
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrachyantheraoside A8, a new natural nor-oleanane triterpenoid as a kidney-type glutaminase inhibitor from Stauntonia brachyanthera†
Rong Li‡
a,
Peifeng Wei‡b,
Yue Wanga,
Ying Liua,
Xuanli Liua and
Dali Meng *a
*a
aKey Laboratory of Structure-Based Drug Design and Discovery, Ministry of Education, School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Wenhua Road 103, Shenyang 110016, PR China. E-mail: mengdl@163.com
bCollege of Pharmacy, Shaanxi University of Chinese Medicine, Xianyang 712000, China
First published on 15th November 2017
Abstract
With the aim of finding a better kidney-type glutaminase (KGA) inhibitor with potential anti-cancer properties, 18 nor-oleanane triterpenoids from Stauntonia brachyanthera, including 2 new ones, were screened against KGA. The structure-based virtual ligand screening identified 8 compounds, which might have more chance to become inhibitors of KGA with low binding energy. Glutaminase inhibition experiments and a series of cellular level assays, including cytotoxicity, wound healing, transwell invasion, western blot, and cell apoptosis, further confirmed the inhibitory effect of 5 (brachyantheraoside A8). These provided the possibility for 5 to become an inhibitor.
Introduction
Cancer is a deadly class of diseases whose mortality levels have increased every year.1 The development and progression of human cancer involve multiple steps, including sustaining proliferative signaling, evading growth suppressors, and resisting cell death.2 Why is cancer so stubborn? Studies have shown that cancer cells produce lots of energy by glycolysis and glutaminolysis rather than pyruvate oxidation. Glutaminase is a metabolic enzyme responsible for glutaminolysis, a process harnessed by cancer cells to feed their accelerated growth and proliferation.There are two kinds of main glutaminase in humans including kidney-type glutaminase (KGA) and liver-type glutaminase (LGA).3–5 KGA is often upregulated in cancer cells and touted as an attractive drug target. Recent studies have highlighted the importance of KGA as a promising molecular target for various human cancers.6–8 KGA is a good target for cancer therapy, but there are very few selective and potent KGA inhibitors available. 6-Diazo-5-oxo-l-norleucine (DON) is limited due to severe toxicity and weak millimolar inhibitory potency.9–11 Bis-2-(5-phenylacetimido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES), with low toxicity,12 is another KGA inhibitor. However, it's not a drug-like compound because of its poor solubility and low bioavailability.13 As a consequence, it's particularly necessary for us to search a natural KGA inhibitor against cancer.
Nor-oleanane triterpenoids were the main compositions of S. brachyanthera based on our previous work.14–16 Although multiple bioactivities had been confirmed,17–20 KGA inhibitory effect had been unreported for this rare kind of triterpenoids. In this study, a series of nor-oleanane triterpenoids, including two new ones were isolated from S. brachyanthera by various chromatographic methods. Their potential kidney-type glutaminase inhibitor activities were evaluated employing virtual screening, glutaminase assay, cytotoxic experiments in KGA over expressed cells, and a train of verification of cellular level assays including wound healing, transwell invasion, western blot and cell apoptosis.
Experimental
Materials and methods
The nuclear magnetic resonance (NMR) spectra were acquired using Bruker ARX-600 spectrometer (Bruker Biospin, Rheinstetten, Germany). HR-TOF-MS was measured on a micro TOFQ Bruker mass spectrometer. Chemical shifts (δ ppm) are relative to TMS (tetramethylsilane) as an internal standard. Analytical HPLC was performed on a kromasil-ODS column (4.6 × 200 mm, AkzoNobel Global, Bohus, Sweden) consisting with a refractive index detector (Shimadzu RID-10A, Kyoto, Japan). Signs of optical rotation were observed by an optical rotation detector (Jasco LC2000P, Tokyo, Japan). Preparative HPLC was equipped with an YMC-ODS column (YMC-Pack ODS-A, 250 × 20 mm, 5 μm) and a refractive index detector (Shimadzu RID-10A). Column chromatographic (CC) purifications were performed on silica gel (200–300 mesh, Qingdao Haiyang Chemical Group Corporation, Qingdao, China), as well as Sephadex LH-20 (GE Healthcare, Uppsala, Sweden), and Macroporous resin HPD100 (Cangzhou Bon adsorber Technology Co., Ltd., Cangzhou, China). D,L-Galactosamine, D-glucose, L-fucose, L-arabinose, D-xylose, and L-rhamnose were all purchased from Sigma-Aldrich (Sigma-Aldrich China, Shanghai, China). Silica gel GF254 (Qingdao Haiyang Chemical Group Corporation, Qingdao, China) were used for analytical and preparative TLC. All solvents and chemicals used were analytical grade, while other reagents were of HPLC grade or of the highest grade commercially available.Plant material
The plants of S. brachyanthera were collected in October, 2009 in Hunan Province, and were identified by Prof. Jincai Lu, School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, China. The specimen (no. HLG-0910) was deposited in the School of Traditional Chinese Material Medica, Shenyang pharmaceutical University, China.Extraction and isolation
The air-dried fruits of S. brachyanthera (7.0 kg), whose seeds were separated, were crushed into good pieces and were decocted with EtOH/H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, V/V, 35 L) under refluxing for 4 hours for 4 times, and the total extraction was retrieved after filtered. After a recuperation of the solvent from the mixture in vacuo, the final aqueous fractions (2.7 kg) were dissolved in water and passed through macroporous adsorptive resin (HPD-100), eluted seriatim with H2O, 30% EtOH, 60% EtOH and 90% EtOH, respectively. The 60% EtOH eluates (210 g) were dealt with silica gel column chromatography (CC) (1000 mm × 100 mm i.d.) with a successive CH2Cl2–MeOH system (100:1–0:100, v/v) to give eight fractions (1–8). And Comp. 9–11, 18 (3.4, 6.2, 4.3, 5.7 mg) were gained from the Fr. 4 (7:1–6:1) under the help of ODS; similarly, Fr. 5 (5:1–4:1) provided us with Comp. 4 (42.4 mg) and Comp. 5 (30.5 mg) by recrystallization; Comp. 1 (20.0 mg), 2 (21.5 mg), 3 (14.3 mg), 6 (10.3 mg), and 7 (155.5 mg) were found in Fr. 6 (3:1–2:1). And Fr. 2 (100:7–10:1) and Fr. 7 (1:1) were treated by ODS CC (500 mm × 60 mm i.d.) with MeOH/H2O (v/v, 0%, 30%, 60%, 90%, 100%, respectively) following HPLC system, supplying Comp. 8 and 12–17 (25.3, 10.2, 14.9, 35.4, 8.6, 3.9, 18.7 mg, respectively).
:3, V/V, 35 L) under refluxing for 4 hours for 4 times, and the total extraction was retrieved after filtered. After a recuperation of the solvent from the mixture in vacuo, the final aqueous fractions (2.7 kg) were dissolved in water and passed through macroporous adsorptive resin (HPD-100), eluted seriatim with H2O, 30% EtOH, 60% EtOH and 90% EtOH, respectively. The 60% EtOH eluates (210 g) were dealt with silica gel column chromatography (CC) (1000 mm × 100 mm i.d.) with a successive CH2Cl2–MeOH system (100:1–0:100, v/v) to give eight fractions (1–8). And Comp. 9–11, 18 (3.4, 6.2, 4.3, 5.7 mg) were gained from the Fr. 4 (7:1–6:1) under the help of ODS; similarly, Fr. 5 (5:1–4:1) provided us with Comp. 4 (42.4 mg) and Comp. 5 (30.5 mg) by recrystallization; Comp. 1 (20.0 mg), 2 (21.5 mg), 3 (14.3 mg), 6 (10.3 mg), and 7 (155.5 mg) were found in Fr. 6 (3:1–2:1). And Fr. 2 (100:7–10:1) and Fr. 7 (1:1) were treated by ODS CC (500 mm × 60 mm i.d.) with MeOH/H2O (v/v, 0%, 30%, 60%, 90%, 100%, respectively) following HPLC system, supplying Comp. 8 and 12–17 (25.3, 10.2, 14.9, 35.4, 8.6, 3.9, 18.7 mg, respectively).
Acid hydrolysis
The operation was carried out with a modified method reported.16,20 In brief, the solution of sample (each about 4.0 mg) is homogenized in methanol (2.5 mL), associated with 1 M H2SO4 (2.5 mL). Then the solution was heated at 90 °C for 7 h (kept sealed) and was neutralized with 1 M NaOH to give cloudy solution. An extraction with EtOAc (saturated with H2O, 5 mL × 2) was done when the mixture was cool down, of which the aqueous solution was retrieved under reduced pressure. The H2O fraction was diluted and passed through a Sep-Pak C18 cartridge, which then analyzed by HPLC under the following method: column, Capcell Pak NH2 UG80; solvent, MeCN–H2O (3:1); flow rate, 0.5 mL min−1; detection, RI and OR. The identification of D-glucose, L-arabinose, and L-rhamnose present in the polysaccharide parts were recognized by the polarities with those of authentic sample. It was detected at 589 nm on a polarimeter that the optical rotation of 0.2% (W/V) of D-glucose, L-arabinose, L-rhamnose, and L-fucose consisting with a homologous standard distilled water solution.
Cloning, protein expression, and purification of the KGA
The KGA was cloned into a pET-26 (b) vector fusion with C-terminal His tag, and the protein was expressed in Escherichia coli BL21 (DE3)-RIL-Codon plus cells grown in 6 L LB broth medium. Cells were sonicated in lysis buffer [50 mM Hepes (pH 7.5), 500 mM NaCl, 5 mM imidazole, 10% glycerol, 1 mm DTT and 0.1% (v/v) Triton X-100], and further lysed by French press. The soluble fraction was passed on to a Ni-NTA affinity column, and the bound protein was eluted with 500 mM imidazole. The protein was further purified by Superdex-200 gel filtration column and then concentrated to 20 mg mL−1 in a buffer containing 20 mM Hepes (pH 7.5), 200 mM NaCl, 10% glycerol, and 3 mM DTT.8Glutaminase assay
The enzymatic activity was measured in assay buffer containing 50 mM Tris-Acetate pH 8.6, 150 mM K2HPO4, 0.25 mM EDTA, 0.1 mg mL−1 bovine serum albumin (BSA), 1 mM DTT, 2 mM NADP+ and 0.01% Triton X-100. To measure the inhibition, the inhibitor (prepared in DMSO) was first pre-mixed with glutamine and glutamate dehydrogenase (GDH) and reactions were initiated by the addition of KGA. Final reactions contained 2 nM KGA, 10 mM glutamine, 6 units per mL GDH and 2% DMSO. Generation of NADPH was monitored by fluorescence (Ex. 340/Em. 460 nm) every minute for 15 minutes.21Cell culture and cytotoxicity assay
SW1990 and MD-MB-231, HCC1806 cells were cultured in DMEM, MEM medium (CORNING, USA). Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 air. After incubating for 24 h at 37 °C at 1 × 104 cells per well in 96-well plates (100 μL per well), isolated compounds (6.25, 12.5, 25, 50, 75 and 100 μM) were added for another 48 h. Fresh 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) solution (100 μL) was added to each well to be incubated for 4 h at 37 °C. Afterwards, the growth medium was removed and replaced with 150 μL of DMSO, after blending on a Microplate reader for 10 min. The absorbance of each well was measured at 490 nm by using a Multimode Reader and IC50 values were calculated.Wound healing assay
The anti-migration effect of 5 on HCC1806 cells were measured by wound healing migration assay as described in literature.22 A sufficient number of cells were placed on 6 well plates for 24 h. After the serum-free medium was added to cells for 4 h, cells were further incubated with various concentrations of compounds (0, 10, 20, and 30 μM, respectively) for 48 h. Finally, the sizes of wounds areas were measured.Transwell invasion assay
HCC1806 cells were cultured with serum-free MEM medium for 6 h.23 Invasion of them was assayed using transwell (Corning Costar) with 6.5 mm diameter polycarbonate filters (8 mm pore size) in 24-well culture plates. Briefly, the lower surface of the filter was coated with matrigel, and fresh medium (10% FBS) was placed in the lower wells. 1 × 105 of the cell suspension with different concentrations of compound (0, 10, 20, and 30 μM, respectively) were loaded into each of the upper wells, and the chamber was incubated at 37 °C for 24 h. The cells were filtered and stained with crystal violet. Non-invasion cells on the upper surface were removed by wiping with a cotton swab, and chemotaxis was quantified by counting cells that had migrated to the lower side of the filter.Cell apoptosis assay
To assess apoptosis, the cells were incubated with compounds at different concentrations (0, 10, 20, and 30 μM, respectively) for 24 h, and then stained with propidium iodide (PI) and FITC-conjugated Annexin V for 30 min at 4 °C in darkness. The fluorescence intensity of each dye on each cell was measured utilizing flow cytometry.Western blot assay
The cells were washed with PBS three times and lysed with RIPA lysis buffer containing protease inhibitor. Equal amounts (30 μg) of protein were separated in 10% SDS-polyacrylamide gel and blotted onto a PVDF membrane. The membrane was then blocked with 5% non-fat dry milk in TBST and incubated with various primary antibodies. After overnight incubation with primary antibody, the membrane was hybridized with HRP-conjugated secondary antibody for 1 h and washed three times with TBST. The immunoreactive bands were visualized using the ECL system.Statistical analysis
All computations were made with the statistical software of Statistical Product and Service Solutions (SPSS, version 17.0), and the level of significance was set at P < 0.05.Results
The identification of the new compounds
Compound 4, white amorphous powder (MeOH). It was assigned as C50H78O19 on the basis of HR-ESI-MS with a quasi-molecular iron peak at [M + K + H]2+: m/2z 511.2392 (calcd. 511.2421). The 1H and 13C NMR data (Table 1) showed that 1 had the same aglycone as that of brachyantheraoside A2.18 The comparison of the NMR data of aglycone of 1 with those of brachyantheraoside A2 suggested that the differences between them were the chemical shifts of C-3 and C-28, in which the hydroxyl at C-3 in brachyantheraoside A2 [δH 3.44 (1H, s), δC 78.3] was substituted by a glycoside bond in 1 [δH 3.31 (1H, dd, J = 11.5, 3.7 Hz), δC 88.6], and the ester glycoside bond at C-28 in brachyantheraoside A2 (δC 175.9) was replaced by a carboxylic acid in 1 (δC 180.0). The sugar units at C-3 were determined as three L-arabinose and one L-rhamnose after the acid hydrolysis of 4 by direct HPLC analysis of the hydrolysate using refractive index (RI) and optical rotation (OR) detectors. The linkages of the sugar units were established on the basis of HMBC correlations between H-1′′′ (δ 5.00) of the terminal arabinose and C-4′′ (δ 79.4) of the middle arabinose, H-1′′ (δ 4.97) of the middle arabinose and C-3′ (δ 81.3) of the inner arabinose, H-1′ (δ 4.92) of the inner arabinose and C-3 (δ 88.6) of aglycone, the H-1′′′′ (δ 6.07) of the rhamnose and the C-2′ (δ 74.9) of the inner arabinose. The α-anomeric configurations for the arabinopyranosyl moieties were determined from their coupling constants [J = 5.2 (ara), 6.7 (middle-ara′), 7.4 Hz (terminal-ara′′), respectively]. The configuration of L-rhamnose was determined to be α-form from its 13C NMR chemical shifts of C-3 (δ 73.0) and C-5 (δ 70.1).24 Therefore, the structure of 4 was determined as 3-O-α-L-arabinopyranosyl-(1 → 4)-α-L-arabinopyranosyl-(1 → 3)-α-L-arabinopyranosyl-[α-L-rhamnopyranosyl-(1 → 2)]-30-noroleana-12, 20 (29)-dien-28-oic acid (Fig. 1), which was named as brachyantheraoside A7.| Pos. | 4 | 5 | Pos. | 4 | 5 | ||||
|---|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | ||
| 1 | 0.95, 1.46 | 39.3 | 0.92, 1.47 | 39.3 | 3-ara (inner) | 3-ara (inner) | 3-ara (inner) | 3-ara (inner) | |
| 2 | 1.86, 2.09 | 26.9 | 1.79, 2.00 | 26.9 | 1′ | 4.92, d (5.2) | 105.0 | 4.96, d (4.7) | 104.7 |
| 3 | 3.31, dd (11.5, 3.7) | 88.6 | 3.28, dd (11.7, 4.1) | 88.6 | 2′ | 4.14 | 74.9 | 4.50 | 72.5 |
| 4 | 40.1 | 39.9 | 3′ | 4.28 | 81.3 | 4.63 | 74.7 | ||
| 5 | 0.84 | 56.3 | 0.77 | 56.2 | 4′ | 4.46 | 68.4 | 4.37 | 79.0 |
| 6 | 1.31, 1.47 | 18.8 | 1.31, 1.44 | 18.8 | 5′ | 3.97, 4.26 | 65.2 | 4.35, 4.49 | 62.9 |
| 7 | 1.30, 1.50 | 33.5 | 1.31, 1.47 | 33.4 | 6′ | 4.92, d (5.2) | |||
| 8 | 39.9 | 40.2 | 3-ara′ (middle) | 3-ara′ (middle) | 3-ara′ (middle) | 3-ara′ (middle) | |||
| 9 | 1.62, d (6.2) | 48.4 | 1.60 | 48.3 | 1′′ | 4.97, d (6.7) | 104.9 | 5.09, d (6.2) | 104.3 |
| 10 | 37.4 | 37.3 | 2′′ | 4.36 | 73.1 | 4.50 | 75.5 | ||
| 11 | 1.86, 2.00 | 24.1 | 2.10, 2.15 | 23.8 | 3′′ | 4.62 | 74.3 | 4.20 (1H, m) | 74.3 |
| 12 | 5.49 | 123.3 | 5.44, brs | 123.5 | 4′′ | 4.37 | 79.4 | 4.38 | 68.9 |
| 13 | 144.6 | 143.8 | 5′′ | 3.78, 4.47 | 66.4 | 3.78, 4.36 | 66.4 | ||
| 14 | 42.5 | 42.4 | 6′′ | ||||||
| 15 | 1.21, 2.00 | 28.7 | 1.16, 2.32 | 28.6 | 3-ara′′ (terminal) | 3-ara′′ (terminal) | 3-rha | 3-rha | |
| 16 | 2.04, 2.16 | 24.2 | 1.60, 1.85 | 24.1 | 1′′′ | 5.00, d (7.4) | 107.7 | 6.02 | 102.4 |
| 17 | 47.4 | 47.7 | 2′′′ | 4.46 | 73.4 | 4.58, dd (9.3, 3.1) | 72.9 | ||
| 18 | 3.25, dd (13.3, 4.4) | 48.3 | 3.13, dd (13.3, 4.8) | 47.8 | 3′′′ | 4.63 | 75.1 | 4.72 | 72.7 |
| 19 | 2.16, 2.27 | 42.4 | 2.17, 2.56 | 42.0 | 4′′′ | 4.25 | 70.5 | 4.12 | 74.2 |
| 20 | 149.7 | 148.7 | 5′′′ | 3.73, 4.26 | 67.8 | 4.53 | 70.6 | ||
| 21 | 2.18, 2.28 | 30.3 | 2.07, 2.19 | 30.0 | 6′′′ | 1.65, d (6.2) | 18.9 | ||
| 22 | 1.96, 2.10 | 38.8 | 1.74, 2.00 | 38.0 | 3-rha | 3-rha | 28-glc (inner) | 28-glc (inner) | |
| 23 | 1.21, s | 28.4 | 1.17 | 28.5 | 1′′′′ | 6.07 | 102.5 | 6.24, d, (8.2) | 96.1 |
| 24 | 1.09, s | 17.3 | 1.08 | 17.3 | 2′′′′ | 4.57 | 72.8 | 4.12 | 74.2 |
| 25 | 0.81, s | 15.9 | 0.87 | 16.0 | 3′′′′ | 4.73 | 73.0 | 4.37 | 79.0 |
| 26 | 0.96, s | 17.9 | 1.09 | 17.8 | 4′′′′ | 4.27 | 74.1 | 4.22 | 71.8 |
| 27 | 1.28, s | 26.5 | 1.24 | 26.3 | 5′′′′ | 4.23 | 70.1 | 4.08 | 78.2 |
| 28 | 4.76, 4.81 | 180.0 | 176.1 | 6′′′′ | 1.63, d (6.2) | 19.0 | 4.33, 4.70 | 69.8 | |
| 29 | 107.5 | 4.73, brs, 4.66, brs | 107.6 | 28-glc′ (terminal) | 28-glc′ (terminal) | ||||
| 30 | 1′′′′′ | 5.03, d (7.5) | 105.7 | ||||||
| 2′′′′′ | 4.03 | 75.5 | |||||||
| 3′′′′′ | 4.20 | 78.8 | |||||||
| 4′′′′′ | 4.33 | 71.2 | |||||||
| 5′′′′′ | 3.89 | 78.7 | |||||||
| 6′′′′′ | 3.81, 4.31 | 64.5 | |||||||
| –COCH3 | |||||||||
 | ||
| Fig. 1 The structures of 1–18 compunds. | ||
Compound 5, white amorphous powder (MeOH). Its molecular was determined as C57H90O25 based on quasi-molecular iron peak at [M + Na]+: m/z 1197.5848 (calcd. for 1197.5771) from HR-EST-MS. The 1H and 13C NMR data (Table 1) showed that 5 had the same aglycone as 4. While, the down-field shifted signal at δC 176.1 of C-28 suggested the linkage of a sugar moiety to the carboxylic acid. Its sugar units were determined as two L-arabinose, two D-glucose, and one L-rhamnose in the molecular after the acid hydrolysis of 2 by direct HPLC analysis of the hydrolysate using RI and OR detectors. The linkages of the sugar units were established on the basis of HMBC correlations between the H-1′′′ (δ 6.02) of the terminal L-rhamnose and the C-2′′ (δ 79.0) of the middle L-arabinose, the H-1′′ (δ 5.09) of the middle L-arabinose and the C-4′ (δ 79.0) of the inner L-arabinose, the H-1′ (δ 4.96) of the inner L-arabinose and the C-3 (δ 88.6) of the aglycon, H-1′′′′′ (δ 5.03) of the terminal glucose and the C-6′′′′ (δ 69.8) of the inner glucose, the H-1′′′′ (δ 6.24) of the inner glucose and the C-28 (δ 176.2) of the aglycon. The α-anomeric configuration for the arabinoses and the β-anomeric configuration for the glucoses were determined from their coupling constants [J = 8.2 Hz (glc), 7.5 Hz (glc′), 4.7 Hz (ara), 6.2 Hz (ara′), respectively], while the α-anomeric configuration for L-rhamnose was determined from its chemical shifts of C-3 (δ 72.7) and C-5 (δ 70.6).24 All of the data above led to the formulation of 5 as 3-O-α-L-rhamnopyranosyl-(1 → 2)-α-L-arabinopyranosyl-(1 → 4)-α-L-arabinopyranosyl-30-norolean-12, 20 (29)-dien-28-oic acid β-D-glucopyranosyl-(1 → 6)-β-D-glucopyranosyl ester (Fig. 1), which was given the name of brachyantheraoside A8.
By comparing their physical and spectroscopic data with those of the literatures reported, the rest compounds were identified as 3-O-β-D-glucopyranosyl-(1 → 3)-α-L-rhamnopy-ranosyl-(1 → 2)-α-L-arabinopyranosyl-30-norolean-12,20 (29)-dien-28-oic acid,25 brachyantheraoside A5,26 yemuoside YM11,27 yemuoside YM9,28 brachyantheraoside A2,18 brachyantheraoside A4,29 brachyantheraoside B9,18 brachyantheraoside D1,19 brachyanthera acid,18 brachyantheraoside B3,30 brachyantheraoside B6,18 brachyantheraoside C2, brachyantheraoside B1,30 brachyantheraoside B8,30 eupteleasaponin VIII,31 brachyanthera acid B.19
Virtual screening and glutaminase assay
The structure-based virtual ligand screening was showed in Table 2. There were eight compounds exhibited inhibitory activity towards KGA (mf Score values: −233 to −167; KGA IC50 values: 95.04 to 6.10) to varying degrees and the others had no activity. Comp. 5 was the highest binding affinity to KGA with the most negative free binding energy (mf Score value = −233). The mf Score values of 12 and 13 were −21 and −59, which represented low binding affinities to KGA. In the enzyme inhibition assay, three compounds, 1, 4, 5, expressed significant inhibitory effects to KGA, with the IC50 values of 10.97, 15.38, and 6.10, respectively.| No. | mf Scroe (kcal mol−1) | KGA IC50 (μM) | IC50 (μM) | ||
|---|---|---|---|---|---|
| HCC1806 | MD-MB-231 | SW1990 | |||
| 1 | −212 | 10.97 ± 0.69 | 29.72 ± 1.41 | 27.73 ± 2.51 | 28.05 ± 1.10 |
| 2 | −205 | 29.67 ± 1.21 | >100 | >100 | >100 |
| 3 | −207 | 32.18 ± 0.95 | >100 | >100 | >100 |
| 4 | −208 | 15.38 ± 1.44 | 78.65 ± 2.55 | 97.33 ± 2.91 | 86.75 ± 2.49 |
| 5 | −233 | 6.10 ± 0.57 | 24.94 ± 1.43 | 29.23 ± 0.37 | 30.70 ± 1.48 |
| 6 | −194 | 47.83 ± 2.01 | >100 | >100 | >100 |
| 7 | −175 | 80.51 ± 1.30 | >100 | >100 | >100 |
| 8 | −167 | 95.04 ± 2.31 | >100 | >100 | >100 |
| 9 | −161 | >100 | — | — | — |
| 10 | −159 | >100 | — | — | — |
| 11 | −134 | >100 | — | — | — |
| 12 | −21 | >100 | — | — | — |
| 13 | −59 | >100 | — | — | — |
| 14 | −159 | >100 | — | — | — |
| 15 | −154 | >100 | — | — | — |
| 16 | −152 | >100 | — | — | — |
| 17 | −142 | >100 | — | — | — |
| 18 | −142 | >100 | — | — | — |
Cell experiments
The cytotoxic activities of eight compounds, which exhibited better effects from virtual screening and glutaminase assay, were further evaluated on three different human tumour cell lines that were all KGA over expressed (HCC1806, MD-MB-231, SW1990 cell lines) (American Type Culture Collection, Rockville, MD, USA) employing MTT assay (Table 2). It could be found that 1, 4, and 5 could decrease the cells viabilities in different degree. Among them, 5 expressed stronger antitumor activity, especially for HCC1806 cell (IC50 value = 24.94 μM). As a result, it was selected to perform the following test to confirm its inhibitory effect on KGA.The effects of 5 on HCC1806 cells migratory ability were evaluated using a wound healing assay. Wound healing rates of four kinds of drug concentration (0, 10, 20, 30 μM) were 84.93%, 57.89%, 47.65%, and 22.82%, respectively, indicating that the cell migration ability was significantly inhibited when 5 (30 μM) was added to cell (Fig. 2A and B).
 | ||
| Fig. 2 Effect of brachyantheraoside A8 (5) on HCC1806 cell migration and invasion. (A) Comp. 5 inhibited HCC1806 cells migration. (B) The migration ability in HCC1806 cells treated with 5 were analyzed by wound healing assay. (C) Microscope images of invading HCC1806 cells that have migrated through the Matrigel induced by 0, 10, 20, and 30 μM of 5. (D) Cell invasion assay result of 5 against HCC1806 cells. *P < 0.05 represents significant differences compared with control. | ||
It is well commonly accepted that the tumor invasion of basement membranes is one of the crucial steps in the complex multistep event resulting to the successful formation of a metastasis. Therefore, the transwell invasion assay for HCC1806 cells was studied. Similarly, the result revealed that the number of cell migration decreased obviously after treated with 5 (10, 20, and 30 μM, Fig. 2C and D). Compared with control group, inhibition ratios reached to 53.76%, 85.11%, and 96.39%, respectively, which suggested the anti-migratory activity of 5 was in a concentration-dependent manner.
Based on the above results, annexin V and PI staining were performed to distinguish early and late apoptotic cells. The rate of apoptosis was increased after treatment with 5 for 48 h (Fig. 3). The rates of apoptosis in HCC1806 cells treated with 10, 20, and 30 μM of 5 were 1.63%, 3.53%, and 5.89%, respectively, and were higher than the control cells (0.45%).
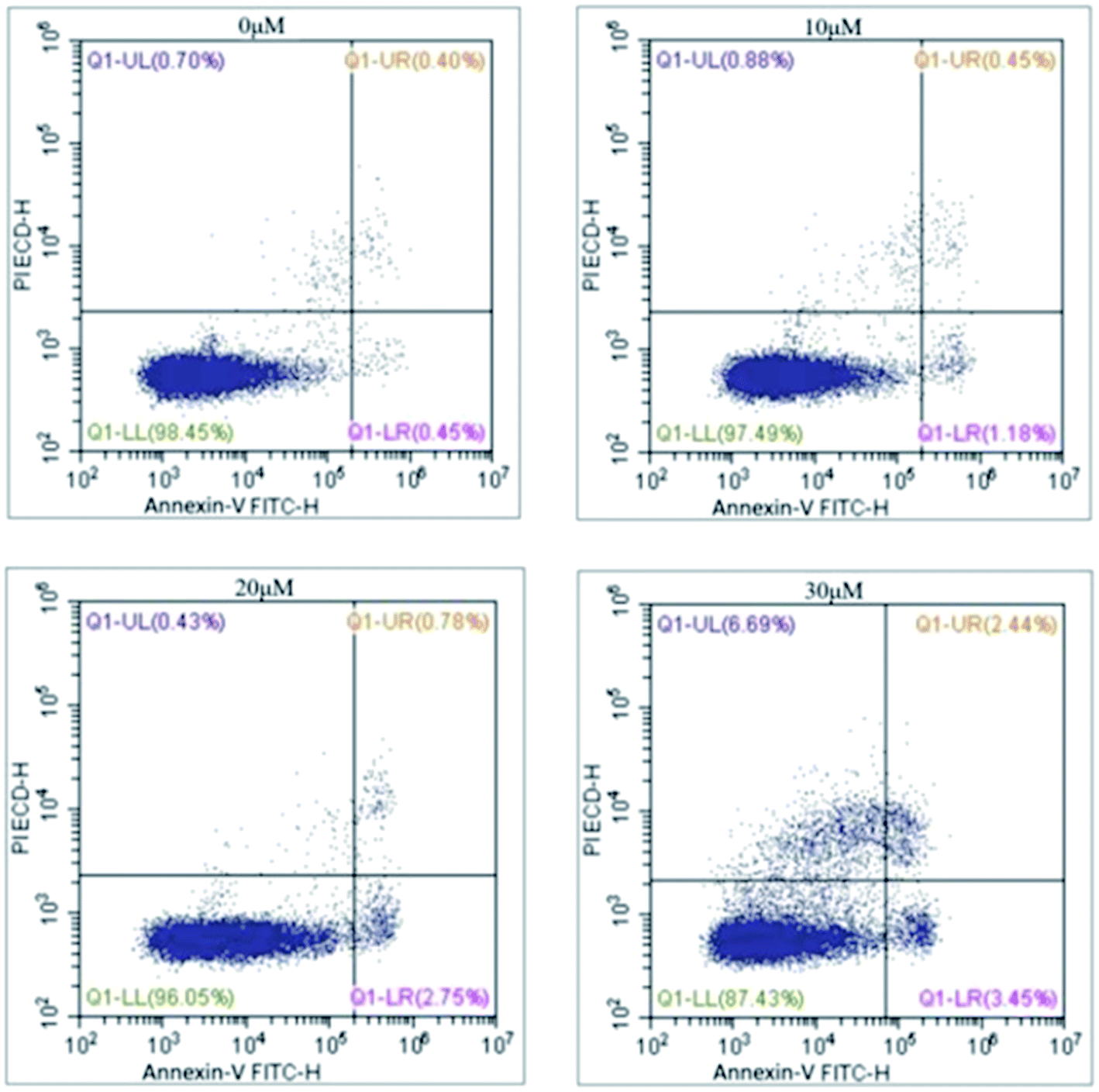 | ||
| Fig. 3 Comp. 5 induced apoptosis in dose-dependent manner. | ||
Meanwhile, Bcl-2 family proteins were further investigated in HCC1806 cells to track the signal factor associated with apoptosis. The increasing expression of Bax and decreasing expression of Bcl-2 were found in the drug group compared with the control group in current results. At the same time, the ratio of Bax to Bcl-2 was also significantly enhanced (Fig. 4).
 | ||
| Fig. 4 Western blot analysis of Bcl-2 and Bax in HCC1806 cell treated with different concentration of 5 for 48 h. β-actin was used as internal control. | ||
Discussion
Although surgical resection and chemotherapy can cure restricted primary tumors, metastatic disease is largely incurable because of its systemic nature and the resistance of spreading tumor cells to existing therapeutic agents. Cancer cells have an increasing reliance on glycolysis for their bioenergetic and biosynthetic requirements, which is always coupled with an up-regulated amount of KGA.32,33 This is because glutamine can make meets to tumor cells. Whereas, one vital step in the utilization of glutamine is its transformation to glutamate by the mitochondrial enzyme, glutaminase KGA. Suppression of the broadly expressed KGA with inhibitors can be a potential method to kill tumors. Plant-based agents have played an important role in the treatment of cancers and natural products have been considered to be an important source for antitumor drug discovery and development.34 Therefore, it's essential to seek a natural KGA inhibitor.In this study, a series of nor-oleanane triterpenoids was screened via structure-based virtual ligand. It could be found that the mf Score values of nor-oleanane triterpenoid saponins were much lower than those of others. That's to say, nor-oleanane triterpenoid saponins had potential inhibitory activity to KGA. On the other hand, it could be considered that acetyl might be the most suspicious functional groups, which made 12 and 13 with low mf Score values. The enzyme inhibition experiments and cell assays further confirmed inhibitory effects of nor-oleanane triterpenoid saponins on KGA, showing that they could be potent inhibitors of KGA and might be used as leading compounds to develop new anti-tumor agents. In the glutaminase assay, various of nor-oleanane triterpenoids had demonstrated different levels of inhibitory to KGA. However, when the double bond in C-29 was replaced by hydroxyl, the inhibitory effects will decrease dramatically, which were also found in the cytotoxicity assay, indicating the importance of substitution of a double bond in C-29 to the effect.
The inhibitory effects of compounds against HCC1806, MD-MB-231, SW1990, cells were further investigated using MTT method. These three kinds of cells were inducing over-expression of KGA, so they were chosen to judge the correlation of compounds towards KGA. HCC1806 cell activity was severely inhibited by 5 with the IC50 value of 6.10 μM. Further study verified that 5 exerted robust cytotoxic effects in three cells, as evidenced by the reduction in cell viability, inhibition of cell colonization and migratory abilities. The migration and invasive abilities of cancer are the initial steps for tumor metastasis. From Fig. 2 and 3, compared with control group, wound healing rate was slowed to 22.82%, and inhibition ratio reached to 96.39%, respectively, which obviously indicated that 5 (30 μM) significantly suppressed the migration and invasion of HCC1806 cells in vitro.
Previous studies have shown that tumorigenesis is associated with malfunction of cell apoptosis.35 However, apoptosis is one of the critical mechanisms of the anti-tumor agents. It had been also observed that the apoptosis rate increased in a dose-dependent manner in the HCC1806 cell lines after 5 was added. Tumor cell apoptosis has been regulated by a series of apoptosis-related proteins. Bcl-2 family proteins play an important role in the regulation of the mitochondria-mediated pathway of apoptosis. Bcl-2 family proteins include anti-apoptotic such as Bcl-2 and Bcl-xl and proapoptotic proteins containing Bax and Bid. Bax and Bcl-2 are the major factors that control the apoptosis.36 The ratio of Bax to Bcl-2 was significantly enhanced from Fig. 3. The results indicated that 5-induced cell death is under control of Bax/Bcl-2.
In summary, brachyantheraoside A8 (5) significantly inhibited the growth of HCC1806 cell and induced apoptosis through the modulating Bax/Bcl-2 ratio in a dose-dependent manner. Thus, comp. 5 might be an effective drug against HCC1806, and further investigation including in vivo evaluation need to be carried out. In the current, breast cancer and pancreatic cancer threatened human health all the time. Therefore, this studies contributed to the development of human health.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81073154, 81374061, 81573694), the Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University, and the Program for Innovation team of Liaoning province (Grant No. LT2015027).References
- A. C. Mamede, S. D. Tavares, A. M. Abrantes, J. Trindade, J. M. Maia and M. F. Botelho, The role of vitamins in cancer, Nutr. Cancer, 2011, 63, 479–494 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 2011, 35, 646–674 CrossRef PubMed.
- M. Watford, V. Chellaraj, A. Ismat, P. Brown and P. Raman, Hepatic glutamine metabolism, Nutrition, 2002, 18, 301 CrossRef CAS PubMed.
- B. I. Labow, W. W. Souba and S. F. Abcouwer, Mechanisms governing the expression of the enzymes of glutamine metabolism-glutaminase and glutamine synthetase, J. Nutr., 2001, 131, 2467–2474 Search PubMed.
- J. C. Aledo, P. M. Gómez-Fabre, L. Olalla and J. Márquez, Identification of two human glutaminase loci and tissue-specific expression of the two related genes, Mamm. Genome., 2000, 11, 1107 CrossRef CAS PubMed.
- P. Gao, I. Tchernyshyov, T. C. Chang, Y. S. Lee, K. Kita, T. Ochi, K. I. Zeller, A. M. De Marzo, J. E. Van Eyk, J. T. Mendell and C. V. Dang, c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism, Nature, 2009, 458, 762–765 CrossRef CAS PubMed.
- J. B. Wang, J. W. Erickson, R. Fuji, S. Ramachandran, P. Gao, R. Dinavahi, K. F. Wilson, A. L. B. Ambrosio, S. M. G. Dias, C. V. Dang and R. A. Cerione, Targeting mitochondrial glutaminase activity inhibits oncogenic transformation, Cancer Cell, 2010, 18, 207–219 CrossRef CAS PubMed.
- K. Thangavelua, C. Q. Pan, T. Karlberg, G. Balaji, M. Uttamchandani, V. Suresh, H. Schüler, B. C. Low and J. Sivaraman, Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7705–7710 CrossRef PubMed.
- R. A. Shapiro, V. M. Clark and N. P. Curthoys, Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site, J. Biol. Chem., 1979, 254, 2835–2838 CAS.
- K. Thangavelu, Q. Y. Chong, B. C. Low and J. Sivaraman, Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA), Sci. Rep., 2014, 4, 3827 CrossRef CAS PubMed.
- R. Catane, D. D. Von Hoff, D. L. Glaubiger and F. M. Muggia, Azaserine, DON, and azotomycin: three diazo analogs of L-glutamine with clinical antitumor activity, Cancer Treat. Rep., 1979, 63, 1033–1038 CAS.
- Y. Xiang, Z. E. Stine, J. Xia and C. V. Dang, Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis, J. Clin. Invest., 2015, 125, 2293–2306 Search PubMed.
- K. Shukla, D. V. Ferraris, A. G. Thomas, M. Stathis and B. Duvall, Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors, J. Med. Chem., 2012, 55, 10551–10563 CrossRef CAS PubMed.
- L. Wang, Q. Zhu, D. L. Meng, N. Li and X. Li, Isolation and identification of chemical constituents from peels of Stauntonia brachyanthera Hand.-Mazz, J. Shenyang Pharm. Univ., 2012, 29, 272–275 CAS.
- C. Chen, Z. X. Li, D. L. Meng, N. Li and X. Li, Isolation and identification of chemical constituents from stems of Stauntonia brachyanthera Hand-Mazz, J. Shenyang Pharm. Univ., 2011, 28, 419 Search PubMed.
- C. Chen and D. L. Meng, Chemical constituents from Stauntonia brachyanthera Hand-Mazz, Biochem. Syst. Ecol., 2013, 48, 182–185 CrossRef CAS.
- Z. Wang, S. M. Mei, L. Zhen and C. Y. Xiao, Pharmacognostical study of Dong nationality drug Stauntonia brachyanthera, Chin. J. Ethnomed. Ethnopharm., 2007, 4, 031 Search PubMed.
- D. L. Meng, L. H. Xu, C. Chen, D. Yan, Z. Z. Fang and Y. F. Cao, A new resource of hepatic protectant, nor-oleanane triterpenoid saponins from the fruit of Stauntonia brachyanthera, J. Funct. Foods, 2015, 16, 28–39 CrossRef CAS.
- X. L. Liu, S. Li and D. L. Meng, Anti-gout nor-oleanane triterpenoids from the leaves of Stauntonia brachyanthera, Bioorg. Med. Chem. Lett., 2016, 26, 2874–2879 CrossRef CAS PubMed.
- J. Zhao, J. Guo, Y. Zhang, D. L. Meng and Z. Sha, Chemical constituents from the roots and stems of Stauntonia brachyanthera Hand-Mazz and their bioactivities, J. Funct. Foods, 2015, 14, 374–383 CrossRef CAS.
- M. I. Gross, S. D. Demo, J. B. Dennison, L. Chen, T. Chernov-Rogan, B. Goyal, J. R. Janes, G. J. Laidig, E. R. Lewis, J. Li, A. L. MacKinnon, F. Parlati, M. L. M. Rodriguez, P. J. Shwonek, E. B. Sjogren, T. F. Stanton, T. T. Wang, J. F. Yang, F. Zhao and M. K. Bennett, Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer, Mol. Cancer Ther., 2014, 13, 890–901 CrossRef CAS PubMed.
- A. Valster, N. L. Tran, M. Nakada, M. E. Berens, A. Y. Chan and M. Symons, Cell migration and invasion assays, Methods, 2005, 37, 208–215 CrossRef CAS PubMed.
- A. C. Tsai, S. L. Pan, H. L. Sun, C. Y. Wang, C. Y. Peng and S. W. Wang, CHM-1, a New Vascular Targeting Agent, Induces Apoptosis of Human Umbilical Vein Endothelial Cells via p53-mediated Death Receptor 5 Up-regulation, J. Biol. Chem., 2010, 285, 5497–5506 CrossRef CAS PubMed.
- R. Kasai, M. Okihara, J. Asakawa, K. Mizutani and O. Tanaka, 13C NMR study of α-and β-anomeric pairs of d-mannopyranosides and l-rhamnopyranosides, Tetrahedron, 1979, 35, 1427–1432 CrossRef CAS.
- H. B. Wang, D. Q. Yu and X. T. Liang, Structures of two nortriterpenoid saponins from Stauntvnia Chinensis, J. Nat. Prod., 1990, 53, 313–318 CrossRef CAS.
- C. Nathan and A. Ding, Nonresolving inflammation, Cell, 2010, 140, 871–882 CrossRef CAS PubMed.
- H. B. Wang, D. Q. Yu, X. T. Liang, N. Watanab, M. Tamai and S. Omura, Yemuoside YM7, YM11, YM13, and YM14: Four nortriterpenoid saponins from Stauntonia chinensis, Planta Med., 1989, 55, 303–306 CrossRef CAS PubMed.
- Z. Wang, S. M. Mei, L. Zhen and C. Y. Xiao, The preparation method of Stauntonia brachyanthera Hand-Mazz vinegar juice, Chinese Patent, CN101376872A, 2009.
- S. U. Nigwekar and S. S. Waikar, Diuretics in Acute Kidney Injury, Semin. Nephrol., 2011, 31, 523–534 CrossRef CAS PubMed.
- D. Liu, D. D. Wang, W. Yang and D. L. Meng, Potential anti-gout constituents as xanthine oxidase inhibitor from the fruits of Stauntonia brachyanthera, Bioorg. Med. Chem., 2017, 25, 3562–3566 CrossRef CAS PubMed.
- T. Murakami, H. Oominami, H. Matsudo and M. Yoshikawa, Bioactive saponins and glycosides. XVIII. Nortriterpene and triterpene oligoglycosides from the fresh leaves of Euptelea polyandra Sieb. et Zucc. (2): Structures of eupteleasaponins VI,VI acetate,VII,VIII, IX, X, XI, and XII, Chem. Pharm. Bull., 2001, 49, 741–746 CrossRef CAS PubMed.
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- R. J. DeBerardinis, A. Mancuso, E. Daikhin, I. Nissim, M. Yudkoff, S. Wehrli and C. B. Thompson, Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19345 CrossRef CAS PubMed.
- R. N. Kharwar, A. Mishra, S. K. Gond, A. Stierle and D. Stierle, Anticancer compounds derived from fungal endophytes: their importance and future challenges, Nat. Prod. Rep., 2011, 28, 1208–1228 RSC.
- E. Z. Bagci, S. M. Sen and M. C. Camurdan, Analysis of a mathematical model of apoptosis: individual differences and malfunction in programmed cell death, J. Clin. Monit. Comput., 2013, 27, 465–479 CrossRef PubMed.
- H. Jiang, P. J. Zhao, D. Su, J. Feng and S. L. Ma, Paris saponin I induces apoptosis via increasing the Bax/Bcl-2 ratio and caspase-3 expression in gefitinib-resistant non-small cell lung cancer in vitro and in vivo, Mol. Med. Rep., 2014, 9, 2265–2272 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11270j |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |