
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVUV photoionization aerosol mass spectrometric study on the iodine oxide particles formed from O3-initiated photooxidation of diiodomethane (CH2I2)
Nana Weiab,
Changjin Hu *a,
Shanshan Zhouab,
Qiao Maab,
Pavel Mikuškac,
Zbyněk Večeřac,
Yanbo Gaia,
Xiaoxiao Lina,
Xuejun Gua,
Weixiong Zhaoa,
Bo Fanga,
Weijun Zhang*ad,
Jun Chene,
Fuyi Liue,
Xiaobin Shane and
Liusi Shenge
*a,
Shanshan Zhouab,
Qiao Maab,
Pavel Mikuškac,
Zbyněk Večeřac,
Yanbo Gaia,
Xiaoxiao Lina,
Xuejun Gua,
Weixiong Zhaoa,
Bo Fanga,
Weijun Zhang*ad,
Jun Chene,
Fuyi Liue,
Xiaobin Shane and
Liusi Shenge
aLaboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Hefei, 230031 Anhui, China. E-mail: hucj@aiofm.ac.cn; wjzhang@aiofm.ac.cn
bGraduate School, University of Science and Technology of China, Hefei, 230026 Anhui, China
cInstitute of Analytical Chemistry of the Czech Academy of Sciences, Veveři 97, CZ-60200 Brno, Czech Republic
dSchool of Environmental Science and Optoelectronic Technology, University of Science and Technology of China, Hefei, 230026 Anhui, China
eNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, China
First published on 15th December 2017
Abstract
Iodine oxide particles (IOPs) formed from O3-initiated photooxidation of diiodomethane have been investigated based on the combination of a thermal desorption/tunable vacuum ultraviolet time-of-flight photoionization aerosol mass spectrometer (TD-VUV-TOF-PIAMS) with a flow reactor for the first time. Characterization of the home-made flow reactor was performed, which indicates the applicability of its combination with TD-VUV-TOF-PIAMS. Based on that, aerosol mass spectra of IOP formation from photooxidation of CH2I2/O3 were studied on-line taking full advantage of both the virtues of the flow reactor and TD-VUV-TOF-PIAMS. The main chemical components of IOPs, including atomic and molecular iodine (I, I2), iodine oxides (IO, OIO, I2O and I2O3) and hydrogen-containing iodine species (HI, HIO and HIO3), were observed and identified based on the corresponding photoionization energy (PIE) curves, and the probable chemical composition and formation mechanism of IOPs were proposed. The work has not only improved the understanding of the formation mechanism of IOPs, but also demonstrated the capability of TD-VUV-TOF-PIAMS for direct molecular characterization of aerosols in flow reactor experiments, whose potential application in mass spectrometric studies of atmospheric aerosols is anticipated.
1. Introduction
Atmospheric iodine chemistry has received increasing attention during the last few decades1 owing to its important role in the catalytic destruction of ozone2,3 and new particle formation (NPF) in the marine boundary layer (MBL).4 Although having been known for more than one century,5 NPF in the costal atmosphere was postulated to be related with classical binary (H2SO4/H2O) or ternary (H2SO4/NH3/H2O) nucleation for a long time. Only recently, has it been identified that iodine compounds (I2 or alkyl iodides) play a key role in ultrafine particle bursts at the coastal zones, and NPF is formed mainly during formation of iodine oxide particles (IOPs).6–9Nucleation and growth of IOPs, originating from iodine photochemistry, has been investigated in numerous laboratory studies.10–16 Generally, it is believed that I atoms produced from the photolysis of iodocarbons or I2 will be oxidized by O3 leading to the production of IO:1
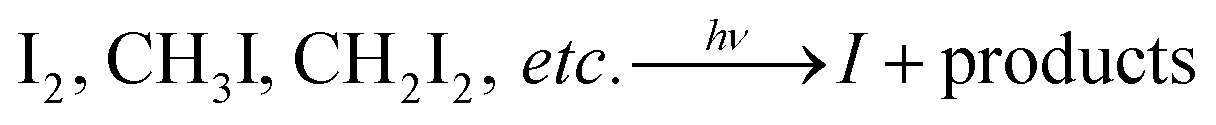 | (R1) |
| I + O3 → IO + O2 | (R2) |
Then OIO will be formed from the IO self-reaction:17,18
| IO + IO → OIO + I | (R3) |
The following recombination of OIO with IO or another OIO, followed by oxidation by O3, will result in the formation of condensable iodine oxides (IxOy, x = 2, 3, 4; y = 3, 4, 5):11,18,19
| OIO + IO → I2O3 | (R4) |
| OIO + OIO → I2O4 | (R5) |
| I2O3 + O3 → I2O4 + O2 | (R6) |
| I2O4 + O3 → I2O5 + O2 | (R7) |
At last, condensation or further polymerization of IxOy will lead to formation of IOPs. So IOPs formation was traditionally believed to occur only when the precursor of iodine, O3, and UV radiation are present simultaneously. Although a number of studies have tried to rationalize the mechanism of IOPs formation, it is still controversial about which is the most probable IOPs chemical composition, I2O4 or I2O5. While both of transmission electron microscope analysis and modelling calculations of IOPs generated from photochemical reaction of I2 and O3 in dry conditions indicated that IOPs are essentially I2O5,11 mass spectrometric study complemented with ab initio quantum calculations showed that I2O4 may be the most plausible candidate to initiate nucleation of IOPs,14 and experimental studies on IOPs formation from the two different IO generating ways (I + O3 and O + I2) based on flow tube also showed that polymerization of I2O4 play a pivotal role in IOPs formation.13 Therefore unambiguous evidence of chemical compositions of IOPs is still a major research challenge in understanding the mechanism of IOPs formation.
