ACMS magnetometers. All the samples were restrained in eicosane to prevent torqueing. The diamagnetic corrections for compounds were estimated from Pascal's constants as well as sample holder calibration.Crystallographic studies
Single-crystal X-ray diffraction measurements of 1–10 were collected on a Rigaku-RAPID diffractometer equipped with graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares methods on F2 using SHELXTL-97 and SHELXTL-97 programs. All non-hydrogen atoms were refined with anisotropic parameters, while hydrogen atoms were placed in calculated positions and refined using a riding model. Due to the low residual electron density, the position of free solvents cannot be determined by single crystal X-ray diffraction techniques, and therefore the SQUEEZE routine in the PLATON software package was applied. Crystallographic data parameters for 1–10 are listed in Tables S1 and S2.† CCDC 1544673–1544682† contain supplementary crystallographic data for compounds 1–10.Results and discussion
Synthesis and IR spectra
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H and CN group, the 2836 cm−1 peak can be assigned to the C–H stretching vibration of O–CH3 group. Furthermore, the peak at 1236 cm−1 represents the C–O stretching vibration of phenol hydroxyl group.
C–H and CN group, the 2836 cm−1 peak can be assigned to the C–H stretching vibration of O–CH3 group. Furthermore, the peak at 1236 cm−1 represents the C–O stretching vibration of phenol hydroxyl group.
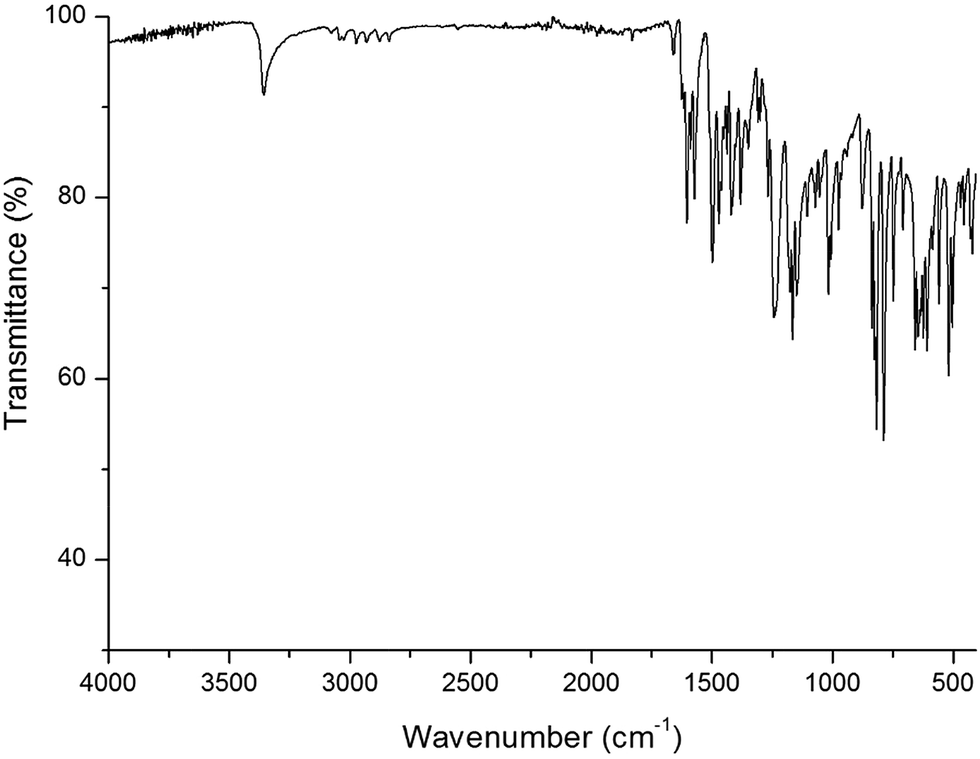 | ||
| Fig. 1 The IR spectrum of ligand HL. | ||
[Gd4(μ3-OH)2L6(acac)4]·2.5CH3CN (1·Gd). Yield based on Gd: 38%. IR (KBr, cm−1): 3693 (s), 3631 (w), 2916 (w), 2836 (w), 1602 (s), 1573 (w), 1515 (m), 1499 (m), 1464 (m), 1384 (m), 1357 (w), 1310 (m), 1253 (w), 1165 (m), 1090 (m), 1019 (w), 833 (m), 919 (w), 881 (w), 832 (w), 787 (w), 760 (w), 525 (w). Anal. calcd for C127H115.5N14.5O22Gd4: C, 53.98; H, 4.10; N, 6.94. Found: C, 54.13; H, 4.25; N, 6.78.
[Tb4(μ3-OH)2L6(acac)4]·2.5CH3CN (2·Tb). Yield based on Tb: 42%. IR (KBr, cm−1): 3693 (s), 3631 (w), 2915 (w), 2836 (w), 1601 (s), 1573 (w), 1515 (m), 1498 (m), 1464 (m), 1384 (m), 1356 (w), 1310 (m), 1252 (w), 1165 (m), 1090 (m), 1019 (w), 833 (m), 918 (w), 881 (w), 831 (w), 787 (w), 760 (w), 524 (w). Anal. calcd for C127H115.5N14.5O22Tb4: C, 53.85; H, 4.09; N, 6.92. Found: C, 53.63; H, 3.92; N, 7.07.
[Dy4(μ3-OH)2L6(acac)4]·2.5CH3CN (3·Dy). Yield based on Dy: 37%. IR (KBr, cm−1): 3693 (s), 3630 (w), 2916 (w), 2836 (w), 1601 (s), 1573 (w), 1514 (m), 1499 (m), 1464 (m), 1384 (m), 1356 (w), 1310 (m), 1253 (w), 1166 (m), 1090 (m), 1019 (w), 832 (m), 919 (w), 881 (w), 832 (w), 788 (w), 760 (w), 525 (w). Anal. calcd for C127H115.5N14.5O22Dy4: C, 53.58; H, 4.07; N, 6.89. Found: C, 53.71; H, 4.19; N, 6.72.
[Ho4(μ3-OH)2L6(acac)4]·2.5CH3CN (4·Ho). Yield based on Ho: 57%. IR (KBr, cm−1): 3693 (s), 3631 (w), 2916 (w), 2835 (w), 1602 (s), 1573 (w), 1514 (m), 1498 (m), 1464 (m), 1384 (m), 1357 (w), 1311 (m), 1253 (w), 1165 (m), 1090 (m), 1018 (w), 833 (m), 919 (w), 881 (w), 831 (w), 787 (w), 760 (w), 525 (w). Anal. calcd for C127H115.5N14.5O22Ho4: C, 53.40; H, 4.06; N, 6.86. Found: C, 53.25; H, 4.15; N, 6.72.
[Yb4(μ3-OH)2L6(acac)4]·2.5CH3CN (5·Yb). Yield based on Yb: 61%. IR (KBr, cm−1): 3693 (s), 3632 (w), 2916 (w), 2836 (w), 1602 (s), 1572 (w), 1515 (m), 1499 (m), 1464 (m), 1383 (m), 1357 (w), 1310 (m), 1252 (w), 1165 (m), 1091 (m), 1019 (w), 833 (m), 919 (w), 881 (w), 833 (w), 787 (w), 760 (w), 525 (w). Anal. calcd for C127H115.5N14.5O22Yb4: C, 52.80; H, 4.01; N, 6.79. Found: C, 52.66; H, 3.89; N, 6.88.
[Eu4(μ3-OH)2L6(tmhd)4]·CH3CN·CH3CH2OH (6·Eu). Yield based on Eu: 35%. IR (KBr, cm−1): 3434 (s), 2958 (w), 2922 (w), 2838 (w), 1603 (s), 1573 (m), 1515 (m), 1464 (m), 1387 (m), 1309 (m), 1251 (m), 1165 (m), 1134 (w), 1091 (m), 1023 (m), 921 (w), 833 (m), 786 (m), 760 (m), 718 (w), 657 (w), 591 (w), 525 (w). Anal. calcd for C154H174N14O24Eu4: C, 57.57; H, 5.46; N, 6.10. Found: C, 57.67; H, 5.27; N, 6.22.
[Gd4(μ3-OH)2L6(tmhd)4]·CH3CN·CH3CH2OH (7·Gd). Yield based on Gd: 44%. IR (KBr, cm−1): 3433 (s), 2958 (w), 2922 (w), 2837 (w), 1603 (s), 1572 (m), 1516 (m), 1464 (m), 1387 (m), 1310 (m), 1251 (m), 1165 (m), 1135 (w), 1091 (m), 1023 (m), 920 (w), 833 (m), 786 (m), 761 (m), 718 (w), 656 (w), 590 (w), 525 (w). Anal. calcd for C154H174N14O24Gd4: C, 57.19; H, 5.42; N, 6.06. Found: C, 57.06; H, 5.53; N, 6.13.
[Tb4(μ3-OH)2L6(tmhd)4]·CH3CN·CH3CH2OH (8·Tb). Yield based on Tb: 46%. Elemental analysis (%) calcd for IR (KBr, cm−1): 3433 (s), 2958 (w), 2921 (w), 2837 (w), 1602 (s), 1572 (m), 1515 (m), 1464 (m), 1388 (m), 1310 (m), 1251 (m), 1164 (m), 1135 (w), 1091 (m), 1023 (m), 920 (w), 833 (m), 787 (m), 761 (m), 718 (w), 655 (w), 590 (w), 525 (w). Anal. calcd for C154H174N14O24Tb4: C, 57.07; H, 5.41; N, 6.05. Found: C, 56.88; H, 5.56; N, 5.94.
[Dy4(μ3-OH)2L6(tmhd)4]·2CH3CN (9·Dy). Yield based on Dy: 51%. Elemental analysis (%) calcd for IR (KBr, cm−1): 3433 (s), 2958 (w), 2921 (w), 2837 (w), 1602 (s), 1572 (m), 1516 (m), 1464 (m), 1387 (m), 1311 (m), 1251 (m), 1164 (m), 1135 (w), 1091 (m), 1023 (m), 921 (w), 833 (m), 786 (m), 762 (m), 718 (w), 656 (w), 591 (w), 525 (w). Anal. calcd for C154H168N16O22Dy4: C, 57.00; H, 5.22; N, 6.91. Found: C, 56.86; H, 5.34; N, 6.79.
[Dy4(μ3-OH)2L6(beac)4]·CH3CN (10·Dy). Yield based on Dy: 38%. Elemental analysis (%) calcd for. IR (KBr, cm−1): 3633 (s), 2963 (w), 2929 (w), 2869 (w), 1605 (s), 1570 (m), 1518 (m), 1500 (m), 1465 (m), 1404 (m), 1385 (m), 1357 (m), 1313 (m), 1263 (w), 1242 (w), 1174 (w), 1092 (m), 1055 (m), 1017 (w), 967 (w), 919 (w), 880 (w), 831 (w), 784 (w), 754 (w), 655 (w), 529 (w), 403 (w). Anal. calcd for C150H128N16O22Dy4: C, 57.07; H, 4.09; N, 7.10. Found: C, 56.92; H, 4.01; N, 7.26.
