
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissociation reaction of B2H6 on TiN surfaces during atomic layer deposition: first-principles study†
Hwanyeol Parka,
Sungwoo Lee a,
Ho Jun Kimb,
Euijoon Yoon*ac and
Gun-Do Lee*ac
a,
Ho Jun Kimb,
Euijoon Yoon*ac and
Gun-Do Lee*ac
aDepartment of Materials Science and Engineering, Seoul National University, Seoul 08826, Korea. E-mail: eyoon@snu.ac.kr; gdlee@snu.ac.kr
bMemory Thin Film Technology Team, Samsung Electronics, Hwaseong-si 445-701, South Korea
cResearch Institute of Advanced Materials, Inter-University Semiconductor Research Center, Seoul National University, Seoul 08826, South Korea
First published on 8th December 2017
Abstract
In the fabrication process of memory devices, a void-free tungsten (W) gate process with good conformability is very important for improving the conductivity of the W gate, leading to enhancement of device performance. As the downscaling continues to progress, void-free W deposition becomes more difficult due to the experimental limitations of conformal film deposition even with atomic layer deposition (ALD) W processes. In ALD W processes, it is known that the B2H6 dosing process plays a key role in deposition of the ALD W layer with low resistivity and in removal of residual fluorine (F) atoms. To comprehend the detailed ALD W process, we have investigated the dissociation reaction of B2H6 on three different TiN surfaces, TiN (001), Ti-terminated TiN (111), and N-terminated TiN (111), using first-principles density functional theory (DFT) calculations. N-terminated TiN (111) shows the lowest overall reaction energy for B2H6. These results imply that severe problems, such as a seam or void, in filling the W metal gate for memory devices could be attributed to the difference in the deposition rate of W films on TiN surfaces. From this study, it was found that the control of the texture of the TiN film is essential for improving the subsequent W nucleation.
1. Introduction
As thin film deposition techniques have advanced following Moore's law for decades, increasingly smaller sizes and higher aspect ratios (AR) for improving device performance have required highly uniform and conformal films.1 Nitride materials, such as silicon nitride and titanium nitride, have been deposited using conventional deposition techniques such as plasma-enhanced chemical vapor deposition (PECVD)2,3 and low-pressure chemical vapor deposition (LPCVD).4,5 However, the down-scaling of memory devices has required another deposition technique such as atomic layer deposition (ALD)6–8 to resolve the step coverage issues of highly integrated devices. As a thin film deposition process, ALD is the most prevalent method due to the demand for excellent step coverage and conformality of deposited thin films. The ALD processes use well-controlled sequential surface reactions to obtain uniform and conformal films.9,10Tungsten (W) is a good material for use as a metal gate with low resistivity in memory devices.11 A thin film of W can be deposited using ALD by alternatively exposing W precursors such as WF6 and reducing agents such as disilane (Si2H6) or diborane (B2H6). First, the successful deposition of W via ALD has been carried out using tungsten hexafluoride (WF6) and Si2H6 in an ABAB… sequence. It was reported that the Si2H6 reactant could play only a sacrificial role to remove residual fluorine (F) from the surface.12 Later, the ALD W process using B2H6 and silane precursors was also intensively investigated.12,13 Recently, the comparative study of ALD W using two different precursors, SiH4 and B2H6, was reported by Guilei Wang et al.14 They concluded that ALD W films using B2H6 showed much lower residual F content and a lower resistivity than those using SiH4, and a better ALD W film as a gate filling metal could be obtained. These ALD W films have been typically utilized as nucleation layers for a metal gate in memory devices before the deposition of the bulk CVD-W film.15–17
Despite much effort in improving ALD W processes, as memory devices become smaller and smaller, the limitation of conformality at ultrahigh aspect ratio (UHAR) contact can induce potential problems such as a seam or void in the final W-plug, leading to an increase in contact resistance.18,19 Further downscaling the memory devices necessitates the theoretical comprehension of the ALD W process due to the experimentally limited observations on the sub-nanometer scale. Although a few experimental studies on ALD W have been investigated, there has been no theoretical report on the reactivity of B2H6.