The application of mass spectrometric techniques to the measurement and characterization of aerosols represents a significant advance in the field of atmospheric science. Over the past decades, aerosol mass spectrometry techniques have been developed considerably, offering off-line or on-line chemical analysis for individual particles or on ensembles.20,21 Although their application in laboratory and field has resulted in many important improvements in understanding of atmospheric aerosols and their processing,22–24 these traditional aerosol mass spectrometry techniques suffered for extensive fragmentation of organic constituents due to “hard ionization” (for example, electron ionization or multiphoton ionization), which prevents specification of chemical components of atmospheric aerosols. Inspiringly, aerosol mass spectrometry coupled with vacuum ultraviolet (VUV) photoionization based on synchrotron radiation (SR) sources has been proved to be an effective “soft” ionization method for real-time, molecular component analysis of organic particles.25–27 SR light sources is featured with high photon flux and tunability. High photon flux makes it has better detection sensitivity than traditional VUV lamp for ultrafine particle analysis. Tunability makes it capable of analyzing broad range of constituents of atmospheric aerosols in two ways. One is it can offer fragment-free or less fragment mass spectra by single-photon ionization. The second is it can identify different chemical composition ambiguously by their ionization energies (IE) or appearance energies (AE) while not only by m/z values through tuning ionization energy. Making the advantage of these features, AMS coupled with a SR source has been successfully applied to laboratory studies of secondary organic aerosols (SOAs) formation and reaction kinetics.28–32 However, in the laboratory studies based on the combination of smog chamber with SR-VUV photoionization AMS, the feature of identifying compounds by IE or AE cannot be made the full advantage.30–32 The reason is that the acquisition of photoionization efficiency (PIE) curves (eventually to obtain IE or AE) needs tens of minutes to several hours depending on the scanning range and scanning step of SR light, while at the same time, the concentrations of the compounds (aerosol particles or gaseous reagents and products) in smog chamber is always changing according to the reaction progress. So it is inevitable that the observed PIE curves are the superimposition of the evolution curve of the compounds to be measured with the inherent PIE curves just depending on the energy of the exciting SR light, which inhabits the accurate identification of the compounds especially for the weak signals of mass spectra. In contrast to that in smog chamber, the concentration of the compounds to be measured in a fixed sampling site only depends on the total flow rate for a fixed flow reactor,33,34 which is particularly suitable for PIE curve measurement as aforementioned.
In this work, our motivation was to make full advantages of both the virtues of flow reactor and thermal desorption/tunable vacuum ultraviolet time-of-flight photoionization aerosol mass spectrometry (TD-VUV-TOF-PIAMS) to study the formation mechanism of IOPs. As CH2I2 is the most abundant iodine-containing compounds from biogenic emissions35 and plays an important role in the formation of iodine oxides in marine boundary layer,36 photooxidation of CH2I2/O3 was chosen as the reaction system to produce IOPs. Firstly, characterization of home-made flow reactor was performed. Then aerosol mass spectra of IOPs forming from photooxidation of CH2I2/O3 were investigated based on the combination of the flow reactor and the TD-VUV-TOF-PIAMS, and the main chemical components of IOPs were identified based on the PIE curves. Finally, the probable chemical composition and formation mechanism of IOPs, as well as the potential application of the combination of the flow reactor and the TD-VUV-TOF-PIAMS based on SR were discussed.
2. Experimental methods
Chemical analysis of IOPs formation from photooxidation of CH2I2 at the presence of O3 was performed based on the combination of the TD-VUV-TOF-PIAMS and the home-made flow reactor (Fig. 1) at the Atomic and Molecular Physics Beamline of the National Synchrotron Radiation Laboratory (NSRL) in Hefei (China).37 | ||
| Fig. 1 Schematic diagram of the flow reactor and the TD-VUV-TOF-PIAMS used for chemical analysis of IOPs. | ||
As shown in Fig. 1, the flow reactor used in this study is similar in spirit to the setup in Kamens's group in University of North Carolina.33 Briefly, it consisted of two 1 m straight sections of a 2.5 cm diameter quartz glass tube and one U-shaped connection tube. While there are 16 sampling ports distributed every 10 cm along the straight sections, only the exit of the tube acted as the sampling ports in this study. Generally, the total flow rates through the tube were in the range of 1.2–2.0 l min−1 corresponding to a Reynolds number of 146–243, which indicates the laminar flow conditions in the flow reactor. Similar to that in our smog chamber experiments,38,39 the flow reactor was continuously flushed with dry zero air until the background particle concentration less than 20 cm−3 prior to each run. The zero air also acted as precursor to produce O3 as well as the bath gas of CH2I2. The flow rates of CH2I2 and O3 were controlled respectively with mass flow meters, and their concentration in the flow tube were monitored separately with a gas chromatograph-electron capture detector (GC-180 ECD, Agilent 7820) and O3 analyzer (TEI model 49i). A sintered glass mixing plate filled with glass beads at the entry of the reactor ensured immediate mixing of CH2I2 and ozone. As UV radiation was always kept on during the experiments and the photolysis of CH2I2 is a fast process, the time of the mixing of CH2I2 and ozone is taken as the zero point of the reaction of I + O3. At the exit of the flow reactor, an annular diffusion denuder40 was used to remove the residual O3 and CH2I2 to make sure the end of the gaseous reaction of CH2I2 + O3. The concentrations and size distributions of IOPs formed from the photooxidation of CH2I2/O3 were monitored on-line with a Scanning Mobility Particle Sizer (SMPS, TSI, model 3936, TSI, USA) consisting of a differential mobility analyzer (DMA, TSI model 3080) and a condensation particle counter (CPC, TSI model 3775).
Real-time chemical analysis of IOPs were performed with the TD-VUV-TOF-PIAMS developed at the Atomic and Molecular Physics Beamline of the NSRL.30 IOPs formed in the flow reactor were tightly focused and sampled into the mass spectrometer through a typical aerodynamic lens assembly41–43 with a 200 μm-diameter orifice. After passing through a three-stage differential pumping system, the particles hit on an 8 mm diameter temperature-controlled copper tip located at the ionization region of the main chamber and were flash thermally vaporized at an appropriate temperature (in the range of 293–873 K). In this work, the same vaporization temperature (453 K) was chosen in all tests. The first reason is for high quality of mass spectrum of IOPs. The second one is this temperature is less than the decomposition temperature of I2O4 (460 K),44,45 so this choice can keep the high intensity of the signal of mass spectrum while avoid the decomposition of I2O4 assumed they exist in IOPs. And the last one, keeping the constant vaporization temperature will make us comparing the IE values from different tests without considering the temperature effect on IE. The plume of the vaporized particles was then photoionized with the tunable VUV beam from synchrotron radiation. The undulator-based spherical grating monochromator (SGM) beamline was operated with the lowest-energy grating, generating photons with energy from 7.5 to 22.5 eV. Generally, a photon flux of 1014 photons per second can be produced and the resolving power of 2700 (E/ΔE) at 15.9 eV can be achieved with adjusting the entrance- and exit-slit width of the grazing-incidence monochromator. Ions formed in the photoionization region were then detected with a reflectron TOFMS. When the undulator is fixed, photoionization mass spectra of IOPs can be investigated at a fixed ionization energy. While scanning the undulator continuously, PIE curves can be extracted resulting in the characterization of the ions at molecular level, which is crucial to learn the chemical component of IOPs and the formation mechanism of IOPs.