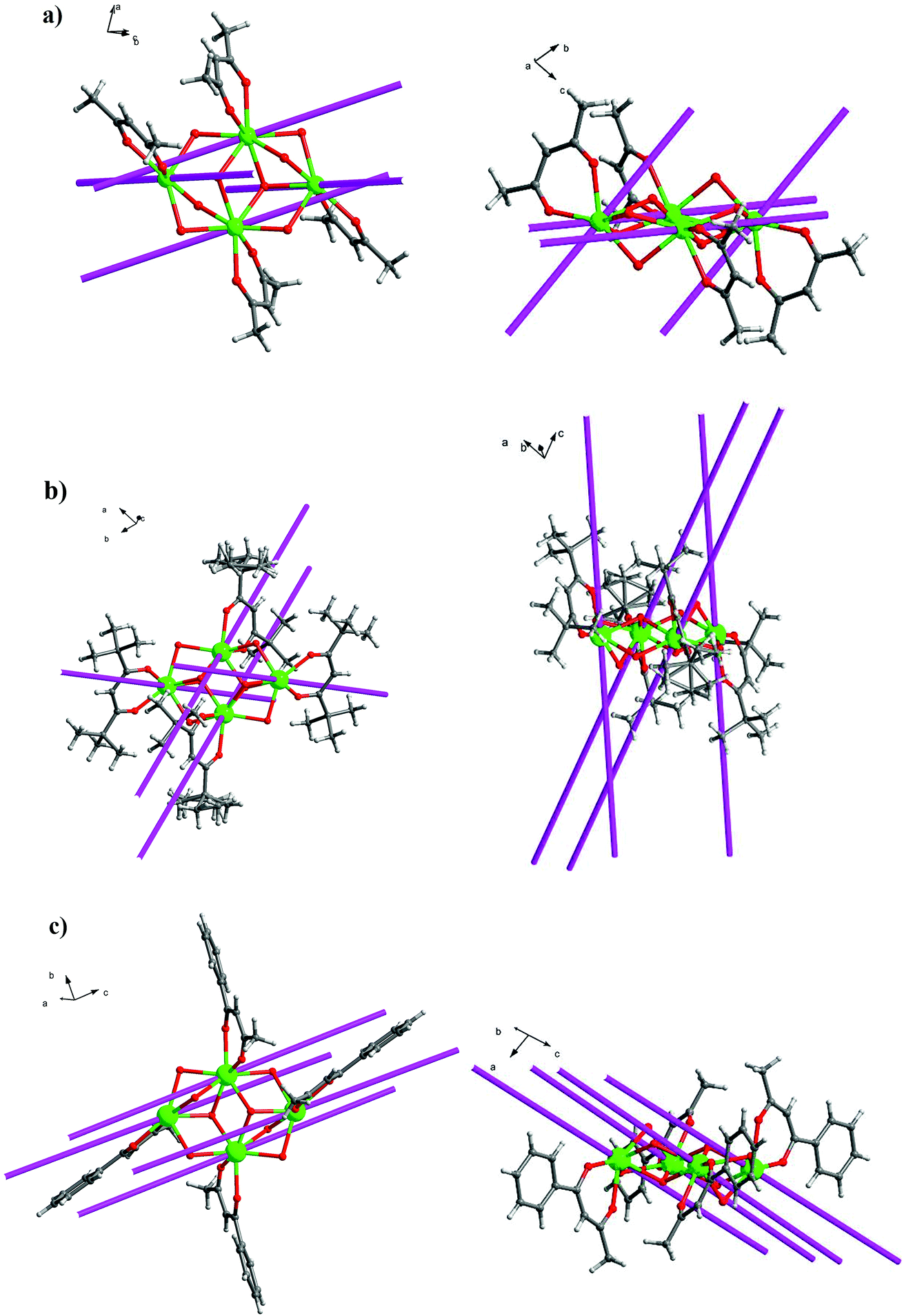
Crystal structure of 1–5
Single X-ray crystal structure analysis reveal that compounds 1–5 are isostructural and crystallize in the triclinic crystal system, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Thus, compound 3·Dy as a representative example is selected to describe in detail. As shown in Fig. 2 and 5, each molecule of 3·Dy contains a butterfly shaped [Dy4O2] core, in which two μ3-OH atoms lie 0.9064(1) Å above and below the [Dy4] plane, bridging the body ions (Dy1 and Dy1A) to the wingtip ions (Dy2 and Dy2A), and the four Dy ions are further linked together by four phenoxide oxygen atoms (O3, O5, O3A and O5A). The [Dy4] core is compose of two scalene triangular, and the distance of Dy1⋯Dy2, Dy1A⋯Dy2 and Dy1⋯Dy1A are 3.8702(2), 3.5171(0) and 3.8668(3) Å, respectively. The periphery of [Dy4] core is surrounded by six L− and four bidentate acac−. All the L− adopt μ2–η2η1 bonding mode (Scheme S1†), they coordinate with Dy3+ ions through two donor atoms (phenoxo-O atom and N atom), and the phenoxo-O atom can also act as a bridge between two Dy3+ ions. Hence, one HL can bridge two Dy3+ ions. Moreover, due to the strong π–π interactions between the quinoline part of L−, four L− ligands are almost in parallel arrangement and locate at different side of the Dy3+ ions. Meanwhile, the acac− adopt η1:η1 bonding mode, blocking the extension of the Dy3+ ions, sequentially resulting into the formation of isolated tetranuclear Dy3+ cluster. In the asymmetric unit of 3·Dy, the eight-coordinated Dy1 ion is coordinated by seven O atoms (two O atoms from acac−, two μ3-OH and three phenoxide oxygen atoms) and one N atom, while the coordination environment of Dy2 ion is completed by six O atoms (two O atoms from acac−, one μ3-OH and three phenoxide oxygen atoms) and two N atoms from L−. Both of them display distorted bicapped trigonal prism coordination geometry (Fig. S1†). All the lengths of Dy–O bonds fall into the range of 2.285(2)–2.411(2) Å, and the range of Dy–N distance are 2.514(3)–2.568(3) Å. Additionally, the Ln---Ln distances gradually shorten with the decrease in the ionic radius of the Ln3+ ions, while the Ln–O–Ln bond angles gradually increase accordingly.
space group. Thus, compound 3·Dy as a representative example is selected to describe in detail. As shown in Fig. 2 and 5, each molecule of 3·Dy contains a butterfly shaped [Dy4O2] core, in which two μ3-OH atoms lie 0.9064(1) Å above and below the [Dy4] plane, bridging the body ions (Dy1 and Dy1A) to the wingtip ions (Dy2 and Dy2A), and the four Dy ions are further linked together by four phenoxide oxygen atoms (O3, O5, O3A and O5A). The [Dy4] core is compose of two scalene triangular, and the distance of Dy1⋯Dy2, Dy1A⋯Dy2 and Dy1⋯Dy1A are 3.8702(2), 3.5171(0) and 3.8668(3) Å, respectively. The periphery of [Dy4] core is surrounded by six L− and four bidentate acac−. All the L− adopt μ2–η2η1 bonding mode (Scheme S1†), they coordinate with Dy3+ ions through two donor atoms (phenoxo-O atom and N atom), and the phenoxo-O atom can also act as a bridge between two Dy3+ ions. Hence, one HL can bridge two Dy3+ ions. Moreover, due to the strong π–π interactions between the quinoline part of L−, four L− ligands are almost in parallel arrangement and locate at different side of the Dy3+ ions. Meanwhile, the acac− adopt η1:η1 bonding mode, blocking the extension of the Dy3+ ions, sequentially resulting into the formation of isolated tetranuclear Dy3+ cluster. In the asymmetric unit of 3·Dy, the eight-coordinated Dy1 ion is coordinated by seven O atoms (two O atoms from acac−, two μ3-OH and three phenoxide oxygen atoms) and one N atom, while the coordination environment of Dy2 ion is completed by six O atoms (two O atoms from acac−, one μ3-OH and three phenoxide oxygen atoms) and two N atoms from L−. Both of them display distorted bicapped trigonal prism coordination geometry (Fig. S1†). All the lengths of Dy–O bonds fall into the range of 2.285(2)–2.411(2) Å, and the range of Dy–N distance are 2.514(3)–2.568(3) Å. Additionally, the Ln---Ln distances gradually shorten with the decrease in the ionic radius of the Ln3+ ions, while the Ln–O–Ln bond angles gradually increase accordingly.
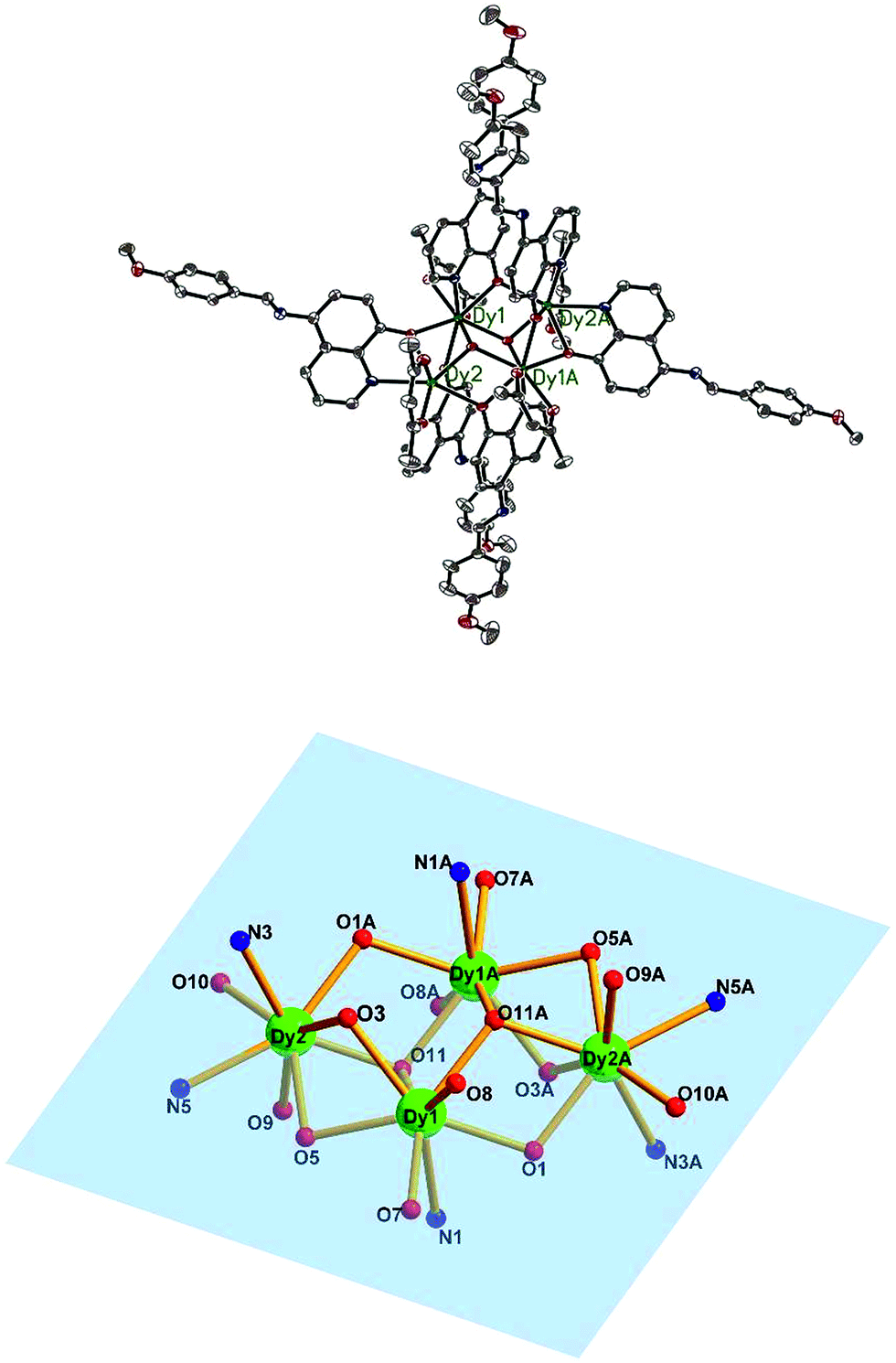 | ||
| Fig. 2 The molecular structure of compound 3·Dy (top) and the butterfly shaped tetranuclear Dy(III) center in 3·Dy (bottom). Hydrogen atoms are omitted for clarity (Dy green; C gray; N blue; O red). | ||
Crystal structure of 6–10
Compounds 6–9 are also isostructural and isolated tetranuclear clusters, crystallizing in the P space group, triclinic crystal system. While 10·Dy crystallizes in the P21/c space group, in monoclinic crystal system, it also displays a butterfly motif. So compound 9·Dy and 10·Dy are selected as an example to describe the structure. Comparably, the structure of 9·Dy and 10·Dy are similar with 3·Dy, and they all consists of a butterfly shaped [Dy4O2] core and six coordinated ligands L− (Fig. 3 and 4). Differently, the coordinated bidentate β-diketonates in 9·Dy and 10·Dy are tmhd− and beac−, respectively, instead of acac− (Fig. 5). The coordination sphere around Dy1 and Dy2 in 9·Dy and 10·Dy are analogous to that in 3·Dy, exhibiting distorted bicapped trigonal prism coordination geometry (Fig. S1 in ESI†). The Dy–O bond lengths of 9·Dy and 10·Dy are in the range of 2.285(3)–2.415(3) Å and 2.323(6)–2.467(7) Å, and Dy–N bond distances of them are 2.533(4)–2.555(4) Å and 2.529(1)–2.546(2) Å. As for the distance between Dy ions in them, for 9·Dy, the distance of Dy1⋯Dy2, Dy1A⋯Dy2 and Dy2⋯Dy2A are 3.5504(3), 3.8696(3) and 3.8963(4) Å. For 10·Dy, the distance of Dy1⋯Dy2, Dy1A⋯Dy2 and Dy1⋯Dy1A are 3.8712(9), 3.6107(1) and 3.8816(1) Å. The crystal packing of compounds 3·Dy, 9·Dy and 10·Dy show that the molecules contact each other through strong π–π interactions, thus generating an infinite supramolecular chains in them (Fig. S2 in ESI†). Finally, these supramolecular chain in 3·Dy, 9·Dy and 10·Dy are stacked through weak intermolecular H-bonding interactions between free solvent molecules locating in their crystal lattices.
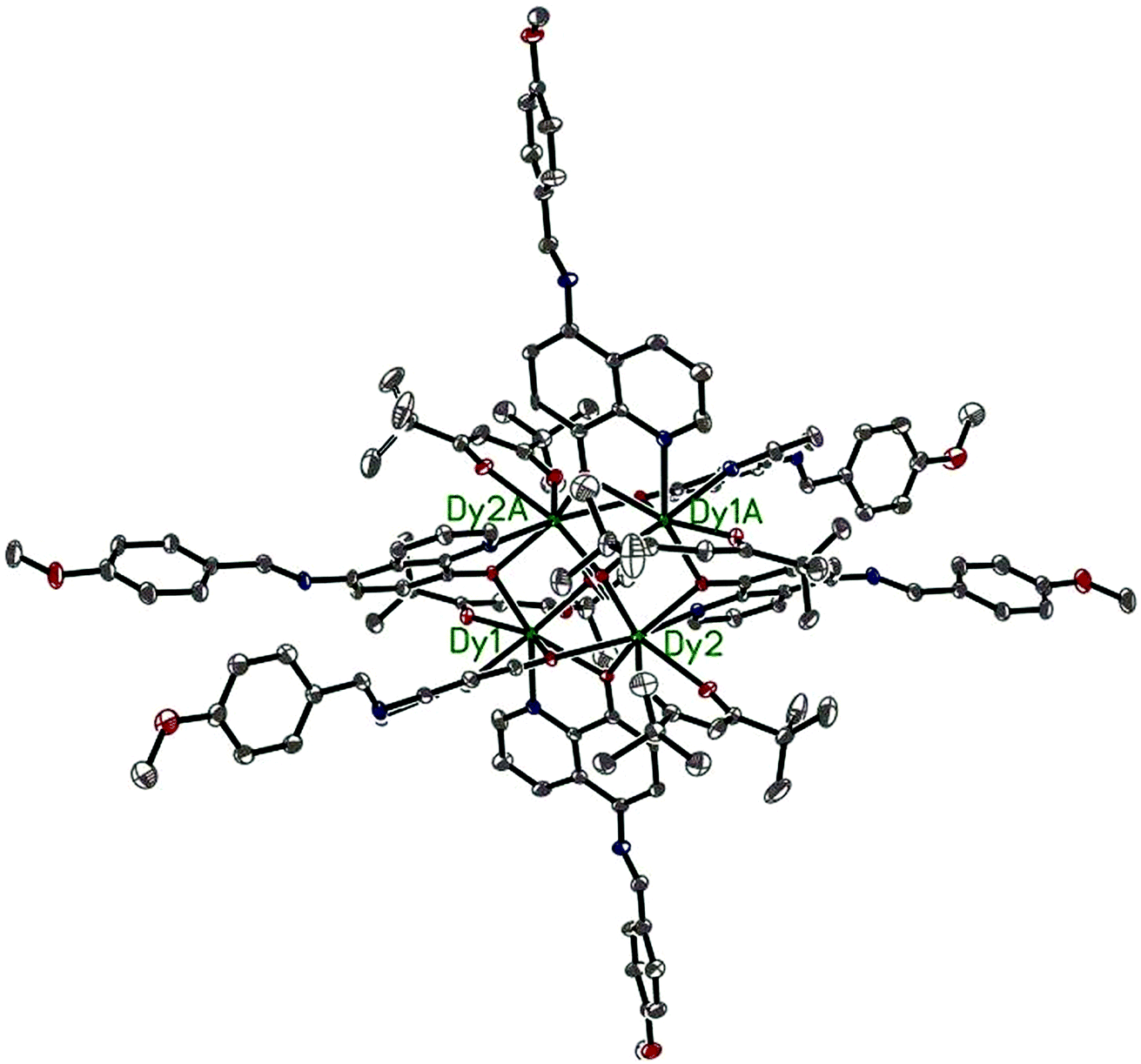 | ||
| Fig. 3 The molecular structure of compound 9·Dy. Hydrogen atoms are omitted for clarity (Dy green; C gray; N blue; O red). | ||
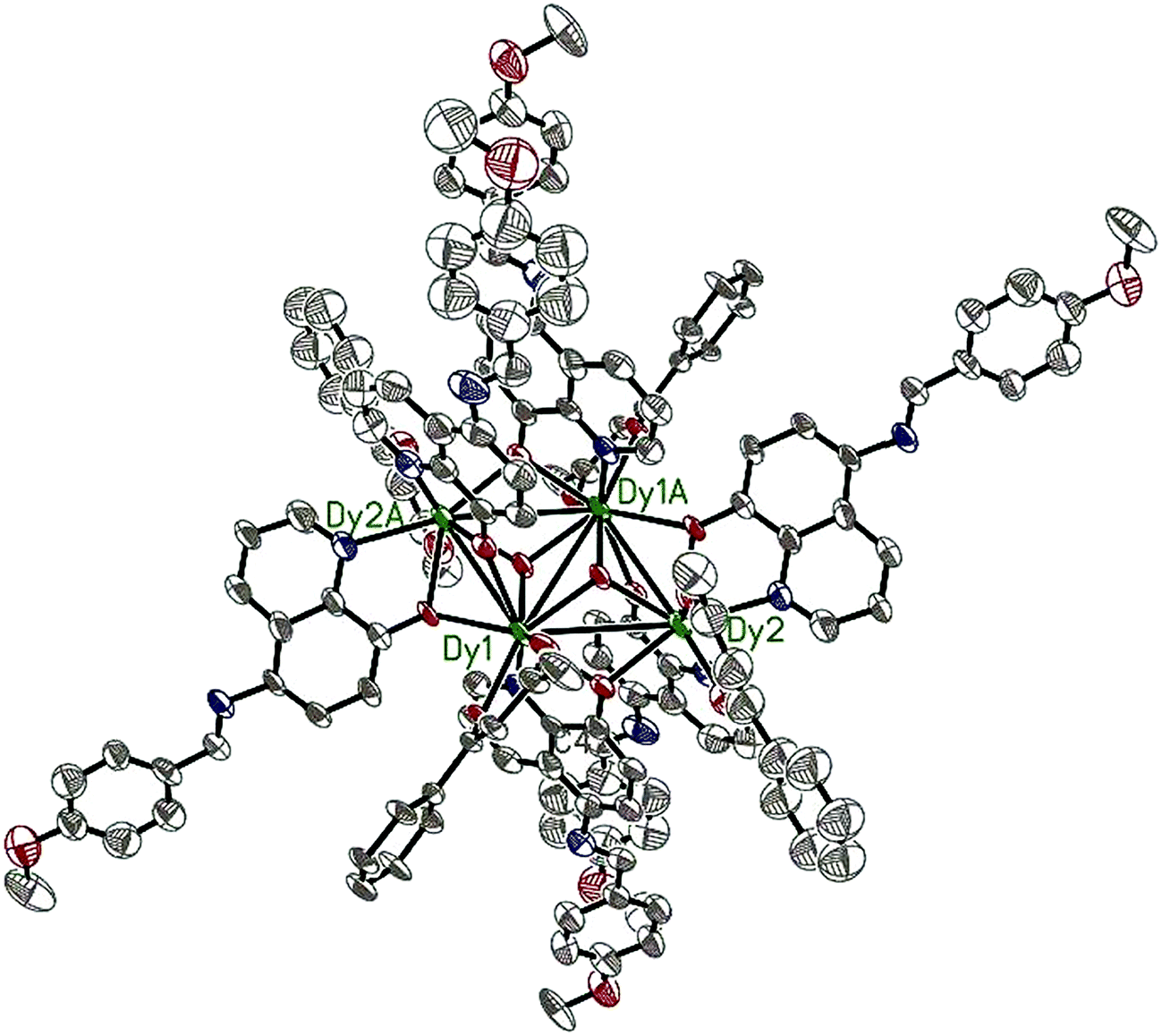 | ||
| Fig. 4 The molecular structure of compound 10·Dy. Hydrogen atoms are omitted for clarity (Dy green; C gray; N blue; O red). | ||
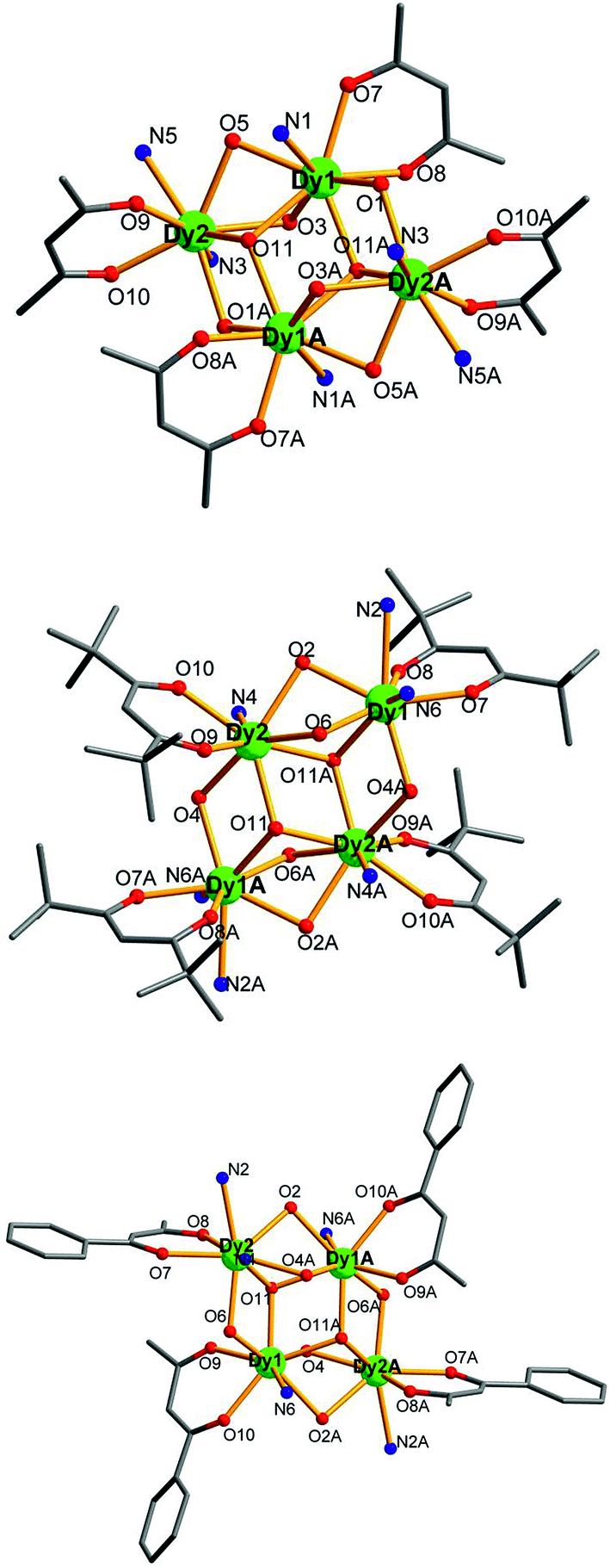 | ||
| Fig. 5 Perspective view of the crystal structures of compounds 3·Dy (top), 9·Dy (middle) and 10·Dy (bottom), with the H atoms and partial HL omitted for clarity. | ||
The structural differences among 3·Dy, 9·Dy and 10·Dy can be attributed to the strengths and symmetries of the three β-diketonate coligands. Meanwhile, the introduction of different substituent groups, to some extent, may changes the ligand field strength of Ln ion centers, thus further affecting their magnetic behaviors.
Magnetic properties
The solid-state temperature-dependent magnetic susceptibilities of compounds 1–10 were investigated in the range of 2–300 K under an applied magnetic field of 1000 Oe using the crystalline aggregates (Fig. 6).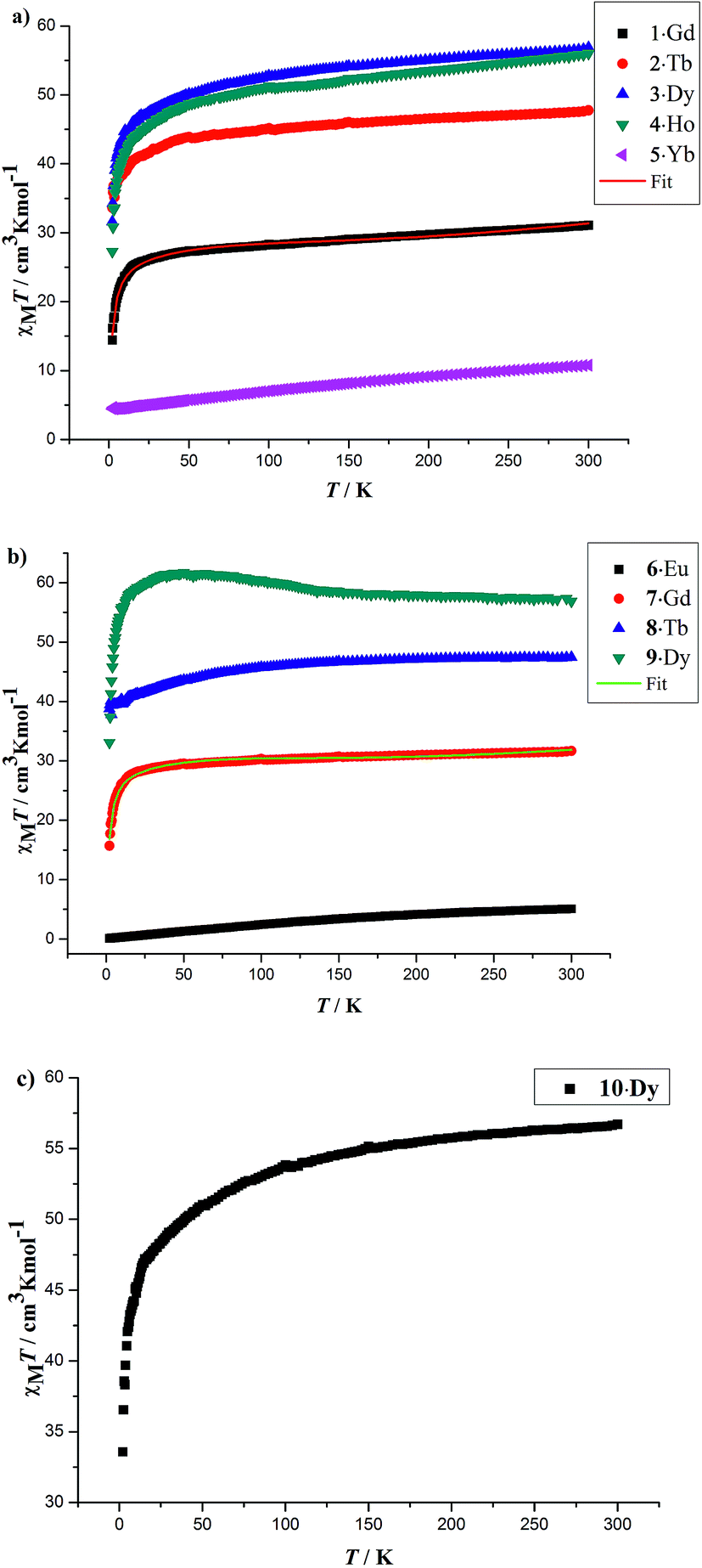 | ||
| Fig. 6 Temperature dependence of the χMT products at 1000 Oe for 1–5 (a), 6–9 (b) and 10 (c) in the range of 2–300 K. The solid red and green lines are the best fit obtained. | ||
At room temperature, the χMT values for compounds 1–5 are 31.08 (1·Gd), 47.74 (2·Tb), 56.87 (3·Dy), 56.02 (4·Ho) and 10.78 (5·Yb) cm3 K mol−1, which are very close to the theoretical values of 31.52, 47.28, 56.68, 56.28 and 10.28 cm3 K mol−1, calculated based on four isolated Gd3+ (8S7/2, g = 2), Tb3+ (7F6, g = 3/2), Dy3+ (6H15/2, g = 4/3), Ho3+ (5I8, g = 5/4), Yb3+ (2F7/2, g = 8/7), respectively. Upon cooling, the χMT values of 1–4 decrease slowly from 300–50 K, and then decline rapidly to minimum values of 14.40, 33.62, 31.60, 27.27 cm3 K mol−1 at 2 K. In the case of 5·Yb, the χMT value gradually decreases to 4.44 cm3 K mol−1. These magnetic behaviors are generally ascribed to the thermal depopulation of the Ln3+ excited Stark sublevels resulting from the spin–orbit coupling and/or weak antiferromagnetic interactions between Ln3+ ions.15 For compounds 6·Eu, the χMT values is 5.07 cm3 K mol−1 at room temperature, the value of which is much higher than the expected value 0 cm3 K mol−1 for four uncoupled Eu3+ (7F0, g = 0), which mainly results from the population of the first and even higher excited states.16 The χMT value of 6·Eu gradually decrease to 0.10 cm3 K mol−1 at 2 K, basically originating from thermal depopulation of the excited levels. For 7–9, the χMT value at room temperature are 31.67, 47.45 and 56.86 cm3 K mol−1, which are all consistent with the expected values of 31.52, 47.28 and 56.60 cm3 K mol−1 for four isolated Ln3+ ions (8S7/2, g = 2 for Gd3+ ions; 7F6, g = 3/2 for Tb3+ ions; 6H15/2, g = 4/3 for Dy3+ ions). For 7·Gd, different from 1·Gd, the χMT value almost keeps constant up to 20 K, then a sharply decline can be clearly observed, reaching a minimum value of 15.70 cm3 K mol−1 at 2 K. This trend indicates weak antiferromagnetic interaction exist between adjacent Gd3+ ions. For 8·Tb, the χMT value gradually decreases and drop to the minimum value of 38.81 cm3 K mol−1, which can also be ascribed to weak magnetic exchange between Tb3+ ions and/or the thermal depopulation of the Tb3+ excited Stark sublevels.17 For 9·Dy, a continuous increase of χMT value upon cooling can be observed up to 50 K, subsequently, a sharp decline to a minimum value of 33.07 cm3 K mol−1 is finally seen at 2 K. This behavior indicates that weak ferromagnetic interactions must dominates between Dy3+ ions. For 10·Dy, the plots of χMT vs. T is similar with 1·Gd, the χMT at room temperature 56.72 cm3 K mol−1 are close to the expected value of 56.68 cm3 K mol−1 for four free Dy3+ ions. Upon lowering the temperature, the χMT value gradually decreases and then further drops to 33.58 cm3 K mol−1 at 2 K. This trend may be caused by depopulation of the MJ sublevel and antiferromagnetic interactions between Dy3+ ions. The χMT–T plots of 1·Gd and 7·Gd were nicely fitted by Hamiltonian (1) with the model in Fig. S5,† giving g = 1.96, J = −0.08 cm−1 and g = 2.06, J = −0.16 cm−1 for 1·Gd and 7·Gd, respectively, the small J values suggest that weak antiferromagnetic interactions exist between adjacent Gd ions. The magnetic susceptibilities of 3·Dy, 9·Dy and 10·Dy fitted by Curie–Weiss law give the Weiss constants θ = −5.41 for 3·Dy, θ = 1.93 for 9·Dy and θ = −1.27 for 10·Dy (Fig. S6 in ESI†), the positive/negative parameter θ values further confirm the ferromagnetic/antiferromagnetic coupling between Dy3+. Surprisingly, 3·Dy, 9·Dy and 10·Dy, featuring similar Dy4 clusters, display different magnetic interactions.