In this study, we investigated the reactivity of B2H6 with three different TiN surfaces using first principles study based on density functional theory (DFT) calculation to explore the reaction mechanism of the underlying TiN layers during the B2H6 dosing process in the ALD W deposition because the underlying surfaces can have significant effects on the characteristics of the subsequent W nucleation layers.15,20 TiN films have been widely used as a glue/barrier layer for subsequent W nucleation.21 Although transition metal nitrides always have a problem of oxidation at elevated temperatures,22 TiN film used in fabrication process of a memory device does not exposed to the oxidation W film is deposited right after deposition of TiN films in ALD process under vacuum system. Three different planes of TiN surfaces, TiN (001), Ti-terminated TiN (111), and N-terminated TiN (111) were taken into account because poly-crystalline TiN layers with (001) and (111) preferred orientations were mainly observed in deposition of TiN films.23,24 The decomposition reaction pathways and reaction energetics on three different TiN surfaces were investigated. It is expected that comparative analysis of the reaction mechanism of B2H6 with different TiN surfaces would give us insight into how important the underlying TiN surfaces could be for improving the quality of the subsequent W layer during the B2H6 dosing process in ALD W deposition.
2. Computational methods
In our theoretical results, all DFT calculations were performed using Vienna Ab initio Simulation Package (VASP) program with the Perdew–Burke–Ernzerh of (PBE) functional in the generalized gradient approximation (GGA).25,26 TiN (001) and TiN (111) surfaces with B1–NaCl structure were used as the reactive surfaces with the B2H6 precursor. The optimized lattice parameter of TiN was a0 = 4.21 Å, which is in good agreement with the experimental value (a0 = 4.24 Å).27 For the TiN (001) surface, a 4-layer slab of (2 × 2) supercell was considered. For comparison, the TiN surfaces with Ti-terminated and N-terminated (111) orientations were considered with a 5-layer slab of (2 × 2) supercell. For all TiN surfaces, such as TiN (001), Ti-terminated TiN (111), and N-terminated TiN (111), vacuum