3. Results and discussion
3.1 Characterization of the flow reactor
As aforementioned, the evolution of the components in smog chamber is always going on following the initiation of the reaction, which impedes the effective utilization of PIE curves when smog chamber is combined with SR photoionization AMS. Different from the smog chamber, flow reactor is characterized by the fixed reaction situation if the sampling port and the total flow rate are fixed, no matter how long the sampling duration time. So prior to the combination with the TD-VUV-TOF-PIAMS at NSRL, the stabilization of the home-made flow reactor was tested.Three test experiments were performed according to the stabilization of the particle number concentration, the size distributions and the mass concentration of IOPs formed from photooxidation of CH2I2/O3 system in the flow reactor under different reaction conditions. As shown in Table 1 and Fig. 2, given the initial CH2I2 and O3 concentrations ([CH2I2]0 ∼ 0.5 ppm, [O3]0 2.3–3.2 ppm), when total flow rates in flow reactor were kept less than 2L min−1, the main diameter distribution of IOPs formed was accumulation mode.46 For example, in test #2, the mode diameter of IOPs was 66.1 nm. And it is obvious that the production of IOPs was always kept stable given the constant precursor, oxidant and flow rate (in other words, fixed reaction time). In test #1 and #2, the variation of mean number concentration was less than 10%, the variation of central diameter was less than 2% and the variation of mean mass concentration, which is the key parameter to the bulk measurement with TD-VUV-TOF-PIAMS, was less than 5% during one hour test time. In test #3, the total flow rate in flow reactor was kept at 4L min−1, corresponding to 16.5 seconds reaction time, and the concentration of O3 was around one sixth of that in test #2. In this case, Aitken mode46 of IOPs produced in flow reactor, central diameter of which is about 14.5 nm, can be observed. Even for Aitken mode particles in test #3, the variation of mean number concentration is only 12% and the variation of mean mass concentration is 4% during about one hour test.
| Test no. | Flow ratea (L min−1) | [CH2I2]0 (ppm) | [O3]0 (ppm) | Mean number conc. (# 106/cm−3) | Mode diameter (nm) | Mean mass conc.c (103 μg m−3) |
|---|---|---|---|---|---|---|
| a Flow rate = total inlet flow rate (CH2I2 + O3) = total outlet flow rate (SMPS sampling rate + pump rate).b Nano DMA was used in test #3.c All particles are presumed to be compact and spherical, and the particle density was set at 5.0 g cm−3 for all sizes.13 | ||||||
| #1 | 1.8 | 0.56 ± 0.03 | 2.32 ± 0.12 | 8.53 ± 0.39 | 57.3 ± 0.9 | 8.62 ± 0.41 |
| #2 | 1.3 | 0.52 ± 0.03 | 3.22 ± 0.16 | 7.62 ± 0.80 | 66.1 ± 0.2 | 11.97 ± 0.26 |
| #3b | 4.0 | 1.26 ± 0.25 | 0.50 ± 0.1 | 45.37 ± 5.35 | 14.5 ± 0.3 | 0.69 ± 0.03 |
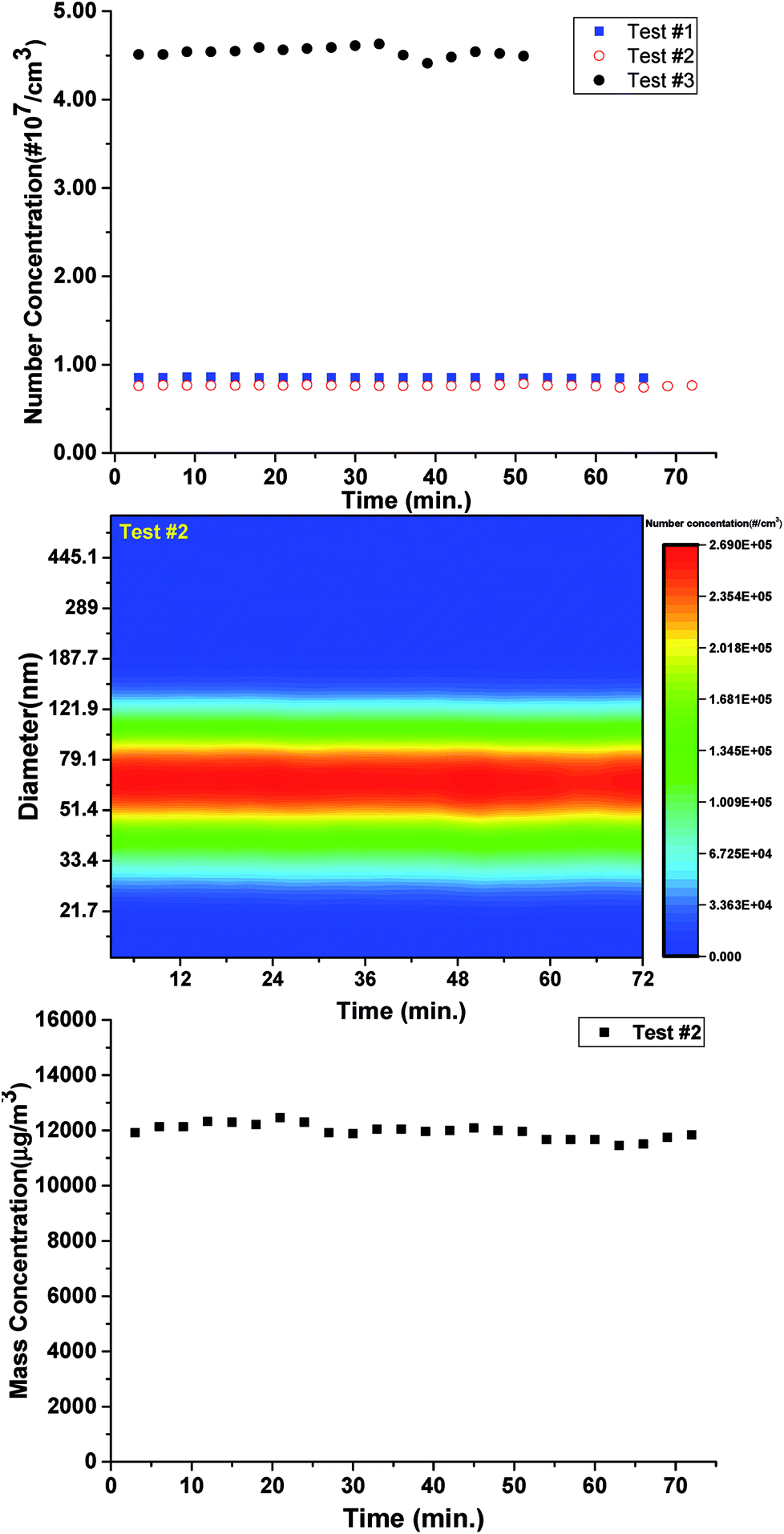 | ||
| Fig. 2 The variation of the particle number concentration (a), the size distributions (b), and the mass concentration (c) of IOPs in the flow reactor according to the duration time. | ||
All of the tests show the perfect stabilization of the flow reactor, which indicates its applicability to be combined with TD-VUV-TOF-PIAMS based on SR to make full use of PIE curves. In this work, however, only IOPs with accumulation mode have been investigated with aerosol photoionization mass spectrum.
3.2 Mass spectrometric study on IOPs
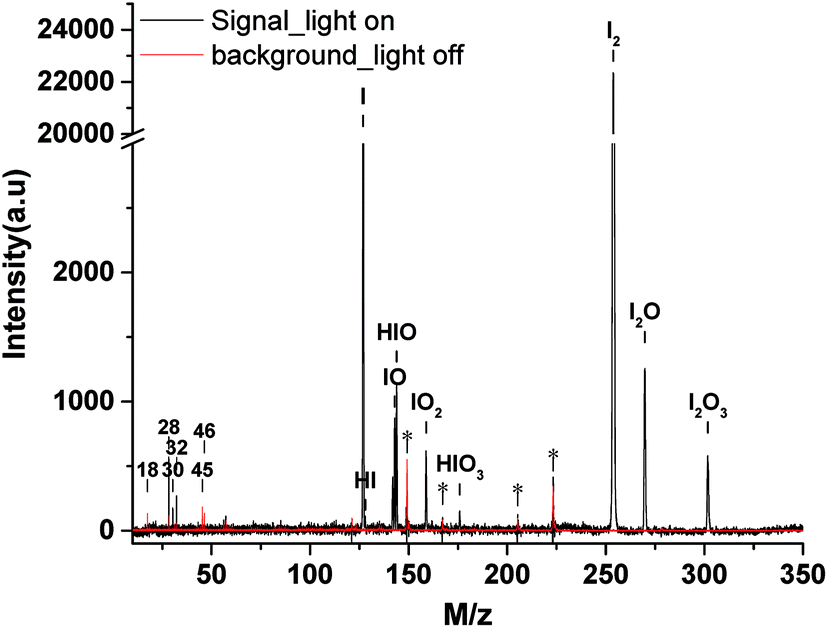 | ||
| Fig. 3 The mass spectra of IOPs formed from photooxidation of CH2I2/O3 (black line) and the background spectra (red line) at the photon energy of 11.0 eV. (a.u., arbitrary units). | ||
A typical thermal desorption photoionization mass spectrum of IOPs, as well as the background, at photon energy of 11.0 eV is also shown in Fig. 3, where the vaporization temperature of heater is 453 K. As the major components of air, it is not strange to observe the signals of H2O (m/z 18), N2 (m/z 28), NO (m/z 30) and O2 (m/z 32) in the background spectrum regarding to their high photoionization efficiencies and cross-section.47 The peaks of ethanol (m/z 46), which was generally used as solvent to clean the orifice of the aerodynamic lens in this work, and its photo-fragment (m/z 45) can also be identified in the background spectrum. In the range of m/z = 100 to m/z = 300, there are some unidentified while always existed features, which are labeled with stars as shown in Fig. 3. Compared with the background spectra in the range of m/z = 100 to m/z = 300, there are many new features resulted from the thermal desorption and photoionization of IOPs, corresponding to m/z = 127, 128, 143, 144, 159, 176, 254, 270 and 302.
As a “footprint” of a molecule or a radical, ionization energy or appearance energy can be used to identify molecular composition unambiguously. It is well known that photoionization mass spectrometry based on SR is characteristic of obtaining PIE curves by scanning photon energy, and IE or AE can be determined from PIE curves. In order to determined the corresponding molecular composition of the peaks observed in mass spectra of IOPs in Fig. 3, the PIE curves of these components were obtained by gating and integrating the corresponding peaks while scanning the photon energy in the range of 8.5–11.0 eV. To minimize the influence of the blocking of the orifice of aerodynamic lens by IOPs, the larger energy interval (0.1 eV) was applied in full range scanning in this work, while small energy interval (0.02 or 0.03 eV) was adopted around the threshold energy regions of IE or AE to obtain more precise value. Several typical PIE curves, corresponding to m/z = 127, 143, 159, 254 and 302, are illustrated in Fig. 4, where Fig. 4a–d were acquired at 0.1 eV energy interval and Fig. 4e and f at 0.02 or 0.03 eV energy interval, and all the data points have been normalized by the corresponding photon intensities monitored simultaneously with a silicon photodiode (SXUV100, International Radiation Detector, Inc.). Both of Fig. 4a and f correspond to m/z = 143 component in mass spectra of IOPs in Fig. 3, which shows how the more precise value of IE can be gotten.