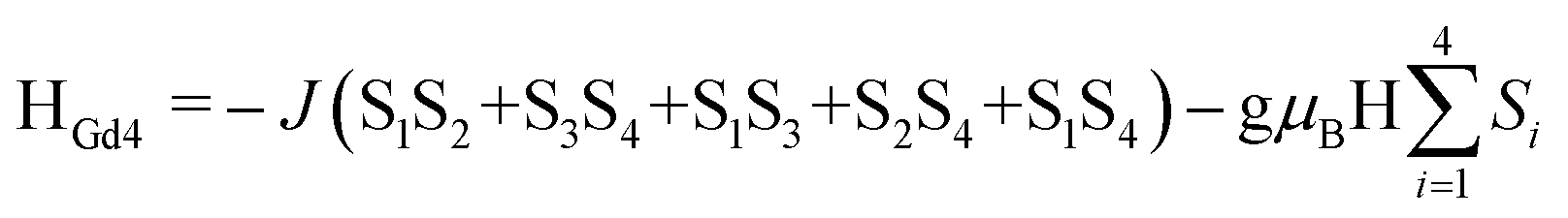 | (1) |
To further investigate the magnetic dynamics of 3·Dy, 9·Dy and 10·Dy, the temperature and frequency dependencies of the alternating current (ac) susceptibilities were performed under 0 direct current and a 3 Oe ac magnetic field (Fig. 7 and 8). Both of the in-phase (χ′) and out-of-phase (χ′′) signals show strong temperature and frequency dependence as well as remarkable peaks, revealing that all the three tetra-nuclear dysprosium compounds exhibit typical features associated with SMM behaviors. Differently, the χ′ and χ′′ maximum values of 3·Dy are in higher temperature region than that of 9·Dy and 10·Dy. In addition, the parameter φ = (ΔTp/Tp)/Δ(log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) v) derived from the temperature dependence of in-phase ac susceptibility are 0.21, 0.28 and 0.22 for 3·Dy, 9·Dy and 10·Dy, which are consistent with that of a superparamagnetic, excluding the possibility of spin glass behaviors.18
v) derived from the temperature dependence of in-phase ac susceptibility are 0.21, 0.28 and 0.22 for 3·Dy, 9·Dy and 10·Dy, which are consistent with that of a superparamagnetic, excluding the possibility of spin glass behaviors.18
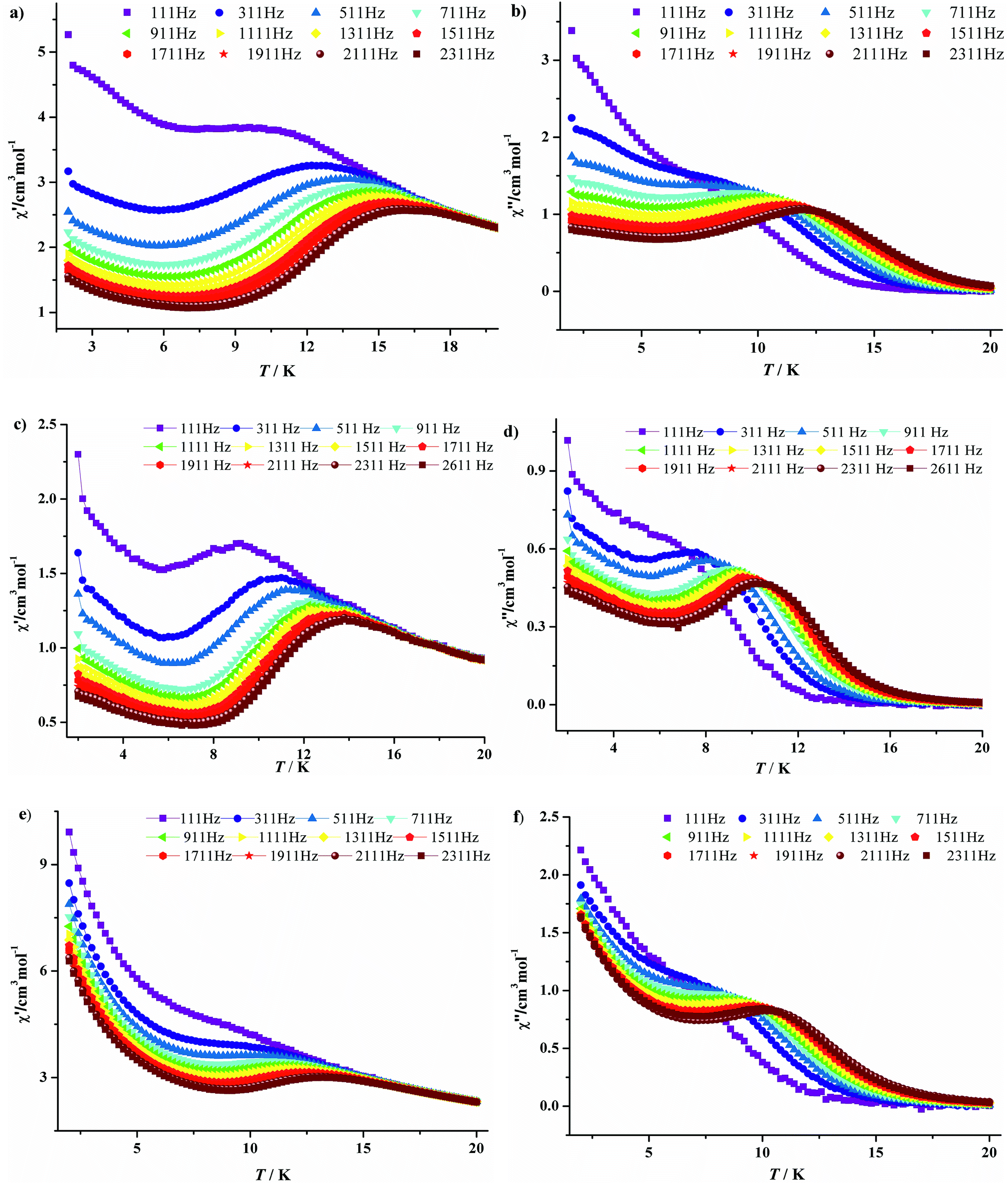 | ||
| Fig. 7 Temperature-dependent in-phase (χ′) and out-phase (χ′′) ac susceptibilities plots at different frequencies for 3·Dy (a, b), 9·Dy (c, d) and 10·Dy (e, f). | ||
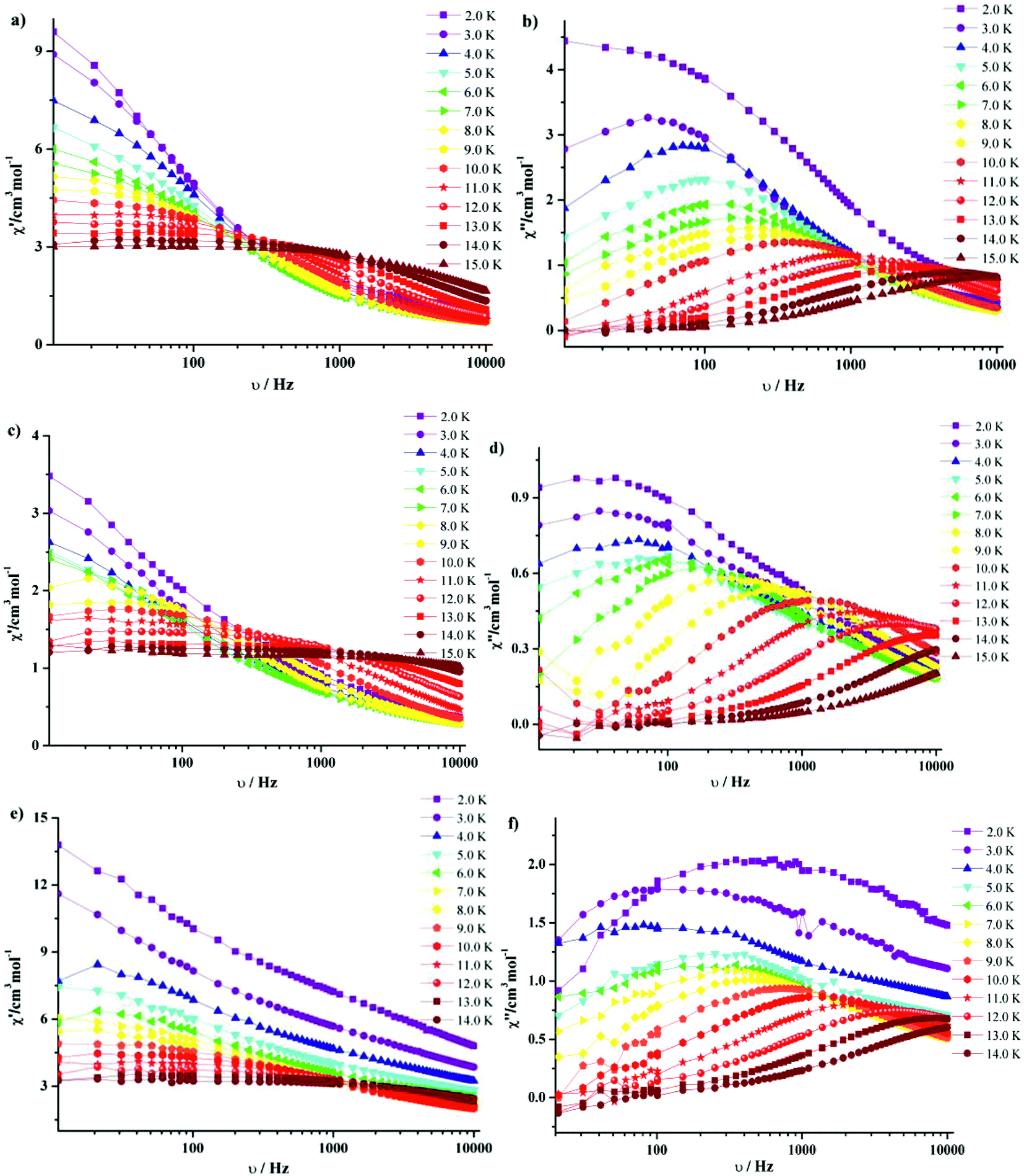 | ||
| Fig. 8 Frequency dependence of in-phase (χ′) and out-phase (χ′′) ac susceptibilities at different temperatures for 3·Dy (a, b), 9·Dy (c, d) and 10·Dy (e, f). | ||
It is noteworthy that all the temperature dependencies of ac susceptibilities for 3·Dy, 9·Dy and 10·Dy increase in the lower temperature region, which might be indicative of quantum tunneling effect.19 To check whether the relaxation processes of them are thermally activated, the relaxation time τ of compounds 3·Dy, 9·Dy and 10·Dy can be derived from the out-of-phase (χ′′) peaks in the form of lnτ plotted as a function of 1/T (Fig. 9). It is interesting that obvious curvatures in the lnτ vs. 1/T plots of 3·Dy, 9·Dy and 10·Dy are observed, indicating a combination of relaxation pathways are operative. As seen from Fig. 8, the relaxation times of them are all temperature dependent in the whole temperature range, suggesting the relaxation processes are controlled by Orbach and Raman mechanisms rather than the quantum tunneling effects, similar phenomenon is reported by a few SMMs.20 Given this, the magnetic data of 3·Dy, 9·Dy and 10·Dy can be fitted following the eqn (2) in 2–15 K temperature range.
|
lnτ = −ln[AT + B + CTn + τ0−1exp(−ΔE/kBT)]
| (2) |
exp(−ΔE/kBT) correspond to the direct, Raman and Orbach processes, respectively. The relaxation energy barriers and pre-exponential factors τ0 of 3·Dy, 9·Dy and 10·Dy can be obtained by fitting τ values based on the Arrhenius law [τ = τ0exp (ΔE/kBT)] in the high temperature ranges (Fig. 9 black lines), the fitting results give ΔE/kB = 99.29(3) K and τ0 = 1.65 × 10−9 s for 3·Dy above 11 K, ΔE/kB = 64.56(4) K and τ0 = 2.09 × 10−7 s for 9·Dy above 9 K and ΔE/kB = 61.05(0) K and τ0 = 7.99 × 10−7 s for 10·Dy above 8 K. Assuming the value of B is zero, based on the above parameters ΔE/kB and τ0, we successfully fitted the plots lnτ vs. 1/T in the whole temperature range using eqn (2) with the fitting parameters n = 3.48, A = 0.4721 s−1 K−3.48, C = 0.8559 for 3, n = 5.64, A = 0.3625 s−1 K−5.64, C = 0.1582 for 9·Dy, n = 5.77, A = 0.4351 s−1 K−5.77, C = 0.139 for 10·Dy. The small values of A and C indicate that the relaxation process is dominated by the Orbach and Raman mechanisms at high and low temperature, respectively. Interestingly, compounds 3·Dy, 9·Dy and 10·Dy display different relaxation energy barriers, and the value of which are relatively larger in the Dy4 compounds with butterfly shaped arrangements, especially for 3·Dy. For example, the energy barriers (ΔE/kB) of them are larger than that of [Dy4(μ3-OH)2(L′)6(tmhd)4]·CH3CN (HL′ = 5-(4-methylbenzylidene)-8-hydroxylquinoline; Htmhd = 2,2,6,6-tetramethyl-3,5-heptanedione) (46.4 K),22 [Dy4(μ3-OH)2(L3)4(HL3)2] (H2L3 = (E)-2-(2-hydroxy-3-methoxybenzylideneamino)phenol) (3.7 K),23 but far lower than the value of [Dy4(μ3-OH)2(bmh)2(msh)4Cl2] (H2bmh = bis(2-hydroxy-3-methoxybenzylidene); Hmsh = 3-methoxysalicylaldehyde hydrazone) (170 K).5a The different slow relaxation properties of 3·Dy, 9·Dy and 10·Dy mainly ascribed to the introduction of different β-diketonates, resulting in the slight change of structures in them.
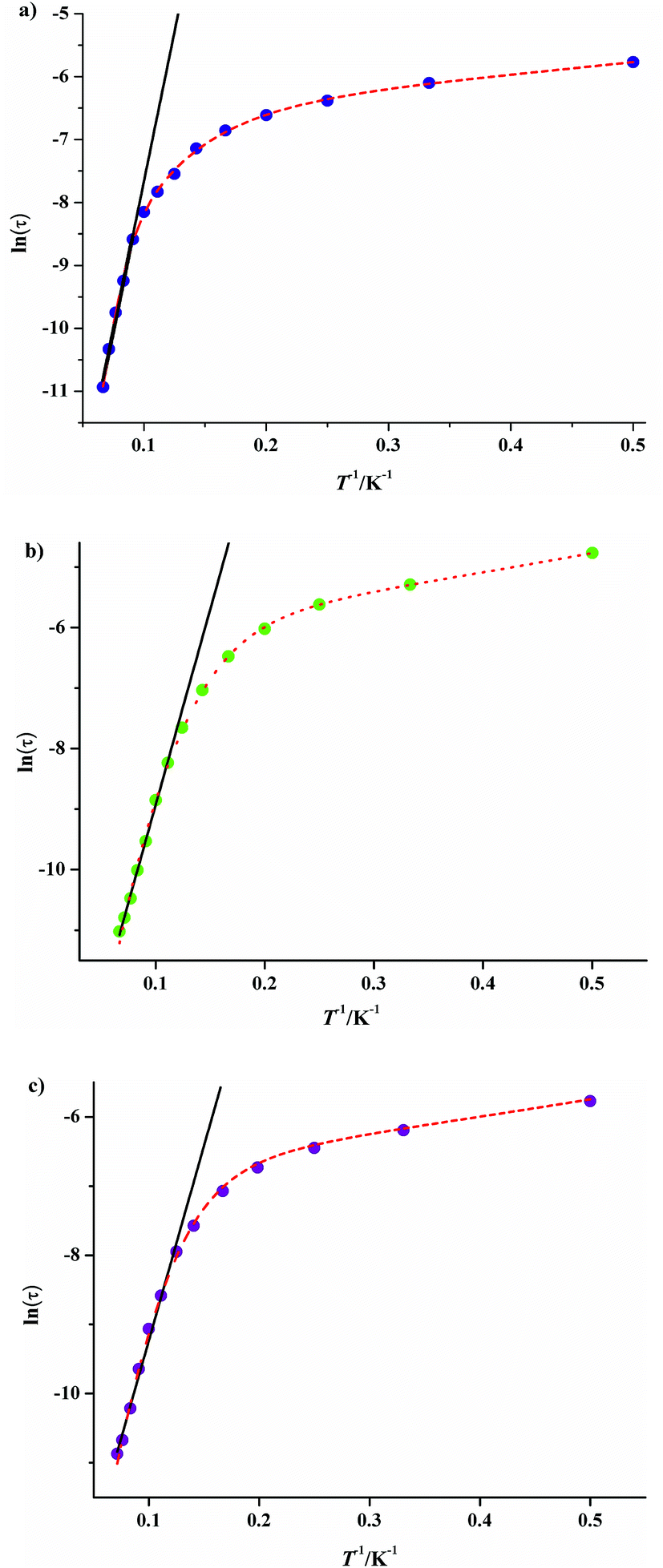 | ||
| Fig. 9 Temperature dependence of the magnetic relaxation time for 3·Dy (a), 9·Dy (b) and 10·Dy (c), ln(τ) vs. T−1 under Hdc = 0; the solid black lines represent the pure Arrhenius fitting at higher temperature region and the red dashed lines represent the fitting based on eqn (2). | ||
The Cole–Cole diagrams of χ′′ vs. χ′ with nearly semi-circle shapes for compounds 3·Dy, 9·Dy and 10·Dy are fitted to the generalized Debye model, giving different distribution coefficient α values (Fig. 10). For 3·Dy, α = 0.25–0.38 was found in the temperature of 2–6 K, while α = 0.27–0.52 (between 2–4 K) and α = 0.30–0.66 (between 2 and 4 K) are obtained for 9·Dy and 10·Dy, respectively. The relative larger α value for 10·Dy indicates that compound 10·Dy possesses a wider distribution of the relaxation time than 3·Dy and 9·Dy. It is noteworthy that three butterfly-shaped tetra-nuclear Dy compounds with similar arrangements exhibit different magnetic behaviors, which is mainly ascribed to the introduction of different β-diketonates, resulting in the change of crystal fields around center Dy3+ ions. In order to investigate the structure–property relationship of 3·Dy, 9·Dy and 10·Dy, some main structural parameters are listed in Table 1, such as the average distance of d(Dy---phenoxide-O) (lengths between Dy3+ ions and phenoxide oxygen atoms), d(Dy---μ3-OH) (lengths between Dy3+ ions and μ3-OH atoms), d(Dy---β-diketonate-O) (lengths between Dy3+ ions and oxygen atoms of β-diketonates), d(Dy–N) (length of Dy–N) and d(Dy---Dy) (length of Dy---Dy). Interestingly, the values of d(Dy---phenoxide-O) and d(Dy---μ3-OH) in the tetranuclear clusters of 3·Dy, 9·Dy and 10·Dy display a continuous increase trend, thus resulting into the shortest distance of Dy---Dy in 3·Dy and the longest distance of Dy---Dy in 10·Dy. This tendency is consistent with the fact that stronger interactions occur between adjacent metal ions in 3·Dy than 9·Dy and 10·Dy, as shown in Fig. 6 and S6.† By comparison, the different lengths of d(Dy---β-diketonate-O) observed in 3·Dy, 9·Dy and 10·Dy may be ascribed to the strong steric-hindrance effect of β-diketonates with different substituent groups. The shortest bond lengths of Dy–N/O in 3·Dy suggests the existence of stronger ligand field around the local Dy3+ ions sites. Moreover, as reflected in Fig. 7, the higher block temperature of ac susceptibilities curves observed in 3·Dy than that in 9·Dy and 10·Dy can be also attributed to the stronger ligand field in 3·Dy. In addition, in order to further investigate the effect of axial ligands on the magnetic properties, the anisotropy axes of the Dy(III) centers in compounds 3·Dy, 9·Dy and 10·Dy have been calculated using the MAGELLAN program (Fig. 11).24 The results reveal that the orientations of the anisotropy axes for compounds 3·Dy, 9·Dy and 10·Dy are different from each other, their angles between the tetranuclear Dy(III) planes are 39.480° and 88.533° for 3·Dy, 26.251° and 50.692° for 9·Dy, 65.515° and 63.916° for 10·Dy, respectively. It is obvious that the β-diketonates dominate orientation of the anisotropy axes for compounds 3·Dy, 9·Dy and 10·Dy. In view of the above analysis, we conclude that the change of substituent groups of axial ligands play an important role in modulating magnetic behaviors.
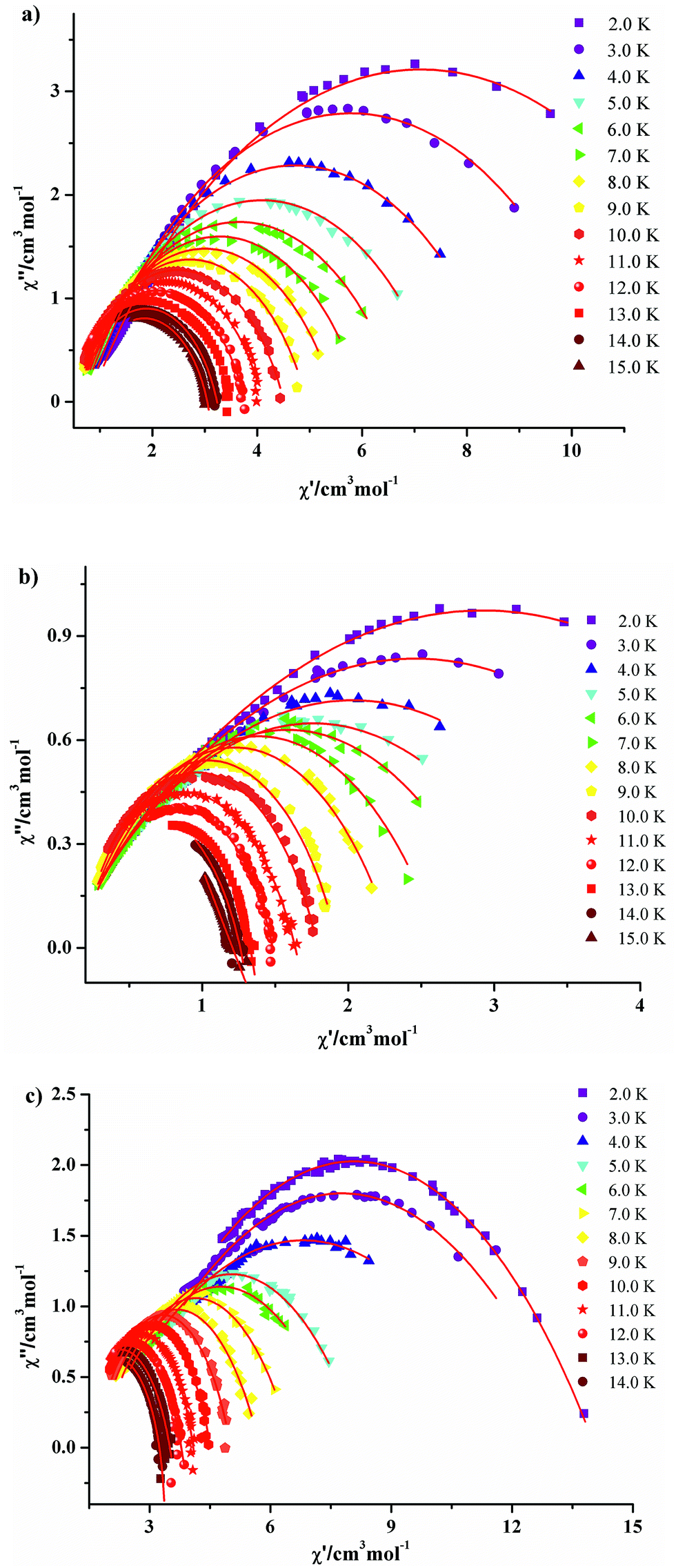 | ||
| Fig. 10 Cole–Cole plots measured under zero-dc field for 3·Dy (a), 9·Dy (b) and 10·Dy (c). The solid lines are the best fits obtained with a generalized Debye model. | ||
| Average distances | Compounds | ||
|---|---|---|---|
| 3·Dy | 9·Dy | 10·Dy | |
| d(Dy---phenoxide-O) (Å) | 2.3801 | 2.3805 | 2.4167 |
| d(Dy---μ3-OH) (Å) | 2.3520 | 2.3649 | 2.3706 |
| d(Dy---β-diketonate-O) (Å) | 2.3068 | 2.3141 | 2.3513 |
| d(Dy---N) (Å) | 2.5318 | 2.5466 | 2.5303 |
| d(Dy–Dy) (Å) | 3.7282 | 3.7473 | 3.7689 |
 | ||
| Fig. 11 The magnetic anisotropy easy axes (pink lines) of Dy3+ ions in compounds 3·Dy (a), 9·Dy (b) and 10·Dy (c), respectively. | ||
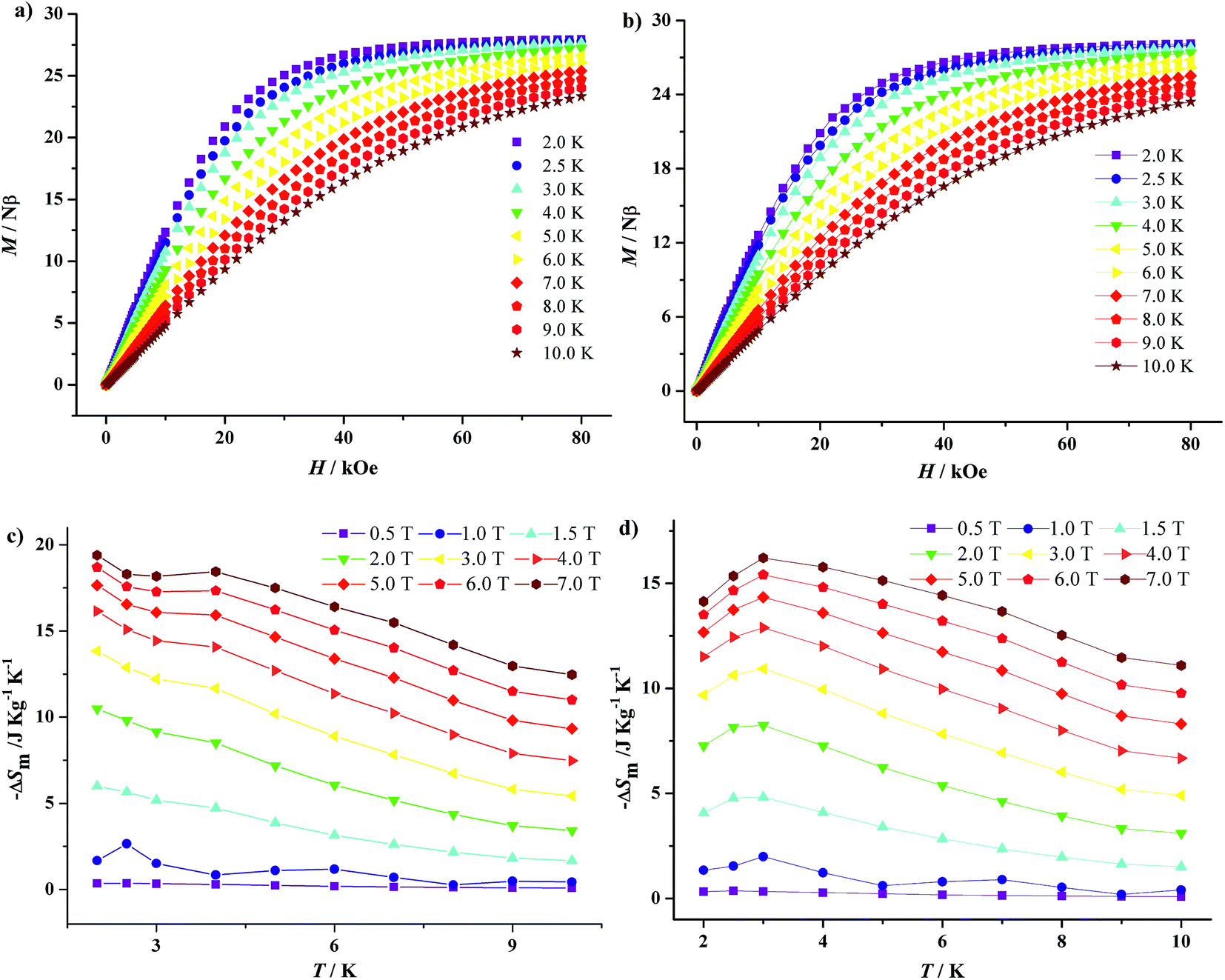
Gadolinium with large spin and negligible magnetic anisotropy is a promising candidate to construct magnetic refrigeration materials. Thus, the field-dependent isothermal magnetization curves of 1·Gd and 7·Gd are measured in the range of H = 0–8 T and T = 2–10 K (Fig. 12a and b). The magnetization of 1·Gd and 7·Gd display a gradual increase with the increasing field, and reach saturation values of 27.94 Nβ for 1·Gd and 28.12 Nβ for 7·Gd at 7 T and 2 K, being close to the theoretical value of 28 Nβ for four Gd3+ (S = 7/2, g = 2) ions.
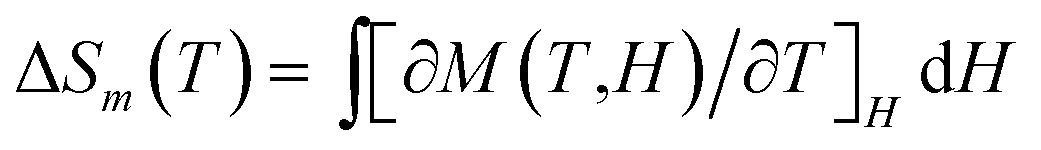 | (3) |
 | ||
| Fig. 12 Magnetization (M) vs. field (H) for 1·Gd (a) and 7·Gd (b) in the temperature range of 2–10 K; temperature-dependencies of −ΔSm for 1·Gd (c) and 7·Gd (d) obtained from the magnetization data at T = 2–10 K and H = 0–7 T. | ||
Additionally, according to the Maxwell eqn (3),25 the isothermal magnetic entropy change (ΔSm) of 1·Gd and 7·Gd can be obtained (Fig. 12c and d). For 1·Gd, the maximum value of −ΔSm is 19.39(2) J K−1 kg−1 at 2 K for 7 T without saturation, which is smaller than the expected value of 24.46 J K−1 kg−1 for four uncoupled Gd3+ ions calculated by function of −ΔSm = 4Rln(2S + 1) (R is the gas constant). For 7·Gd, the value of −ΔSm almost keep unchanged with decreasing temperature below 1 T, then it starts to increase rapidly with the external field exceeding 1 T, finally reaching a maximum value of 16.21(3) J K−1 kg−1 at 3 K for 7 T. The theoretical value is 21.45 J K−1 kg−1, the value of which is larger than the experimental one. The gap between the experimental and theoretical values for 1·Gd and 7·Gd mainly stemmed from the existence of antiferromagnetic interactions in them.26 For Gd-based compounds, the magnetocaloric effect is affected by Mw/NGd and θ. As shown in Table 2, compound 1·Gd and 7·Gd have different values of Mw/NGd and θ, the values of Mw/NGd and |θ| for 1·Gd are smaller than 7·Gd, indicating that 1·Gd have a larger magnetic density and weaker magnetic coupling. Thus, the −ΔSm value of 1·Gd is larger than that of 7·Gd. However, the −ΔSm values of them are both smaller than tetra-nuclear Gd3+ compound [Gd4(OAc)4(acac)8(H2O)4] (37.7 J K−1 kg−1 at 2 K for 7 T) with smaller value of Mw/NGd radio (432.53) and weak ferromagnetic interactions θ (0.23).27 Hence, the selection of low molecular weight ligands that lead to weak interaction is a promising way for constructing Gd3+ based magnetic refrigeration materials.
| Compounds | Mw/NGd | Magnetic interaction [θ, K] | −ΔSmaxm [J kg−1K−1] |
|---|---|---|---|
| 1·Gd | 680.8 | −2.92 | 19.39(2) |
| 7·Gd | 808.5 | −3.35 | 16.21(3) |
Conclusion
In summary, ten new tetranuclear lanthanide compounds have been successfully designed by using Schiff base ligand HL and three different β-diketonate auxiliary ligands. Compounds 1–10 all consist of butterfly-shaped planar tetranuclear clusters, and the Ln4O2 cores are encapsulated inside the same coordination pockets [O14N6]. Magnetic investigations reveal that compounds 1·Gd and 7·Gd display different magnetocaloric effects (−ΔSmaxm = 19.39(2) J K−1 kg−1 for 1·Gd and 16.21(3) J K−1 kg−1 for 7·Gd) due to their divergence in Mw/NGd ratios and magnetic interaction. In addition, compounds 3·Dy, 9·Dy and 10·Dy display different magnetic dynamic behaviors in spite of their similar structures. A simple comparative investigation uncovers that the introduction of different substituent groups in β-diketonate auxiliary ligands result in the change of magnetic anisotropy of Dy3+ centers in them, and thus give rise to the difference of magnetic relaxation behaviors in this system. Given the versatility of the β-diketonates, we plan to regulate their structures and magnetic properties in other types of polynuclear Ln-clusters to further investigate the inner structure–properties relationship more deeply. This work provides an important path in the fine-tuning of the magnetic properties of SMMs. We believe that this work not only present to us various polynuclear Ln3+ clusters but also provide an inspiration in modulating magnetic dynamic behaviors of Ln-based SMMs, further stimulating investigation on the slow relaxation mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Natural Science Foundation of China (21701039), Natural Science Foundation of Hebei Province (B2017201055) and Hebei University construction project for comprehensive strength promotion of Midwest colleges and universities.Notes and references
- (a) C. Dossantos, A. Harte, S. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512–2527 CrossRef CAS; (b) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- (a) J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed; (b) S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS PubMed.
- (a) E. Saitoh, H. Miyajima, T. Yamaoka and G. Tatara, Nature, 2004, 432, 203–206 CrossRef CAS PubMed; (b) M. Y. Wu, F. L. Jiang, X. J. Kong, D. Q. Yuan, L. S. Long, S. A. Al-Thabaiti and M. C. Hong, Chem. Sci., 2013, 4, 3104–3109 RSC.
- (a) S. Y. Lin, X. L. Li, H. S. Ke and Z. K. Xu, CrystEngComm, 2015, 17, 9167–9174 RSC; (b) H. L. Gao, L. Jiang, H. Y. Shen, W. M. Wang and J. Z. Cui, Dalton Trans., 2016, 45, 253–264 RSC; (c) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- (a) P. H. Lin, T. J. Burchell, L. Ungur, L. F. Chibotaru, W. Wernsdorfer and M. Murugesu, Angew. Chem., Int. Ed., 2009, 48, 9489–9492 CrossRef CAS PubMed; (b) Q. Y. Liu, Y. L. Li, W. L. Xiong, Y. L. Wang, F. Luo, C. M. Liu and L. L. Chen, CrystEngComm, 2014, 16, 585–590 RSC; (c) F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- (a) Y. X. Wang, W. Shi, H. Li, Y. Song, L. Fang, Y. H. Lan, A. K. Powell, W. Wernsdorfer, L. Ungur, L. F. Chibotaru, M. R. Shen and P. Cheng, Chem. Sci., 2012, 3, 3366–3370 RSC; (b) J. Liu, Y. C. Chen, Z. X. Jiang, J. L. Liu, J. H. Jia, L. F. Wang, Q. W. Li and M. L. Tong, Dalton Trans., 2015, 44, 8150–8155 RSC; (c) S. K. Langley, N. F. Chilton, I. A. Gass, B. Moubaraki and K. S. Murray, Dalton Trans., 2011, 40, 12656–12659 RSC; (d) E. M. Pineda, Y. H. Lan, O. Fuhr, W. Wernsdorfer and M. Ruben, Chem. Sci., 2017, 8, 1178–1185 RSC.
- (a) S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed; (b) L. Zhao, J. F. Wu, H. S. Ke and J. K. Tang, CrystEngComm, 2013, 15, 5301–5306 RSC; (c) Y. L. Wang, N. Zhou, Y. Ma, Z. X. Qin, Q. L. Wang, L. C. Li, P. Cheng and D. Z. Liao, CrystEngComm, 2012, 14, 235–239 RSC.
- (a) J. Y. Ge, H. Y. Wang, J. Li, J. Z. Xie, Y. Song and J. L. Zuo, Dalton Trans., 2017, 46, 3353–3362 RSC; (b) W. Huang, F. X. Shen, S. Q. Wu, L. Liu, D. Wu, Z. Zheng, J. Xu, M. Zhang, X. C. Huang, J. Jiang, F. Pan, Y. Li, K. Zhu and O. Sato, Inorg. Chem., 2016, 55, 5476–5484 CrossRef CAS PubMed.
- (a) Y. W. Li, S. J. Liu, T. L. Hu and X. H. Bu, Dalton Trans., 2014, 43, 11470–11473 RSC; (b) A. Nandy and S. K. Pradhan, Dalton Trans., 2015, 44, 17229–17240 RSC.
- L. F. Zou, L. Zhao, P. Chen, Y. N. Guo, Y. Guo, Y. H. Li and J. K. Tang, Dalton Trans., 2012, 41, 2966–2971 RSC.
- J. Liu, Y. C. Chen, J. L. Liu, V. Vieru, L. Ungur, J. H. Jia, L. F. Chibotaru, Y. H. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- P. Zhang, L. Zhang, S. Y. Lin, S. F. Xue and J. K. Tang, Inorg. Chem., 2013, 52, 4587–4592 CrossRef CAS PubMed.
- W. M. Wang, W. Z. Qiao, H. X. Zhang, S. Y. Wang, Y. Y. Nie, H. M. Chen, Z. Liu, H. L. Gao, J. Z. Cui and B. Zhao, Dalton Trans., 2016, 450, 8182–8191 RSC.
- (a) H. L. Gao, L. Jiang, W. M. Wang, S. Y. Wang, H. X. Zhang and J. Z. Cui, Inorg. Chem., 2016, 55, 8898–8904 CrossRef CAS PubMed; (b) S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 1492–1503 CrossRef CAS.
- I. Nemec, M. Machata, R. Herchel, R. Boča and Z. Trávníček, Dalton Trans., 2012, 41, 14603–14610 RSC.
- Y. L. Hou, G. Xiong, B. Shen, B. Zhao, Z. Chen and J. Z. Cui, Dalton Trans., 2013, 42, 3587–3596 RSC.
- (a) L. Zhang, P. Zhang, L. Zhao, J. F. Wu, M. Guo and J. K. Tang, Dalton Trans., 2016, 45, 10556–10562 RSC; (b) X. L. Li, H. Li, D. M. Chen, C. Wang, J. F. Wu, J. K. Tang, W. Shi and P. Cheng, Dalton Trans., 2015, 44, 20316–20320 RSC.
- D. P. Dong, Y. J. Zhang, H. Zheng, P. F. Zhuang, L. Zhao, Y. Xu, J. X. Hu, T. Liu and C. Y. Duan, Dalton Trans., 2013, 42, 7693–7698 RSC.
- (a) P. H. Lin, W. B. Sun, M. F. Yu, G. M. Li, P. F. Yan and M. Murugesu, Chem. Commun., 2011, 47, 10993–10995 RSC; (b) N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498–5500 CrossRef CAS PubMed.
- (a) D. Zhang, Y. M. Tian, W. B. Sun, H. F. Li, P. Chen, Y. Q. Zhang and P. F. Yan, Dalton Trans., 2016, 45, 2674–2680 RSC; (b) S. Titos-Padilla, J. Ruiz, J. M. Herrera, E. K. Brechin, W. Wernsdorfer, F. Lloret and E. Colacio, Inorg. Chem., 2013, 52, 9620–9626 CrossRef CAS PubMed; (c) W. B. Sun, B. Yan, Y. Q. Zhang, B. W. Wang, Z. M. Wang, J. H. Jia and S. Gao, Inorg. Chem. Front., 2014, 1, 503–509 RSC.
- K. R. Meihaus, S. G. Minasian, W. W. Lukens, S. A. Kozimor, D. K. Shuh, T. Tyliszczak and J. R. Long, J. Am. Chem. Soc., 2014, 136, 6056–6068 CrossRef CAS PubMed.
- Z. L. Wu, Y. G. Ran, X. Y. Wu, Y. P. Xia, M. Fang and W. M. Wang, Polyhedron, 2017, 126, 282–286 CrossRef CAS.
- K. C. Mondal, G. E. Kostakis, Y. H. Lan and A. K. Powell, Polyhedron, 2013, 66, 268–273 CrossRef CAS.
- N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 Search PubMed.
- (a) G. Karotsis, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 9928–9931 CrossRef CAS PubMed; (b) M. Evangelisti, F. Luis, L. De Jongh and M. Affronte, J. Mater. Chem., 2006, 16, 2534–2549 RSC.
- (a) J. B. Peng, Q. C. Zhang, X. J. Kong, Y. P. Ren, L. S. Long, R. B. Huang, L. S. Zheng and Z. Zheng, Angew. Chem., Int. Ed., 2011, 50, 10649–10652 CrossRef CAS PubMed; (b) J. M. Jia, S. J. Liu, Y. Cui, S. D. Han, T. L. Hu and X. H. Bu, Cryst. Growth Des., 2013, 13, 4631–4634 CrossRef CAS.
- F. S. Guo, J. D. Leng, J. L. Liu, Z. S. Meng and M. L. Tong, Inorg. Chem., 2012, 51, 405–413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1544673–1544682. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11750g |
| This journal is © The Royal Society of Chemistry 2017 |