gaps with values of 23.7 Å, 25.4 Å, and 25.6 Å, respectively, in the z direction were included to avoid interactions between adjacent slabs.Valence orbitals were described by a plane-wave basis set with the cutoff energy of 400 eV. Electronic energies were calculated with a self-consistent-field (SCF) tolerance of 10−4 eV. Ultrasoft Vanderbilt-type pseudopotentials28 were used to describe the interactions between ions and electrons. A 3 × 3 × 3 Monkhorst k-point mesh for bulk TiN was chosen to ensure that the total energies converged within 1 meV per formula unit. The Brillouin zone for three different TiN surfaces was sampled with a 3 × 3 × 1 Monkhorst–Pack k-point mesh. Geometry optimization was performed by minimizing the forces of all atoms to less than 0.02 eV Å−1. In addition, we have calculated total energies for various configurations to determine the energy barrier for dissociative adsorption of B2H6 on the TiN surfaces.
To optimize adsorption structures, we considered three orientations and three positions of B2H6 on the three different TiN surfaces. The details of all nine cases are shown in the ESI (Fig. S1–S3†). The optimized adsorption structures with the lowest energy in the ESI (Tables S1–S3†) were used in this paper.
To calculate the transition state, the distance between the two dissociative atoms is slightly separated, and energy relaxation is performed with the constrained distance. The same procedures are carried out until the force between two dissociative atoms becomes almost zero at the saddle point of energy. Those results of transition structure were also checked by the nudged elastic band method.29
3. Results and discussion
3.1. B2H6 dissociative chemisorption on TiN (001)
Fig. 1 shows the optimized structures for the B2H6 reaction pathway of B–B dissociation on the TiN (001) surface. The optimized structure with the lowest adsorption energy of B2H6 on the surface is displayed in Fig. 1a. As shown in Fig. 1c, it was found that borane (BH3) molecules dissociated from B2H6 favorably react with nitrogen atoms on the TiN (001) surface after B–B dissociation due to stronger B–N bonding nature than B–Ti bonding. As shown in Fig. 2, the reaction energy is −1.89 eV, which means that the reaction is exothermic and energetically favorable. The activation energy from Fig. 1a–c is 1.11 eV, and the transition state is shown in Fig. 1b with no obvious surface reconstruction during the reaction. Generally, if any surface reconstruction is occurred during the reaction, activation and reaction energy can be smaller. However, no obvious surface reconstruction was not found for all surfaces used in this study.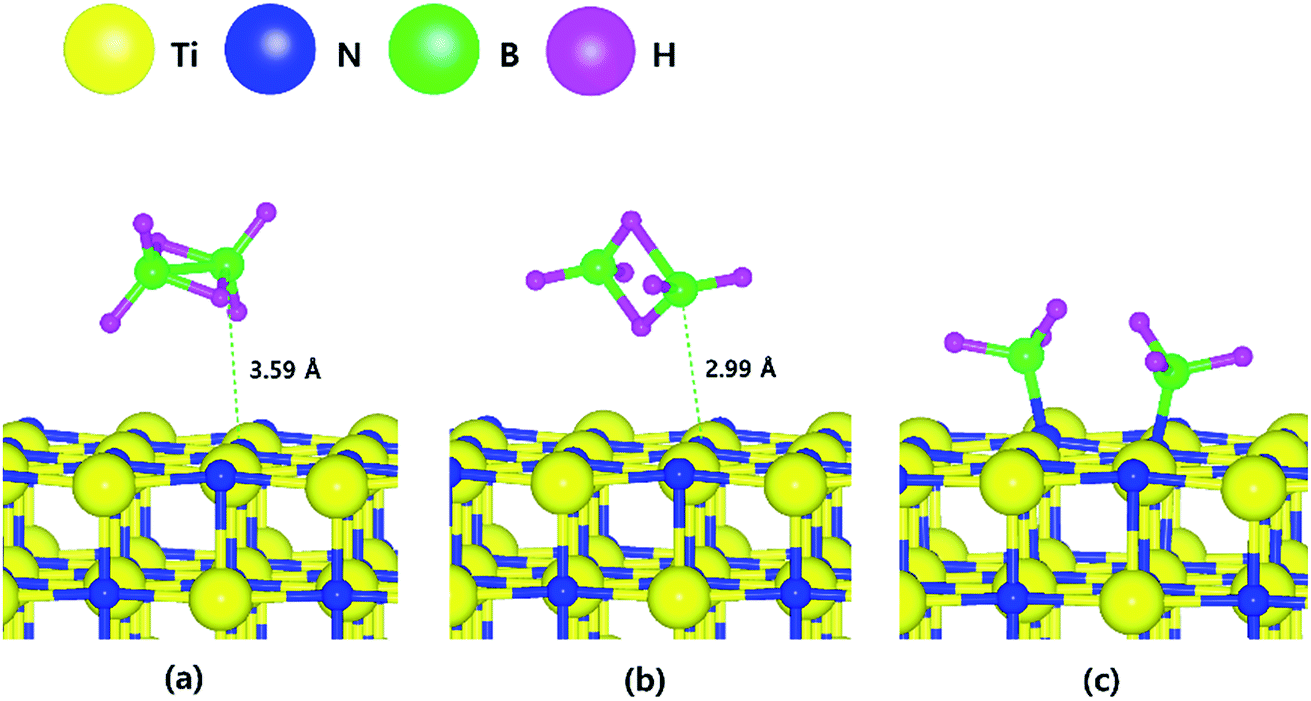 | ||
| Fig. 1 The optimized structures for (a) adsorption state, (b) transition state and (c) reaction state of a B2H6 on the TiN (001) surface. | ||
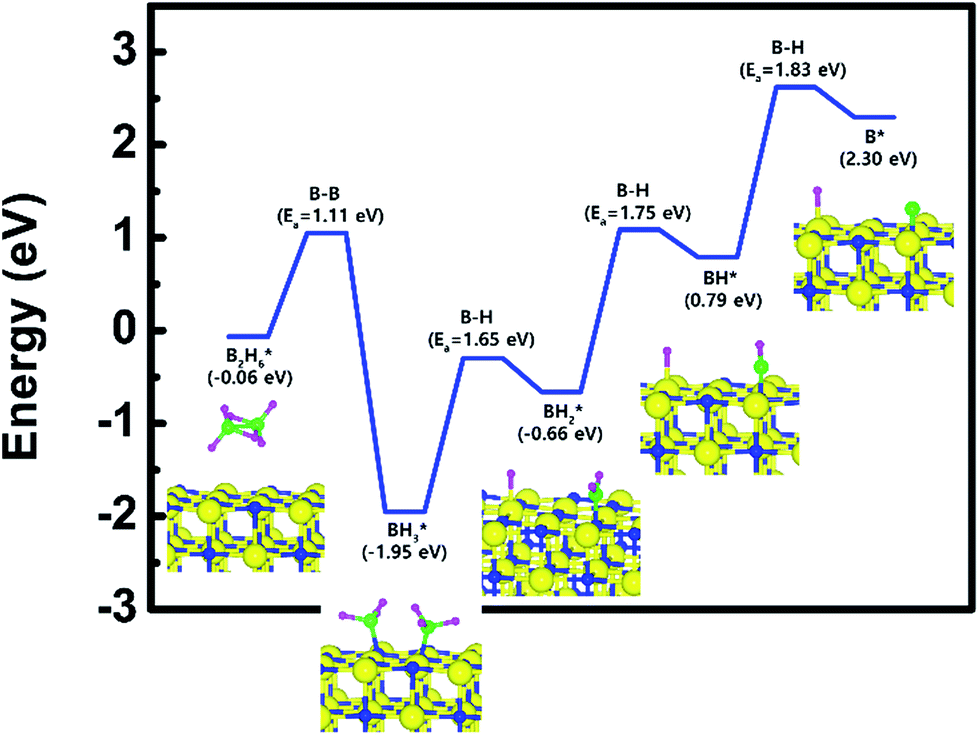 | ||
| Fig. 2 Calculated energy diagram of B2H6 decomposition on the TiN (001) surface. | ||
To complete the overall reaction energetics of B2H6, the calculated energy diagram of B2H6 decomposition on the TiN (001) surface is displayed in Fig. 2. The detailed structures of B2H6 during the overall reaction pathway on the TiN (001) surface for transition state calculations can be found in Fig. S4 (ESI†). During the reaction of the B2H6 precursor on the TiN (001) surface, this calculation shows that the overall reaction process is endothermic, with a calculated overall reaction energy of 2.36 eV. These results indicate that the reaction is thermodynamically unfavorable. Furthermore, B2H6 dissociative chemisorption on TiN (001) is kinetically difficult due to high activation energies that range from a minimum of 1.11 eV to a maximum of 1.83 eV. The low reactivity of B2H6 with the TiN (001) surface might be attributed to the presence of only one dangling bond per atom on the surface.
3.2. B2H6 dissociative chemisorption on Ti-terminated TiN (111)
The decomposition mechanism of B2H6 was also studied on the Ti-terminated TiN (111) surface to estimate the difference between TiN (001) and TiN (111) surfaces. The adsorption and reaction of B2H6 on the Ti-terminated TiN (111) surface are shown in Fig. 3a and c with the transition state shown in Fig. 3b. As shown in Fig. 3c, it was found that dissociated BH3 molecules were adsorbed on the hollow site made by three Ti atoms (site number 3 in Fig. S2†). In Fig. 4, the lowest adsorption energy of B2H6 on the Ti-terminated TiN surface is −4.46 eV, showing that the adsorption is energetically favorable. However, the reaction energy is 0.36 eV, indicating that the reaction is endothermic. The activation energy from Fig. 3a–c is 0.74 eV with the transition state in Fig. 3b, and no obvious surface reconstruction was found during the reaction. There are three more B–H bond breaking steps after a B–B bond breaking step, as shown in Fig. S4 and Table S4 (ESI†).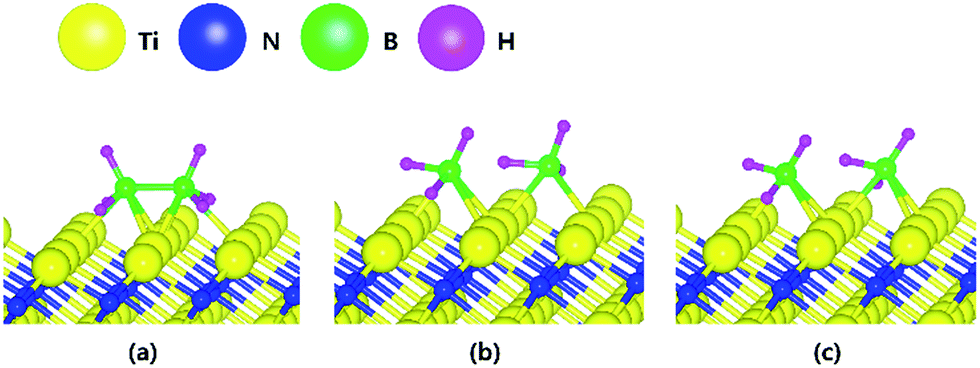 | ||
| Fig. 3 The optimized structures for (a) adsorption state, (b) transition state and (c) reaction state of a B2H6 on the Ti-terminated TiN (111) surface. | ||
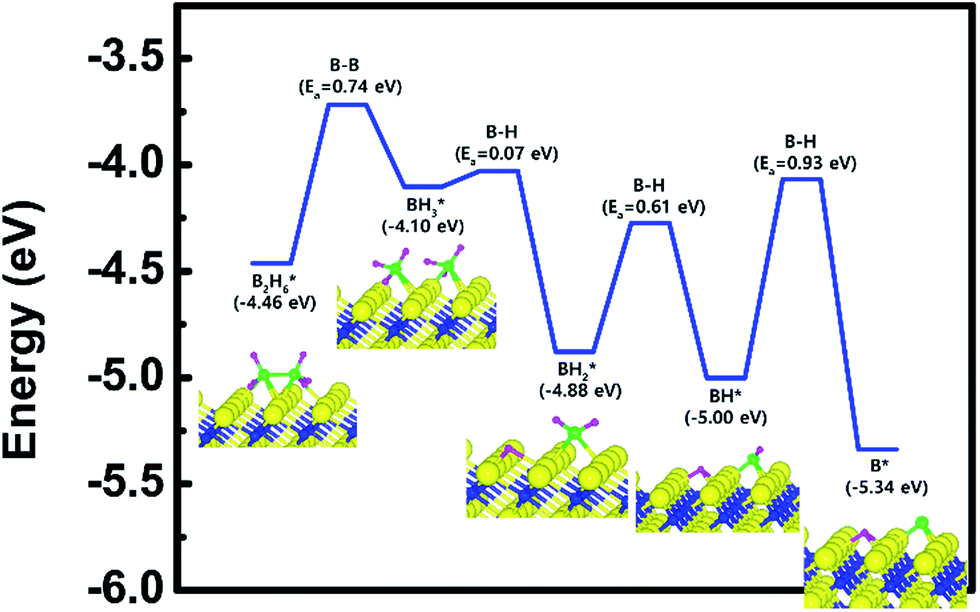 | ||
| Fig. 4 Calculated energy diagram of B2H6 decomposition on the Ti-terminated TiN (111) surface. | ||
The entire energy diagram for the B2H6 decomposition on Ti-terminated TiN (111) is illustrated in Fig. 4, which differs with the diagram for TiN (001) in Fig. 2. It demonstrates that both B–B and B–H bond dissociation steps on the Ti-terminated TiN (111) surfaces are more facile than the TiN (001) surface due to smaller activation energies of dissociation on the Ti-terminated TiN (111) surface. Moreover, the overall reaction of B2H6 is exothermic, with an overall reaction energy of −0.88 eV. These results show that the reaction is energetically favorable. The high reactivity of B2H6 on the Ti-terminated TiN (111) surface may be because the surface has triple dangling bonds per atom, which make the surface even more reactive than the TiN (001) surface. To be more specific, the number of dangling bonds on the Ti-terminated TiN (111) surface is more than that of the TiN (001), so that B2H6 dissociative reaction is more favorable on the former. This analysis is confirmed by higher adsorption of both B and H atoms on Ti-terminated TiN (111) surface than the TiN (001) surface as shown in Table 1. The aforementioned reasons, this surface can also reduce the energy barriers of the B2H6 decomposition as compared to the TiN (001) surface. The activation energies for dissociation of B2H6, BH3, and BH2 are 0.74, 0.07 and 0.61 eV, respectively, which are lower than that of BH (0.93 eV). This implies that the B–H bond dissociation of BH is the rate-determining step along the overall reaction.
| Surface | B | H |
|---|---|---|
| TiN (001) | 4.9 | 2.9 |
| Ti-Terminated TiN (111) | 6.3 | 4.6 |
| N-Terminated TiN (111) | 11.8 | 5.7 |
3.3. B2H6 dissociative chemisorption on N-terminated TiN (111)
The optimized structures for the adsorption, transition state and reaction of B2H6 on the N-terminated TiN (111) surface are depicted in Fig. 5. Fig. 5c shows that both B–B and B–H bond dissociation occur simultaneously during the energy relaxation procedure of the reaction state. In addition, hydrogen molecule (H2) desorption also occurs because two H atoms of B2H6 meet each other at the position away from preferentially adsorbed BHx species with higher binding energy. ESI Movie S1† represent the complete record for reaction process of the B2H6 on N-terminated TiN (111) surface. For more detailed description of the movie, BHx species with relatively higher binding energy than H atom are preferentially adsorbed first and two H atoms in the gas phase are desorbed as H2 apart from the adsorbed BHx species instead of adsorption on the surface.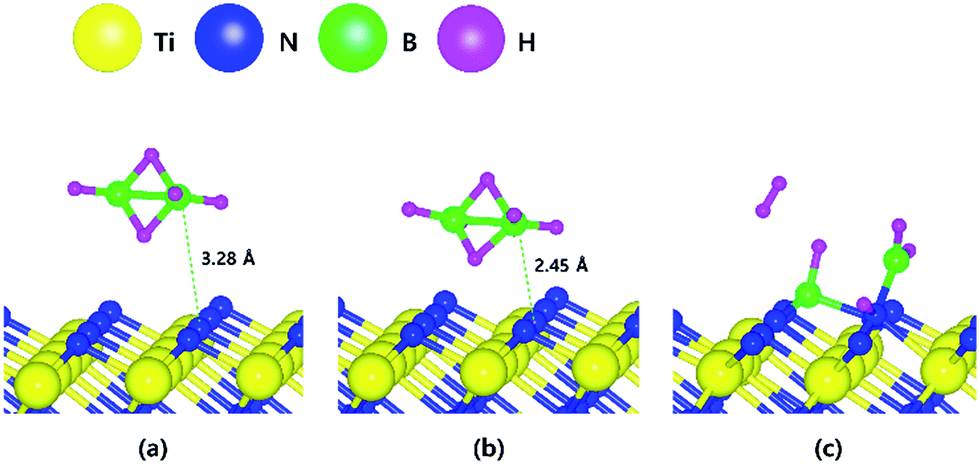 | ||
| Fig. 5 The optimized structures for (a) adsorption state, (b) transition state and (c) reaction state of a B2H6 on the N-terminated TiN (111) surface. | ||
As a result, the remaining species with boron on the surface are BH and BH2. The lowest energies for adsorption and reaction are −0.03 eV and −7.17 eV with a low energy barrier of 0.39 eV. No obvious surface reconstruction was found during the reaction. There are two more B–H bond breaking steps after the first bond breaking step, as depicted in Fig. S6 and Table S6 (ESI†). These results show that three sequential B–H bond breaking steps occur and leave the B atom bound to three N atoms as shown in Fig. S6 (ESI†).
The mechanism of B2H6 decomposition was also studied on the N-terminated TiN (111) surface to estimate the differences with the previously described TiN surfaces. Fig. 6 shows the entire energy diagram for the B2H6 decomposition on N-terminated TiN (111). We found that both B–B and B–H bond breaking on the N-terminated TiN (111) surfaces were much more facile than those of both TiN (001) and Ti-terminated TiN (111) surfaces, as shown in Fig. 2 and 4. This result is primarily because the binding energies of B and H atoms on the N-terminated TiN (111) surface are the highest among the three different TiN surfaces, as shown in Table 1. Furthermore, the decomposition of B2H6 on the surface is energetically favorable due to the downhill reactions and B–H bond breaking with very small barrier. Rather than H atoms, B adatoms would be preferably supplied by B2H6 to form the first monolayer during the B2H6 dosing process in the ALD W deposition due to the much higher binding energy of B on the surface, as shown in Table 1. In the next ALD cycle, WF6 precursor is commonly used for W deposition. A BF3 desorption process would occur on the surface because boron adatoms would react with the F atoms of WF6, therefore, a uniform W film could be deposited. Our results indicate that a specific TiN surface, such as N-terminated TiN (111), plays an important role in improving the properties of the subsequent W nucleation layers during the W ALD process.
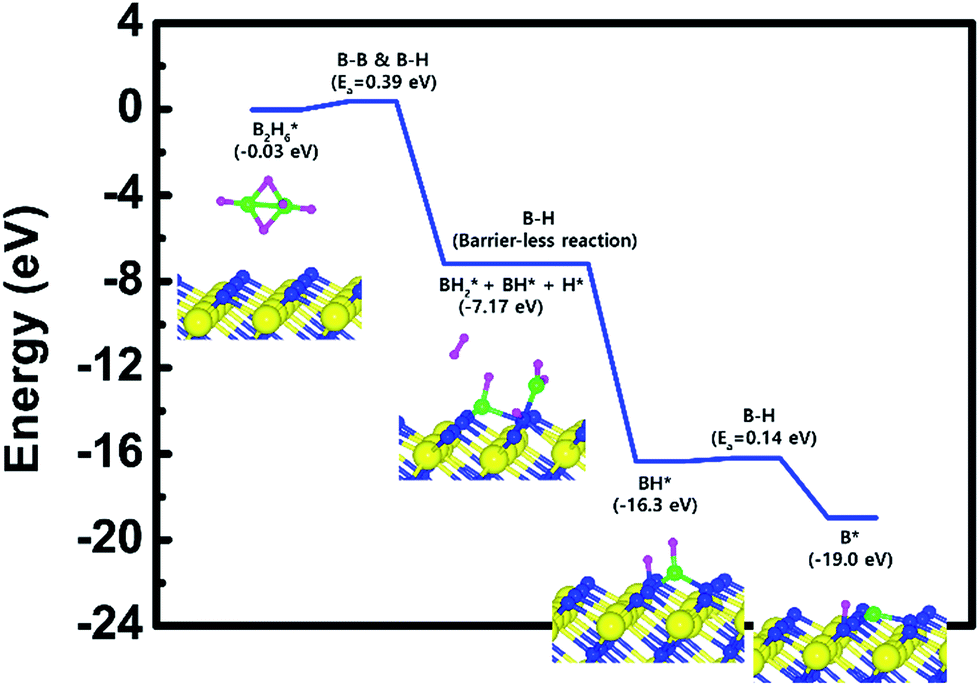 | ||
| Fig. 6 Calculated energy diagram of B2H6 decomposition on the N-terminated TiN (111) surface. | ||
3.4. Discussion
According to the above results, the N-terminated TiN (111) surface is the most efficient in depositing boron-containing layer during the B2H6 dosing process in ALD W deposition. The TiN (001) surface is unfavorable to deposit W film due to residual BH3 species on the surface and, corresponds to a reduction in the number of W sites that can be deposited. We suggest that the reason there are critical problems, such as a seam or void, in filling the W metal gate is the difference in deposition rate on three different TiN surfaces (N-terminated TiN (111) is the fastest and TiN (001) is the slowest). Thus, it is necessary to study the control of preferred orientation in TiN surfaces to enhance the W ALD process.There are several models to explain texture evolution in cubic transition-metal nitrides. Oh and Je proposed that the orientation of poly-crystalline TiN films should initially be (001) due to the lowest surface energy, and with increasing thickness, the (111) texture becomes favored due to the lower elastic modulus in the [111] direction.23,24 The change of the texture in this model is driven by the film/substrate system minimizing the total free energy.
Takeshi Kaizuka et al.30 later reported that a TiN film of (111) preferred orientation with conformal step coverage could be successfully obtained by pre-deposition of the Ti (001) layer before the CVD TiN film deposition. They said that TiN film with the (111) preferred orientation could be induced due to the lattice matching of the Ti film. The combination of their results and our results provides insight into how to design the TiN surfaces to improve the properties of the W films during the W ALD process.
4. Conclusions
We have studied B2H6 decomposition on three different TiN surfaces to understand the detailed reaction mechanisms of B2H6 during the B2H6 dosing process in ALD W deposition. This procedure is essential for depositing dense and conformal W films. In this study, we utilize density functional theory to evaluate the energetics of B2H6 decomposition for overall reactions.The overall reactions of the B2H6 with the Ti-terminated TiN (111) and N-terminated TiN (111) surfaces are energetically favorable, whereas the overall reaction for the TiN (001) is energetically unfavorable. These differences in energetics come from the difference in binding energies of B and H atoms among three different surfaces. N-terminated TiN (111) shows the lowest overall reaction energy compared with three different surfaces due to the highest binding energy of both B and H atoms with the surface, and corresponds to the most reactive surface. From the understanding of the influence of the TiN surfaces during the B2H6 dosing process, the control of the texture of TiN film is required for improvement of the W nucleation layers. These results imply that the understanding of the reactivity of the TiN surfaces gives us insight into improving the W ALD process for future memory devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Euijoon Yoon and Gun-Do Lee acknowledge support from the Supercomputing Center/Korea Institute of Science and Technology Information with supercomputing resources (KSC-2016-C3-0020), from the joint program for Samsung Electronics Co., Ltd. (SEC), from the Brain Korea 21 Plus project for SNU Materials Division for Educating Creative Global Leaders (F15SN02D1702), and from the National Research Foundation of Korea (NRF) grant funded by the Korea government (RIAM NRF-2016R1D1A1A02937045).References
- S. Bin Baek, D. H. Kim and Y. C. Kim, Appl. Surf. Sci., 2012, 258, 6341–6344 CrossRef.
- N. Sharma, M. Hooda and S. K. Sharma, J. Mater., 2014, 2014, 1–8 Search PubMed.
- B. C. Joshi, G. Eranna, D. P. Runthala, B. B. Dixit, O. P. Wadhawan and P. D. Yyas, Indian J. Eng. Mater. Sci., 2000, 7, 303–309 CAS.
- W. Huang, X. Wang, M. Sheng, L. Xu, F. Stubhan, L. Luo, T. Feng, X. Wang, F. Zhang and S. Zou, Mater. Sci. Eng., B, 2003, 98, 248–254 CrossRef.
- A. El Amrani, A. Bekhtari, B. Mahmoudi, A. Lefgoum and H. Menari, Vacuum, 2011, 86, 386–390 CrossRef CAS.
- W.-J. Lee, J.-H. Lee, C. O. Park, Y.-S. Lee, S.-J. Shin and S.-K. Rha, J. Korean Phys. Soc., 2004, 45, 1352–1355 CAS.
- J. W. Klaus, A. W. Ott, A. C. Dillon and S. M. George, Surf. Sci., 1998, 418, L14–L19 CrossRef CAS.
- C. A. Murray, S. D. Elliott, D. Hausmann, J. Henri and A. LaVoie, ACS Appl. Mater. Interfaces, 2014, 6(13), 10534–10541 CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- S. J. Won, J. R. Kim, S. Suh, N. I. Lee, C. S. Hwang and H. J. Kim, ACS Appl. Mater. Interfaces, 2011, 3, 1633–1639 CAS.
- S.-H. Kim, N. Kwak, J. Kim and H. Sohn, J. Electrochem. Soc., 2006, 153(10), 887–893 CrossRef.
- J. Klaus, S. Ferro and S. George, Thin Solid Films, 2000, 360, 145 CrossRef CAS.
- T. Luoh, C.-T. Su, T.-H. Yang, K.-C. Chen and C.-Y. Lu, Microelectron. Eng., 2008, 85, 1739 CrossRef CAS.
- G. Wang, Q. Xu, T. Yang, J. Xiang, J. Xu, J. Gao, C. Li, J. Li, J. Yan, D. Chen, T. Ye, C. Zhao and J. Luoz, ECS J. Solid State Sci. Technol., 2014, 3(4), 82–85 CrossRef.
- S.-H. Kim, S. Yeom, N. Kwak and H. Sohn, J. Electrochem. Soc., 2008, 155, D148 CrossRef CAS.
- S. Smith, K. Aouadi, J. Collins, E. Van der Vegt, M.-T. Basso, M. Juhel and S. Pokrant, Microelectron. Eng., 2005, 82, 261 CrossRef CAS.
- S.-H. Kim, J.-T. Kim, N. Kwak, J. Kim, T.-S. Yoon and H. Sohn, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2007, 25, 1574 CrossRef CAS.
- T. Omstead, G. C. D'Couto, S.-H. Lee, P. Wongsenkaum, J. Collins and K. Levy, Solid State Technol., 2002, 51, 45 Search PubMed.
- S.-H. Kim, E.-S. Hwang, S.-H. Pyi, H.-J. Sun, J.-W. Lee, J.-K. Kim, N. Kawk, H. Sohn and J. Kim, J. Electrochem. Soc., 2005, 152, C408 CrossRef CAS.
- G. Ramanath, H. Kim, H. S. Goindi, M. J. Frederick, C.-S. Shin, R. Goswami, I. Petrov and J. E. Greene, AIP Conf. Proc., 2002, 612, 10 CrossRef CAS.
- C.-H. Kim, I.-C. Rho, S.-H. Kim, Y.-S. Sohn, H.-S. Kang and H.-S. Kim, Electrochem. Solid-State Lett., 2009, 12(3), H80–H83 CrossRef CAS.
- U. Guler, A. V. Kildishev, V. M. Shalaev, A. Boltasseva, D. Stocks and G. Naik, WO Pat., PCT/US14/38561,Ref. 22: Please provide the patent type. 2014.
- U. C. Oh and J. H. Je, J. Appl. Phys., 1993, 74, 1692 CrossRef CAS.
- J. H. Je, D. Y. Noh, H. K. Kim and K. S. Liang, J. Appl. Phys., 1997, 81, 6126 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- N. Schoenberg, Acta Chem. Scand., 1954, 8, 213 CrossRef.
- D. Vanderbilt, Phys. Rev. B, 1992, 46, 6671 CrossRef.
- G. Henkelman and H. Jónsson, J. Phys. Chem., 2000, 113, 9978–9985 CrossRef CAS.
- T. Kaizuka, H. Shinriki, N. Takeyasu and T. Ohta, Jpn. J. Appl. Phys., 1994, 33, 470–474 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11291b |
| This journal is © The Royal Society of Chemistry 2017 |