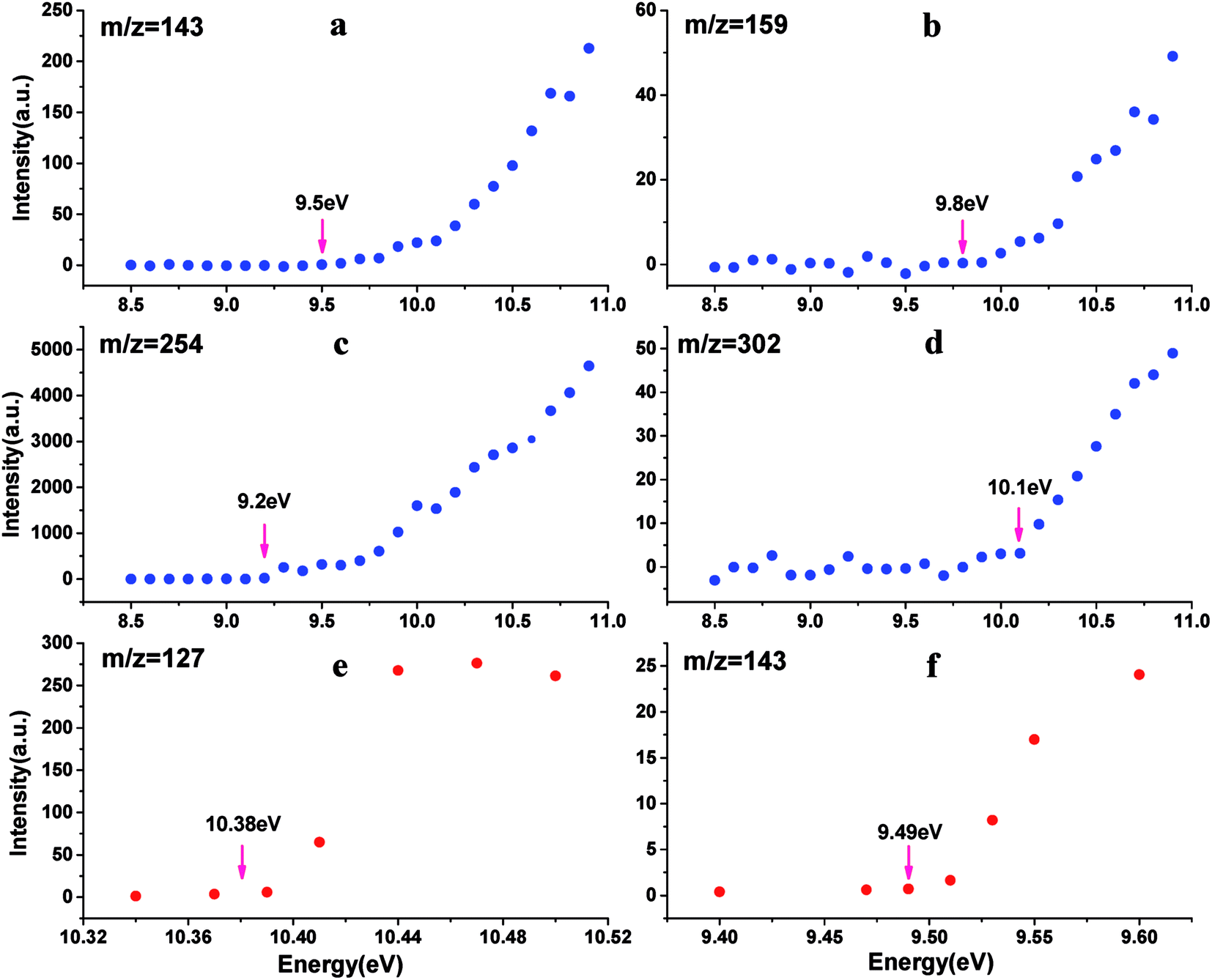 | ||
| Fig. 4 PIE curves for m/z 143 (panel a and f), m/z 159 (panel b), m/z 254 (panel c), m/z 302 (panel d), m/z 127 (panel e) ((a–d), larger energy interval scanning; (e and f), small energy interval scanning). (a.u., arbitrary units). | ||
All of the IEs derived from the PIE curves just like those shown in Fig. 4 are summarized in Table 2 with the references' data. And based on the IEs and the corresponding molecular weight, the assignments of these main components of IOPs were performed. There are three species in the photoionization mass spectra of IOPs. The first are only iodine-containing, such as I+ (m/z 127) and I2+ (m/z 254) with the IEs of 10.38 and 9.16 eV respectively. The second are iodine oxides, such as IO+(m/z 143), OIO+(m/z 159), I2O+(m/z 270) and I2O3+ (m/z 302). While the IEs of IO, OIO and I2O3 were measured as 9.49, 9.80 and 9.90 eV respectively, only the upper limit of IE of I2O could be obtained (<9.30 eV) for the weak signal in low photon energy. The third one are hydrogen-containing iodine species, such as HI+ (m/z 128), HIO+ (m/z 144) and HIO3+ (m/z 176), in which only the IE of HIO were measured as 9.70 eV and the peaks for HI+ and HIO3+ were too weak to acquire PIE curves.
| Ion | m/z | IE (eV) (this work) | IE (eV) (theo.) (ref.)a | IE (eV) (exp.) (ref.)b |
|---|---|---|---|---|
| a ref. 14.b Ref. 48.c N/A not available.d Ref. 49.e Ref. 50.f Ref. 51. | ||||
| I+ | 127 | 10.38 ± 0.02 | 10.35 | 10.451 |
| HI+ | 128 | N/Ac | 10.50 | 10.386 |
| IO+ | 143 | 9.49 ± 0.02 | 9.56 | 9.66d, 9.745e |
| HIO+ | 144 | 9.70 ± 0.03 | N/A | 9.811 |
| OIO+ | 159 | 9.80 ± 0.10 | 9.72, 9.793f | N/A |
| HIO3 | 176 | N/A | N/A | N/A |
| I2+ | 254 | 9.16 ± 0.05 | 9.47 | 9.307 |
| I2O+ | 270 | <9.30 | 9.02 | N/A |
| I2O3+ | 302 | 9.90 ± 0.03 | 9.97 | N/A |
Although some of the chemical components observed here had been observed before in gaseous or particle phase, only a few IEs of these species had been reported. The slow dark reaction of I2 and O3 has been studied experimentally using 118 nm VUV light photoionization TOF-MS by Gómez Martín et al.,14 where atomic and molecular iodine, hydrogen iodide and iodine oxides have been observed in the gas phase products and the corresponding IEs has been estimated based on quantum chemistry calculations. The chemical composition of IOPs formed from photodissociation of CH2I2 has been studied by Hoffmann et al. using on-line atmospheric pressure chemical ionization mass spectrometry,15 and I−, I2−, IO−, IO2− and IO3− have been detected in their works. Based on electron ionization aerosol mass spectrometry, Jimenez et al. have also studied new particle formation from photooxidation of CH2I2, and most of the species observed in this work have also been investigated in their work.16 Regarding to their technique of ionization, assignment of the chemical composition of IOPs were mainly depended on molecular weights not on IEs in these studies. As far as we know, only the IEs of I, I2, IO and HIO in gas phase have been investigated experimentally,48,50,51 and it is the first time to directly investigate the IEs of iodine oxides from IOPs experimentally in this work. Although the IE values obtained in this work show good agreement with literature values, it seems that out values are systematically a little lower than the literature values obtained experimentally in gas phase. For example, IE of I2 form IOPs obtained in this work is 9.16 eV, while that obtained in gas phase is 9.31 eV (see Table 2). Regarding to the temperature of thermal desorption (453 K) used here, the population of excited vibrational states may be the partial reason resulting in the lower IEs in this work. For example, the fraction of I2 in the second vibrational state (υ2) at 453 K is 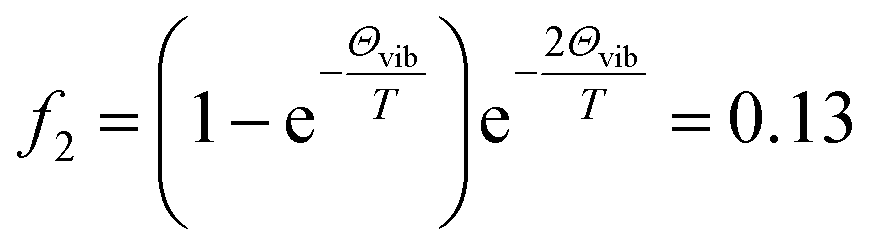 (where Θvib, named vibrational temperature, is 308 K for I2),52 and the ionization of the second vibrational state of I2 will result in about 0.06 eV lower of ionization energy as
(where Θvib, named vibrational temperature, is 308 K for I2),52 and the ionization of the second vibrational state of I2 will result in about 0.06 eV lower of ionization energy as  of I2 is 214.52 cm−1.
of I2 is 214.52 cm−1.
Regarding to the facts that no IOPs were formed and no signal were observed in background mass spectra when the photooxidation of CH2I2/O3 was not initiated, it is presumed that the observed I+ and I2+ may result from the thermal decomposition or fragmentation of iodine oxides and/or hydrogen-containing iodine species in the IOPs.15,16 The main components of iodine oxides in IOPs are still controversial and the knowledge about them is still far deficient. Although highly oxidized iodine oxides, such as I2Oy (y = 1, …, 5) and I3Oy (y = 1, …, 7) have been observed in gaseous products generated from the reaction of I2 and O3,14 the largest iodine oxide observed in IOPs is only I2O3 in this work and the work of Jimenez et al.16 Traditionally, I2O4 and I2O5 are believed to be the two main candidates via gas-phase collisions to nucleate the new IOPs. The crystal structure of I2O4 was approximated as a one-dimensional solid comprising infinite –I–O–IO2–O– chains and weaker interchain I–O bands.53 While in solid I2O5, a I2O5 unit is composed of two IO3 pyramids which have one O atom in common, simply as O2I–O–IO2.54 However, both of the two iodine oxides are known to decompose under thermal induction:
| 5I2O4(150–200 °C) → 4I2O5 + I2 | (R8) |
| 2I2O5(∼400 °C) → 2I2 + 5O2 | (R9) |
As the IOPs were vaporized at 180 °C in this work, the thermal decomposition of I2O4 may be the one potential source of the detected I+ and I2+.
Secondly, although single-photon ionization in TD-VUV-TOF-PIAMS instead of 70 eV electron impact ionization used in traditional AMS was applied in this work, there was no guarantee to avoid the photodissociation of I2O4 or I2O5 during photoionization. Thus the photodissociation of I2O4 and I2O5 may be another reason for no detection of I2O4+ and I2O5+, as well as the detection of I+, IO+, OIO+, I2O+ and I2O3+. A mass spectroscopic study on I2O5 aerosol particles generated from atomizing standard I2O5 sample will be performed in the near future, which is presumed to offer some useful information to verify this possibility. Thirdly, regarding to the weak peak of I2O+ and even weaker signal of I2O3+, it is presumed that the photoionization cross sections of high iodine oxides, such as I2O4 and I2O5, are too small, thus the concentrations of I2O4+ and I2O5+ were too low that were beyond the detection limit of the aero mass spectrometer used in this work.
As for no detection of I2O4 and I2O5, that whether I2O4 or I2O5 is the condensable unit of IOPs is still unclear and cannot be identified directly just depending on mass spectra. However, by combining the results of this work and the other studies,10–18 the identity of I, IO, OIO, I2, I2O and I2O3, that is to say the reaction mechanism of (R1)–(R4) aforementioned in Introduction, has been consolidated.
It is interesting to observe ions of iodine acids, such as HI+, HIO+ and HIO3+, in the mass spectra of IOPs. Similar ion signals of iodine acids were also observed before in AMS of aerosol particles formation from photooxidation of CH2I2 (ref. 16) and from I2O5 dissolved in water55 in laboratory studies, as well as from new particle formation in an iodine-rich, coastal atmospheric environment in recent field research.56 The detection of HI+, HIO+ and HIO3+ indicates that either there are larger oxyacids in IOPs or there are new production of oxyacids on AMS vaporizer.16 It is presumed in one way that when the IOPs impact on AMS vaporizer, I2O4 will thermally decomposes to I2O5 via (R9), and then I2O5 will react with H2O to generate HIO3. It is noteworthy that the peak of H2O are hardly to be observed in the mass spectra of IOPs being compared with the background spectrum (see Fig. 3). However, more deep work are needed to identify whether the disappearance of H2O mainly results from its reaction with I2O5. While in dry conditions, the iodine oxyacids in IOPs may result from some unknown gas-phase chemistry or from the inhomogeneous reaction between OH, HO2 or H2O with iodine oxides in IOPs.16 Regarding to the relative humidity (RH ∼ 23%) and the temperature of the vaporizer (T = 453 K) in this work, both of the aforementioned formation passages of HIO3 are plausible, whichever indicates the presence of the higher iodine oxides.
4. Conclusions
In this work, chemical analysis of IOPs formed from the photooxidation of CH2I2 has been performed on-line based on the combination of the flow reactor system with the TD-VUV-TOF-PIAMS. Atomic and molecular iodine (I, I2), iodine oxides (IO, OIO, I2O and I2O3) and hydrogen-containing iodine species (HI, HIO and HIO3) were observed in aerosol mass spectra, and their corresponding IEs were identified directly in aerosol phase for the first time. The formation mechanism of IO, OIO, I2O and I2O3, as well as the potential source of I and I2 has been consolidated. I2O4 and I2O5 haven't been detected directly in IOPs mass spectra in this work, the detection of HIO and HIO3, however, indicates the presence of the higher iodine oxides in IOPs. Although more work are needed to be carried out for the CH2I2/O3 reaction system itself, the study on chemical composition of IOPs carried in this work has shown that the combination of the flow reactor system with the SR-based TD-VUV-TOF-PIAMS is to be a promising approach for on-line study of chemical components and formation mechanism of atmospheric aerosol by making the full advantage of flow tube (sampling-duration-time-independent) and SR photoionization (identify the chemical composition ambiguously on IE or AE). As for experimental technique, more work is needed to resolve the block of the orifice of aerodynamic lens and to improve the detect limit of the TD-VUV-TOF-PIAMS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the National Natural Science Foundation of China (U1532143, 41575126, 91544228) and the National Key Research and Development Program of China (2016YFC0202205, 2017YFC0209506). The authors also acknowledge support from the Institute of Analytical Chemistry of the CAS, v. v. i., under the Institutional Research Plan No. RVO: 68081715.References
- A. Saiz-Lopez, J. M. C. Plane, A. Baker, L. Carpenter, R. von Glasow, J. C. Gómez Martín, G. Mc Figgans and R. Saunders, Chem. Rev., 2012, 112, 1773 CrossRef CAS PubMed.
- K. A. Read, A. S. Mahajan, L. J. Carpenter, M. J. Evans, B. V. E. Faria, D. E. Heard, J. R. Hopkins, J. D. Lee, S. J. Moller, A. C. Lewis, L. Mendes, J. B. McQuaid, H. Oetjen, A. Saiz-Lopez, M. J. Pilling and J. M. C. Plane, Nature, 2008, 453, 1232 CrossRef CAS PubMed.
- A. Saiz-Lopez, A. S. Mahajan, R. A. Salmon, S. J. B. Bauguitte, A. E. Jones, H. K. Roscoe and J. M. C. Plane, Science, 2007, 317, 348 CrossRef CAS PubMed.
- C. D. O'Dowd, J. L. Jimenez, R. Bahreini, R. C. Flagan, J. H. Seinfeld, K. Hameri, L. Pirjola, M. Kulmala, S. G. Jennings and T. Hoffmann, Nature, 2002, 417, 632 CrossRef PubMed.
- J. A. Aitken, Trans.–R. Soc. Edinburgh, 1897, 3, 15 Search PubMed.
- C. D. O'Dowd, M. Geever, M. K. Hill, M. H. Smith and S. G. Jennings, Geophys. Res. Lett., 1998, 25, 1661 CrossRef.
- C. D. O'Dowd, G. McFiggans, D. J. Creasey, L. Pirjola, C. Hoell, M. H. Smith, B. J. Allan, J. M. C. Plane, D. E. Heard, J. D. Lee, M. J. Pilling and M. Kulmala, Geophys. Res. Lett., 1999, 26, 1707 CrossRef.
- C. D. O'Dowd, K. Hämeri, J. M. Mäkelä, L. Pirjola, M. Kulmala, S. G. Jennings, H. Berresheim, H. C. Hansson, G. Leeuw, G. J. Kunz, A. G. Allen, C. N. Hewitt, A. Jackson, Y. Viisanen and T. Hoffmann, J. Geophys. Res.: Atmos., 2002, 107, 1 Search PubMed.
- J. L. Grenfell, R. M. Harrison, A. G. Allen, J. P. Shi, S. A. Penkett and C. D. O'Dowd, J. Geophys. Res.: Atmos., 1999, 104, 13771 CrossRef CAS.
- J. B. Burkholder, J. Curtius, A. R. Ravishankara and E. R. Lovejoy, Atmos. Chem. Phys., 2004, 4, 4943 CrossRef.
- R. W. Saunders and J. M. C. Plane, Environ. Chem., 2006, 2, 299 CrossRef.
- R. W. Saunders and J. M. C. Plane, J. Aerosol Sci., 2006, 37, 1737 CrossRef CAS.
- R. W. Saunders, R. Kumar, J. C. Gómez Martín, A. S. Mahajan, B. J. Murry and J. M. C. Plane, Z. Phys. Chem., 2010, 224, 1095 CrossRef CAS.
- J. C. Gómez Martín, O. Gálvez, M. T. Baeza-Romero, T. Ingham, J. M. C. Plane and M. A. Blitz, Phys. Chem. Chem. Phys., 2013, 15, 15612 RSC.
- T. Hoffmann, C. D. O'Dowd and J. H. Seinfeld, Geophys. Res. Lett., 2001, 28, 1949 CrossRef CAS.
- J. L. Jimenez, R. Bahreini, D. R. Cocker, H. Zhuang, V. Varutbangkul, R. C. Flagan, J. H. Seinfeld, C. D. O'Dowd and T. J. Hoffmann, J. Geophys. Res.: Atmos., 2003, 108, 4318 CrossRef.
- W. J. Bloss, D. M. Rowley, R. A. Cox and R. L. Jones, J. Phys. Chem. A, 2001, 105, 7840 CrossRef CAS.
- J. C. Gómez Martín, P. Spietz and J. P. Burrows, J. Phys. Chem. A, 2007, 111, 306 CrossRef PubMed.
- N. Kaltsoyannis and J. M. C. Plane, Phys. Chem. Chem. Phys., 2008, 10, 1723 RSC.
- K. A. Pratt and K. A. Prather, Mass Spectrom. Rev., 2011, 31, 1 CrossRef PubMed.
- K. A. Pratt and K. A. Prather, Mass Spectrom. Rev., 2011, 31, 17 CrossRef PubMed.
- E. E. Gard, M. J. Kleeman, D. S. Gross, L. S. Hughes, J. O. Allen, B. D. Morrical, D. P. Fergenson, T. Dienes, M. E. Gälli, R. J. Johnson, G. R. Cass and K. A. Prather, Science, 1998, 279, 1184 CrossRef CAS PubMed.
- D. M. Murphy, D. S. Thomson and M. J. Mahoney, Science, 1998, 282, 1664 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525 CrossRef CAS PubMed.
- E. R. Mysak, K. R. Wilson, M. Jimenez-Cruz, M. Ahmed and T. Baer, Anal. Chem., 2005, 77, 5953 CrossRef CAS PubMed.
- J. N. Shu, K. R. Wilson, M. Ahmed and S. R. Leone, Rev. Sci. Instrum., 2006, 77, 043106 CrossRef.
- W. Z. Fang, L. Gong, X. B. Shan, F. Y. Liu, Z. Y. Wang and L. S. Sheng, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 129 CrossRef CAS.
- E. Gloaguen, E. R. Mysak, S. R. Leone, M. Ahmed and K. R. Wilson, Int. J. Mass Spectrom., 2006, 258, 74 CrossRef CAS.
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, Atmos. Chem. Phys., 2009, 9, 3209 CAS.
- W. Z. Fang, L. Gong, X. B. Shan, F. Y. Liu, Z. Y. Wang and L. S. Sheng, Anal. Chem., 2011, 83, 9024 CAS.
- W. Z. Fang, L. Gong, Q. Zhang, M. Q. Cao, Y. Q. Li and L. S. Sheng, Environ. Sci. Technol., 2012, 46, 3898 CrossRef CAS PubMed.
- W. Z. Fang, L. Gong and L. S. Sheng, Environ. Chem., 2017, 14, 75 CrossRef CAS.
- N. M. Czoschke, M. Jang and R. M. Kamens, Atmos. Environ., 2003, 37, 4287 CrossRef CAS.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Atmos. Meas. Tech., 2011, 4, 445 CrossRef CAS.
- J. M. Mäkelä, T. Hoffmann, C. Holzke, M. Väkevä, T. Suni, T. Mattila, P. P. Aalto, U. Tapper, E. I. Kauppinen and C. D. O'Dowd, J. Geophys. Res., 2002, 107, 1401 CrossRef.
- L. J. Carpenter, W. T. Sturges, S. A. Penkett, P. S. Liss, B. Alicke, K. Hebestreit and U. Platt, J. Geophys. Res., 1999, 104, 1679 CrossRef CAS.
- S. S. Wang, R. H. Kong, X. B. Shan, Z. Y. Wang, Y. W. Zhang, L. S. Sheng, L. Q. Hao and S. K. Zhou, J. Synchrotron Radiat., 2006, 13, 415 CrossRef CAS PubMed.
- C. J. Hu, Q. Ma, Z. Liu, Y. Cheng, L. Q. Hao, N. N. Wei, Y. B. Gai, X. X. Lin, X. J. Gu, W. X. Zhao, M. Q. Huang, Z. Y. Wang and W. J. Zhang, Atmos. Chem. Phys. Discuss. DOI:10.5194/acp-2017-433.
- G. Pan, C. J. Hu, Z. Y. Wang, Y. Cheng, X. H. Zheng, X. J. Gu, W. X. Zhao, W. J. Zhang, J. Chen, F. Y. Liu, X. B. Shan and L. S. Sheng, Rapid Commun. Mass Spectrom., 2012, 26, 189 CrossRef CAS PubMed.
- P. Mikuška, Z. Večeřa, A. Bartošíkova and W. Maenhaut, Anal. Chim. Acta, 2012, 714, 68 CrossRef PubMed.
- P. Liu, P. J. Ziemann, D. B. Kittelson and P. H. McMurry, Aerosol Sci. Technol., 1995, 22, 293 CrossRef CAS.
- P. Liu, P. J. Ziemann, D. B. Kittelson and P. H. McMurry, Aerosol Sci. Technol., 1995, 22, 314 CrossRef CAS.
- X. Zhang, K. A. Smith, D. R. Worsnop, J. Jimenez, J. T. Jayne and C. E. Kolb, Aerosol Sci. Technol., 2002, 36, 617 CrossRef CAS.
- A. Wikjord, P. Taylor, D. Torgerson and L. Hachkowski, Thermochim. Acta, 1980, 36, 367–375 CrossRef CAS.
- H. Fjellvag and A. Kjekshus, Acta Chem. Scand., 1994, 48, 815 CrossRef.
- K. T. Whitby and G. M. Sverdrup, California aerosols: their physical and chemical characteristics, in The Character and Origins of Atmospheric Aerosols: A Digest of Results from the California Aerosol Characterization Experiment (ACHEX), ed. G. M. Hidy, P. K. Mueller, D. Grosjean, B. R. Appel and J. J. Wesolowski, Advances in Environmental Science and Technology, Wiley, New York, 1980, vol. 9, p. 477 Search PubMed.
- N. Wainfan, W. C. Walker and G. L. Weissler, Photoionization efficiencies and cross sections in O2, N2, CO2, Ar, H2O, H2, and CH4, Phys. Rev., 1955, 99, 542 CrossRef CAS.
- NIST Chemistry Webbook, SRD 69, National Institute of Standards and Technology, Gaithersburg, MD, 2017, http://webbook.nist.gov/chemistry/ Search PubMed.
- P. S. Monks', L. J. StieP, D. C. Tardy, J. F. Liebman, Z. Y. Zhang, S. C. Kuo and R. B. Klemm, J. Phys. Chem., 1995, 99, 16566 CrossRef.
- Z. Zhang, P. S. Monks, L. J. Stief, J. F. Liebman, R. E. Huie, S.-C. Kuo and R. B. Klemm, J. Phys. Chem., 1996, 100, 63 CrossRef CAS.
- S. Y. Lee, J. Phys. Chem. A, 2004, 108, 10754 CrossRef CAS.
- D. A. Mcquarrie and J. D. Simon, Physical chemistry: A molecular approach, University Science Books, Sausalito, California, 1997 Search PubMed.
- H. Fjellvag and A. Kjekshus, Acta Chem. Scand., 1994, 48, 815 CrossRef.
- K. Selte, A. Kjekshus, O. Hartmann, D. Holme, A. Lamvik, E. Sunde and N. A. Sørensen, Acta Chem. Scand., 1970, 24, 1912 CrossRef CAS.
- M. W. Chase, J. Phys. Chem. Ref. Data, 1996, 25, 1297 CrossRef CAS.
- M. Sipilä, N. Sarnela, T. Jokinen, H. Henschel, H. Junninen, J. Kontkanen, S. Richters, J. Kangasluoma, A. Franchin, O. Peräkylä, M. P. Rissanen, M. Ehn, H. Vehkamäki, T. Kurten, T. Berndt, T. Petäjä, D. Worsnop, D. Ceburnis, V. M. Kerminen, M. Kulmala and C. O'Dowd, Nature, 2016, 537, 532 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |