
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected cleavage of thiacalix[4]arene sulfoxides†
J. Mikšátkoa,
V. Eignerb and
P. Lhoták *a
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technicka 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz; Fax: +420-220444288; Tel: +420-220445055
bSolid State Department, UCTP, 166 28 Prague 6, Czech Republic
First published on 21st November 2017
Abstract
Thiacalix[4]arenes having one bridge oxidized to a sulfoxide moiety can react with organolithium reagents to form cleaved structures which are otherwise difficult to access. Cleavage of the macrocyclic skeleton is independent of the starting conformation as proven by the same product obtained from the corresponding cone, partial cone or 1,3-alternate derivatives. Quenching of the crude reaction mixture with D2O allowed elucidation of the mechanism which is based on the ligand exchange of the sulfoxide functionality. The general applicability of this reaction was demonstrated using the corresponding distal or proximal disulfoxide derivatives, while the quenching of the reaction mixture using various electrophiles allowed the isolation of the corresponding mono- and di-substituted oligomers. The structure of the unexpected product (a tetrameric derivative) was assigned by single crystal X-ray crystallography.
Introduction
Thiacalix[4]arenes, since their first appearance1 in 1997, have been attracting increasing attention as potential candidates for building blocks and/or molecular scaffolds due to their similarity with the much more well established calix[n]arene family.2 The introduction of sulfur atoms in place of the methylene bridges results in novel features or properties3 that are impossible (or very difficult) in classical calixarene chemistry. Thus, the stronger directing effects of sulfur compared to the CH2 group offer unusual regioselectivity in the electrophilic aromatic substitution of thiacalix[4]arenes as documented by direct meta nitration4 or formylation5 (with respect to phenolic oxygens). Moreover, the regio- and stereo-selective S-alkylation to sulfonium salts6 and chemoselective S-oxidation to various sulfoxides or sulfones7 represent other unique opportunities inaccessible to the more common calixarene counterpart.Recently, we studied the partial S-oxidation of thiacalix[4]arenes using NaBO3·4H2O as the oxidizing agent.8 Interestingly, despite the possible formation of many regio- and stereoisomers, the reactions leading to mono- or disulfoxides were highly regio- and stereoselective depending on the starting conformation used.9
Synthetic availability of the monosulfoxide derivatives led us to attempt further exploration in the derivatization of thiacalix[4]arenes. The sulfoxide is a versatile moiety which has found numerous applications in organic chemistry, inter alia in ortho-directed metallations of aryl sulfoxides. As shown in Scheme 1, the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality is a powerful director in ortho-lithiation10 (or ortho-magnesiation11) which could be used for the introduction of an electrophile moiety into the thiacalixarene skeleton. In this paper we report on our unexpected, yet fortuitous, efforts in the derivatization of the thiacalixarene system.
O functionality is a powerful director in ortho-lithiation10 (or ortho-magnesiation11) which could be used for the introduction of an electrophile moiety into the thiacalixarene skeleton. In this paper we report on our unexpected, yet fortuitous, efforts in the derivatization of the thiacalixarene system.
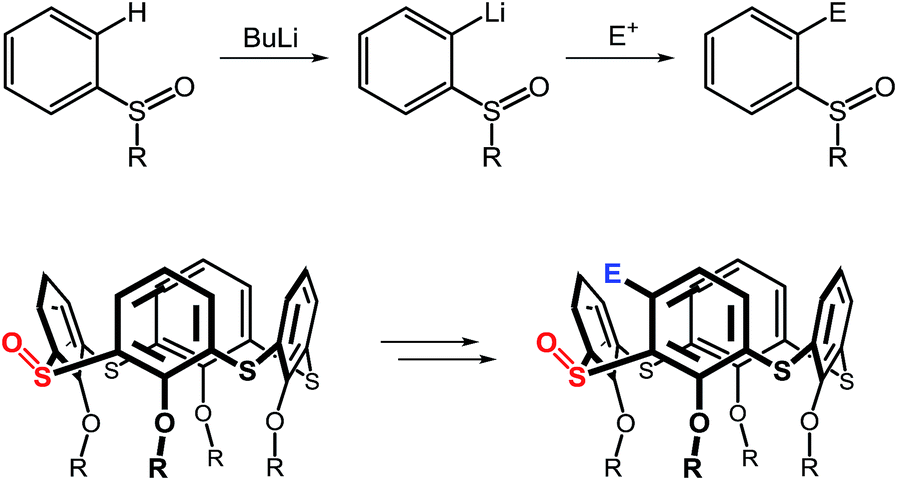 | ||
| Scheme 1 ortho-Lithiation of sulfoxides and possible application in thiacalix[4]arene chemistry. | ||
Results and discussion
The starting monosulfoxides 1–3 were obtained as recently described9 in good yields using the corresponding thiacalixarene conformations and NaBO3·4H2O as the oxidizing agent (CHCl3/AcOH, 60 °C, 4 h). Sulfoxide 1, immobilized in the cone conformation, was subjected to ortho-lithiation reaction (Scheme 2) with n-BuLi (10 equiv.) in THF at −78 °C and after one hour the corresponding electrophile (I2 or DMF) was added. To our surprise, both reactions yielded the same product with no trace of electrophile within the molecule. Instead, we isolated the linear tetramer 4 (90% yield) resulting from opening of the thiacalixarene skeleton with the SO functionality being cleaved off. To evaluate the influence of the starting conformation on the unexpected course of the reaction we carried out the same reaction sequence with the partial cone 2 and the 1,3-alternate 3. In both cases the same product 4 was obtained in high yields (92% and 91%, respectively).
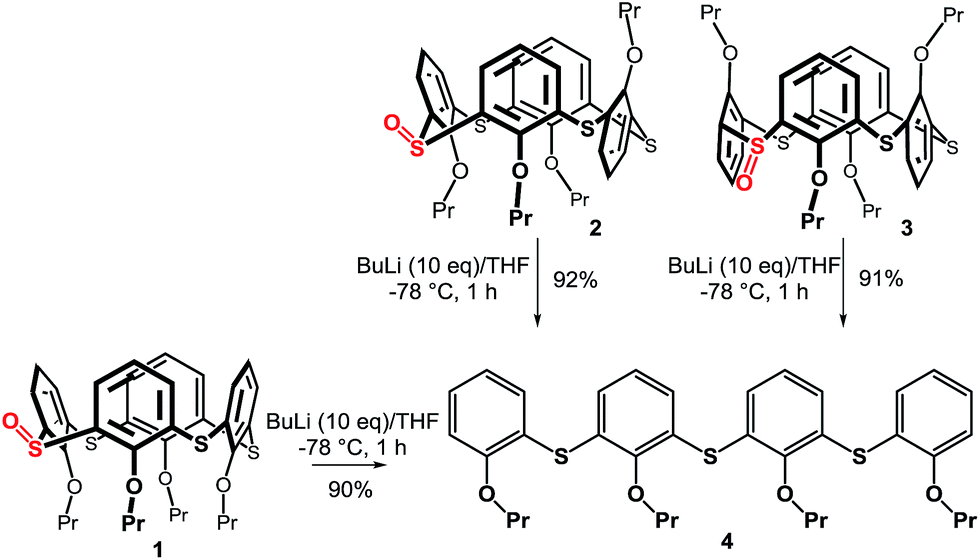 | ||
| Scheme 2 Attempted ortho-lithiation in several conformations of thiacalix[4]arene. | ||
The structure of the products was confirmed by a combination of NMR and MS techniques. The HRMS ESI+ analysis of 4 showed signals at m/z = 657.21393 and 673.18726, which were in good agreement with the [M + Na]+ (657.21374) and [M + K]+ (673.18768) cations predicted for the cleaved product. The 1H NMR spectrum of 4 (CDCl3) revealed the presence of seven aromatic signals and two different propyl groups (triplets at 4.11 and 3.93 ppm from –OCH2– moieties; multiplets at 1.85 and 1.69 ppm from –CH2CH3; triplets at 1.02 and 0.88 ppm from –CH2CH3) indicating the existence of main symmetry plane in the molecule.
The above results indicate that the cleavage reaction was not dependent on the starting conformation of the thiacalixarene. This enabled us to use monosulfoxide 5 for the ensuing study as this derivative is conformationally mobile, and hence, synthetically more easily accessible than sulfoxides 1–3 which have defined conformations. The attempted lithiation of 5 under identical reaction conditions (n-BuLi (10 equiv.) in THF at −78 °C for 1 h, followed by direct hydrolysis) gave oligophenol ether 6 (see ESI† for the X-ray structure) in 92% yield (Scheme 3).
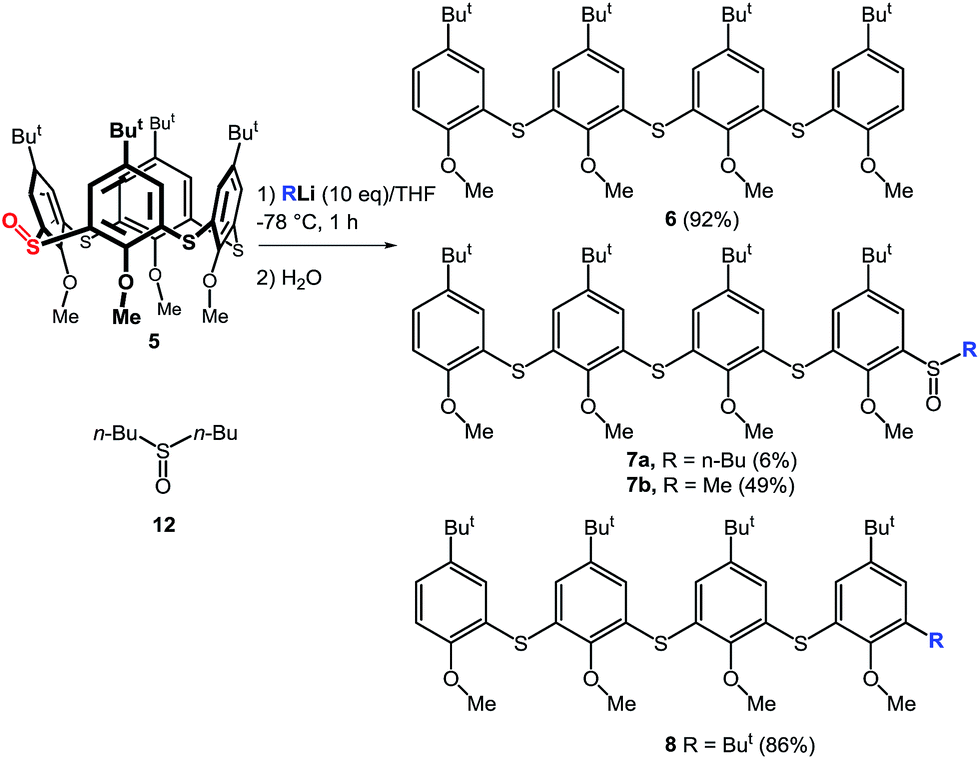 | ||
| Scheme 3 Cleavage of thiacalix[4]arenes by organolithium compounds. | ||
As this type of reactivity was unknown in thiacalixarene chemistry we sought to shed light on the mechanism using a range of reaction conditions. Thus, compound 5 was reacted with various bases (2 equiv.) under standard reaction conditions (THF/−78 °C/1 h, then quenched with H2O) to determine what kind of base is the most suitable. The results clearly showed that n-BuLi provided the highest yield of 6 (43% yield), while s-BuLi and t-BuLi gave 6 in 37% and 5% yields, respectively. Application of Grignard agents (MeMgCl, PhMgCl), NaH or LDA did not invoke any reaction as only starting compound was observed. The use of MeLi gave 6 only in 11% yield, on the other hand, oligomeric sulfoxide 7b was isolated as the main product (49%) from the reaction mixture. The same type of compound 7a was also obtained from the crude reaction mixture with n-BuLi together with dibutyl sulfoxide 12 (Scheme 3). Another type of product was isolated from the reaction with t-BuLi, compound 8, containing a directly appended tert-butyl group.
The structure of 7b was supported by the presence of four singlets from the tert-butyl groups in the 1H NMR spectrum (CDCl3) at 1.27, 1.24, 1.08, and 1.07 ppm, together with four singlets from OCH3– moieties (3.93, 3.92, 3.91, and 3.83 ppm), confirming the absence of any symmetry elements. The methyl group of the sulfoxide moiety could be found as a singlet at 2.80 ppm. The HRMS ESI+ analysis of 7b revealed signals at m/z = 809.33952, 831.32160, and 847.29315, which were in excellent agreement with the predicted values for the [M + H]+ (809.33963), [M + Na]+ (831.32158), and [M + K]+ (847.29552) cations.
The above described results indicate that cleavage of the thiacalixarene derivatives could proceed via a ligand exchange mechanism (Scheme 4) which is documented for the reaction of sulfoxides with organolithium and Grignard reagents.12 These substitution reactions are considered to proceed via an SN2-type process involving the formation of a hypervalent sulfurane,13 as a transition state complex rather than an intermediate (structure B in the tentative mechanism shown in Scheme 5). Substitution of aryl moiety with alkyls (Me, Bu) is plausible because of strain release due to the macrocyclic arrangement around the sulfoxide moiety—thus leading directly to cleaved structure C corresponding to products 7a, 7b. On the other hand, the formation of derivative 8 suggested a ligand coupling procedure (Scheme 4), which has been reported for some sterically demanding sulfoxides.14
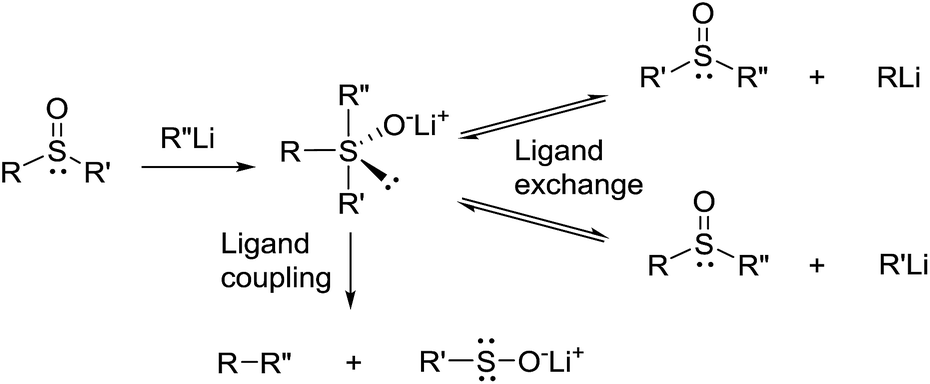 | ||
| Scheme 4 Reactions of sulfoxides with organolithium agents. | ||
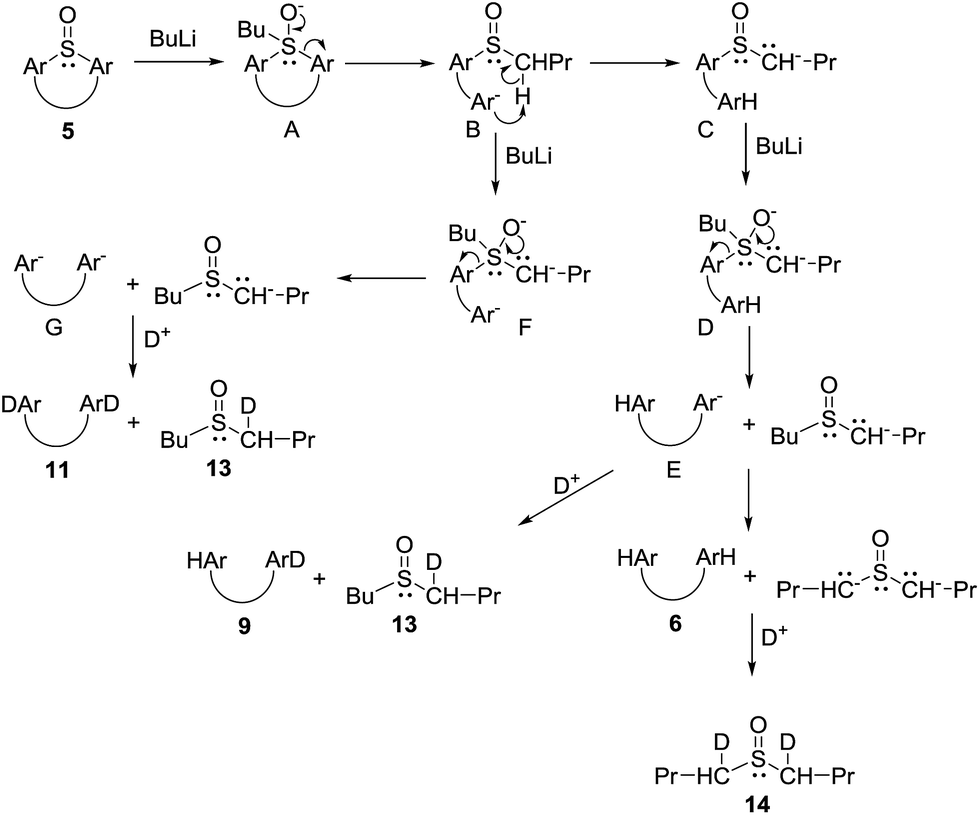 | ||
| Scheme 5 Tentative mechanism for the cleavage reaction of thiacalix[4]arenes by n-BuLi. | ||
To further study the mechanism, we carried out a series of experiments where the reaction mixture was quenched with D2O. Interestingly, the reaction products quenched after stirring for 1 h contained no deuterium on the aromatic moieties instead providing 90% yield of 6. However, HRMS analysis revealed the presence of dideuterated dibutyl sulfoxide 14 (Scheme 6).15 This indicated that the aromatic anions (B and E), formed by the cleavage of the hypervalent sulfuran species (A and D), were quenched by the acidic hydrogens (α-positions of butyl sulfoxide byproduct) before the addition of D2O thus providing only deuterated sulfoxide (Scheme 5).
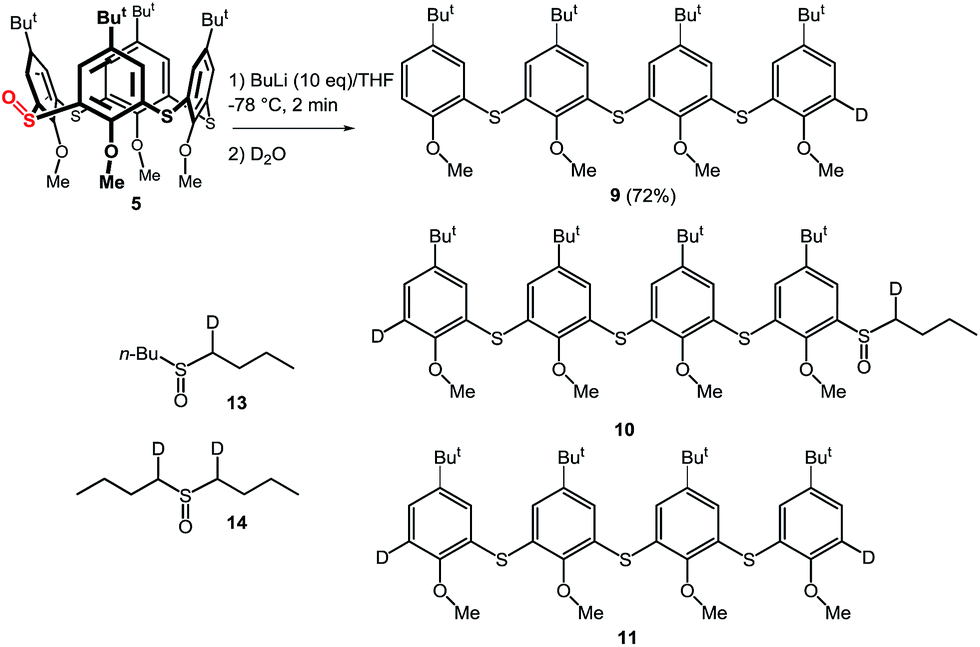 | ||
| Scheme 6 Cleavage of thiacalix[4]arenes–deuteration experiments. | ||
The above reaction is very fast as indicated by quenching (D2O) the reaction mixture after only a 2 min treatment with n-BuLi. Under these conditions a mixture of monodeuterated and dideuterated tetramers 9 and 11 was obtained in 72% yield together with small amount of 10, accompanied by mono- and di-deuterated dibutyl sulfoxide derivatives 13 and 14 (Scheme 6). If the same reaction was quenched after 30 seconds very similar results were obtained. This indicated that the formation of aromatic dianion G or the corresponding hypervalent sulfuran species F is extremely fast and both species gradually disappear from the reaction mixture (Scheme 5).
To show the general applicability of this type of cleavage the corresponding thiacalix[4]arene sulfoxides 15 and 16 (Scheme 7) were subjected to similar reaction conditions (n-BuLi (10 equiv.) in THF at −78 °C for 30 min, followed by direct hydrolysis). As expected, the proximal regioisomer 15 smoothly furnished the appropriate trimer 17 that was isolated in 89% yield after preparative TLC. The concomitant formation of the corresponding p-tert butyl anisol (not isolated) was also observed. Similarly, the distal disulfoxide 16 was cleaved to dimer 18 in 90% isolated yield. Both results strongly suggest that the n-BuLi-induced cleavage of the thiacalix[4]arene skeleton is not limited to only monosulfoxide species, but, in fact, can be successfully applied to a more highly oxidized skeletons.
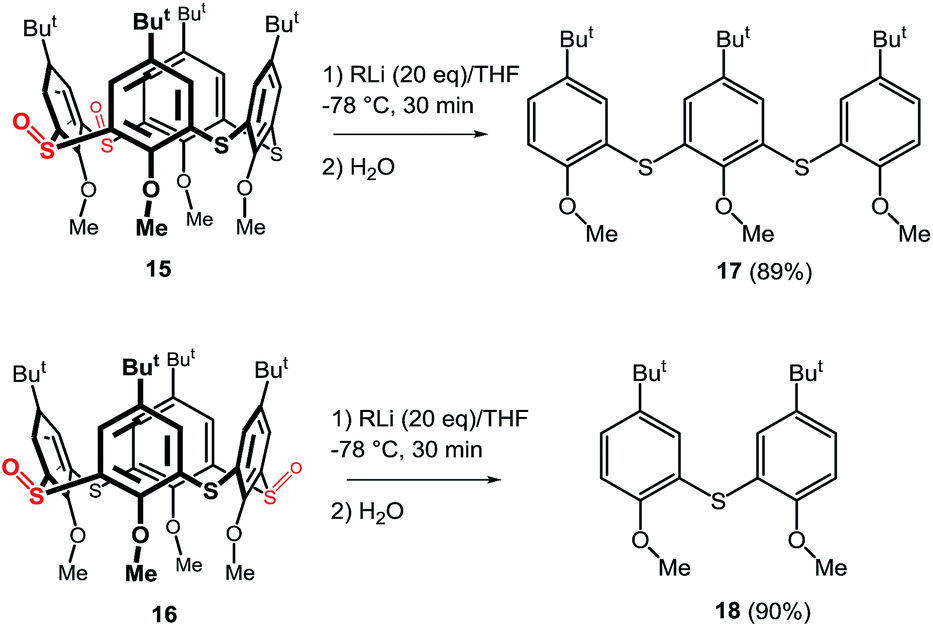 | ||
| Scheme 7 Cleavage of thiacalix[4]arene disulfoxides. | ||
To further show the usefulness of this approach we have carried out a series of experiments where the crude reaction mixture (after the addition of n-BuLi) was quenched by various electrophiles instead of water. Our previous experiments confirmed that the formation of monoanion E or dianion G (Scheme 5) is very fast, while the longer reaction times with BuLi led to their complete disappearance due to the quenching by acidic α-protons of dibutyl sulfoxide formed as a byproduct. Thus, the stirring of starting monosulfoxide 5 with n-BuLi for 20 min led only to the formation of tetramer 6 without any products formed by the reaction with electrophiles added. This indicated that both monoanion E and dianion G are no longer present in the reaction mixture at the time of electrophile addition. Based on this finding we set the reaction time with n-BuLi to only 30 seconds to secure the presence of E and G, than the corresponding electrophile (benzaldehyde, benzophenone, ethyl chloroformate, CO2), was added and the reaction mixture was stirred 10 minutes at −78 °C, the cooling bath was removed, and the reaction mixture was allowed to achieve the room temperature. Using this procedure (Scheme 8) the formation of the corresponding monosubstituted oligomers 19, 21, 23, and 25 was observed in 48%, 43%, 42%, and 44% yield, respectively. In all the cases, the main products (mono-) were accompanied by the corresponding disubstituted oligomers 20, 22, 24 and 26 in approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The only exception is represented by the diacid 26 which was obtained in only very minor amount and was not isolated in enough pure form to allow full characterization.
:1 ratio. The only exception is represented by the diacid 26 which was obtained in only very minor amount and was not isolated in enough pure form to allow full characterization.
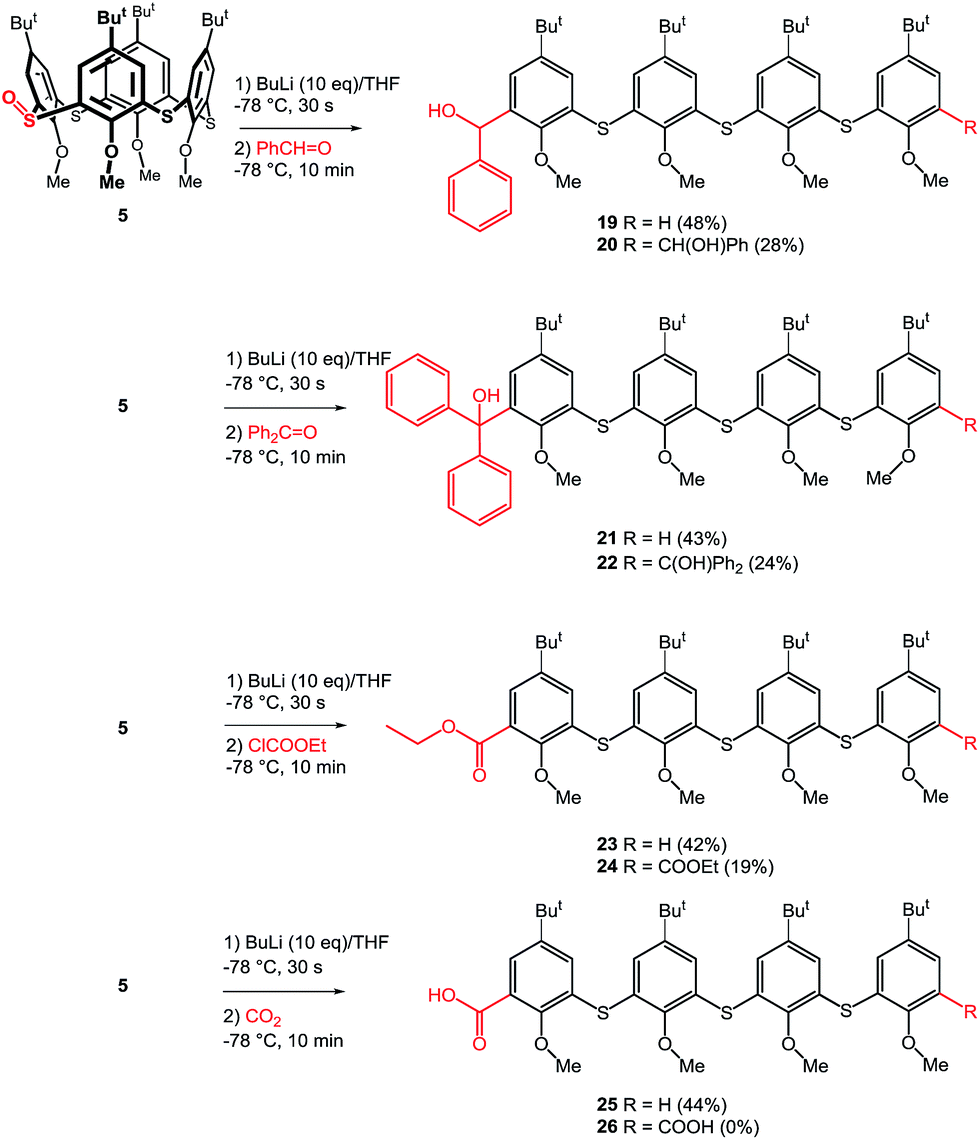 | ||
| Scheme 8 Quenching with various electrophiles. | ||
All the above prepared oligomers are so far unknown compounds which are difficult to access. The tert-butylated compounds 6 and 17 could be potentially obtainable using the step-by-step condensation of the p-tert-butylphenol with SCl2 to form the corresponding tetraphenolic or triphenolic precursors as described by Sone et al.16 On the other hand, the same procedure would be inapplicable for the synthesis of tetramer 4 due to the nonselective reaction of starting phenol in position 2(6) or 4. From this point of view, the cleavage of thiacalixarene sulfoxides offers relatively straightforward and general way for this kind of products, providing the final tetramers in very high yields. Potential utilization of the above tetrameric derivatives can be envisaged in their application as building blocks in the design and synthesis of bigger thiacalixarene-based structures.
Conclusions
In conclusion, thiacalix[4]arenes having one bridge oxidized to the sulfoxide can react with organolithium compounds (best results obtained with n-BuLi) to form cleaved linear structures which are otherwise highly difficult to access. The structure of one such an unexpected product (tetrameric derivative) was assigned by single crystal structure determination. Cleavage of the macrocyclic skeleton is independent of the starting conformation as shown by the same product obtained from the corresponding cone, partial cone or 1,3-alternate derivatives. The quenching of the crude reaction mixture with D2O allowed us to propose a plausible mechanism which is based on ligand exchange on the sulfoxide moiety. The general applicability of this reaction was demonstrated using the corresponding distal or proximal disulfoxide derivatives. Quenching the reaction mixture using various electrophiles allowed for the isolation of the corresponding mono- and di-substituted oligomers. Further synthetic applications of this unusual behavior are currently under study and will be reported in due course.Experimental
General experimental procedures
All chemicals were purchased from commercial sources and used without further purification. Solvents were dried and distilled using conventional methods. All samples were dried in the desiccator over P2O5 under vacuum (1 Torr) at 80 °C for 8 hours. NMR spectra were performed on Agilent 400-MR DDR2 (1H: 400 MHz, 13C: 100 MHz) spectrometer. Chemical shifts (δ) are expressed in ppm and are referred to the residual peak of the solvent or TMS as an internal standard; coupling constants (J) are in Hz. d1-Chloroform (stabilized over silver wool, 99.8% of deuterium) was used for the spectroscopic studies. The signal assignment was supported by 1H–1H COSY, 1H–13C HMQC or 1H–13C HMBC 2D NMR using the standard pulse sequences provided by Bruker. The mass analyses were performed using ESI technique on Q-TOF (Micromass) spectrometer. Melting points were measured on Heiztisch Mikroskop – Polytherm A (Wagner & Munz, Germany) and were not corrected. The IR spectra were measured on an FT-IR spectrometer Nicolet 740 or Bruker IFS66 spectrometers equipped with a heatable Golden Gate Diamante ATR-Unit (SPECAC) in KBr. 100 scans for one spectrum were co-added at a spectral resolution of 4 cm−1. The courses of the reactions were monitored by TLC using TLC aluminium sheets with silica gel 60 F254 (Merck). Thin layer chromatography (TLC) was performed using glass sheets (25 × 25 cm) coated with silica gel 60 F254, followed by visualization with UV light.Synthetic procedures
Data for 6. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.29 (2H, dd, J = 8.61, 2.35 Hz, Ar-H), 7.26 (2H, d, J = 2.35 Hz, Ar-H), 6.98 (2H, d, J = 2.35 Hz, Ar-H), 6.88 (2H, d, J = 2.35 Hz, Ar-H), 6.87 (2H, d, J = 8.22 Hz, Ar-H), 3.93 (6H, s, –OCH3), 3.83 (6H, s, –OCH3), 1.23 (18H, s, t-Bu), 1.07 (18H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 156.2, 154.5, 147.7, 144.1, 130.7, 129.8, 128.7, 127.7, 127.1, 125.8, 121.4, 110.6, 60.8, 55.9, 34.5, 34.1, 31.4, 31.1; IR (KBr) ν 2958, 2904, 2867, 1549, 1489, 1458, 1416, 1392, 1260, 1069, 999, 872, 807 cm−1. HRMS (ESI+): (C44H58O4S3) m/z calcd: 769.33864 [M + Na]+, 785.31288 [M + K]+, found: 769.33960 [M + Na]+, 785.31231 [M + K]+.
Data for 7a. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.75 (1H, d, J = 2.4 Hz, Ar-H), 7.32 (1H, d, J = 2.4 Hz, Ar-H), 7.31 (1H, dd, J = 8.5, 2.4 Hz, Ar-H), 7.28 (1H, d, J = 2.4 Hz, Ar-H), 7.00 (1H, d, J = 2.4 Hz, Ar-H), 6.97 (1H, d, J = 2.4 Hz, Ar-H), 6.89 (1H, d, J = 2.4 Hz, Ar-H), 6.88 (1H, d, J = 8.5 Hz, Ar-H), 6.81 (1H, d, J = 2.4 Hz, Ar-H), 3.93 (3H, s, –OCH3), 3.92 (3H, s, –OCH3), 3.91 (3H, s, –OCH3), 3.83 (3H, s, –OCH3), 3.05 (1H, m, O–CH2–), 2.76 (1H, m, O–CH2–), 1.85 (1H, m, –CH2–), 1.63 (1H, m, –CH2–), 1.44 (2H, m, –CH2CH3), 1.27 (9H, s, t-Bu), 1.24 (9H, s, t-Bu), 1.08 (9H, s, t-Bu), 1.07 (9H, s, t-Bu), 0.93 (3H, t, J = 7.4 Hz, –CH2CH3); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 156.5, 154.9, 154.3, 152.5, 149.4, 148.3, 148.0, 144.3, 137.5, 133.3, 131.1, 130.5, 129.9, 129.0, 128.2, 127.9, 127.9, 127.7, 127.5, 126.7, 126.2, 121.9, 121.1, 110.8, 61.8, 61.0, 61.0, 56.1, 55.1, 35.2, 34.7, 34.6, 34.3, 31.5, 31.3, 31.2, 31.2, 24.5, 22.0, 13.8; IR (KBr) ν 2960, 2888, 1459, 1417, 1262, 1244, 1066, 996, 907, 729, 645 cm−1. HRMS (ESI+): (C48H66O5S4) m/z calcd: 873.36853 [M + Na]+, 889.34247 [M + K]+, found: 873.36877 [M + Na]+, 889.34261 [M + K]+.
Deuteration experiments
Compound 10. HRMS (ESI+): (C48H64D2O5S4) m/z calcd: 853.39914 [M + H]+, 875.38108 [M + Na]+; found: 853.39962 [M + H]+, 875.38134 [M + Na]+.
Compound 11. HRMS (ESI+): C44H56D2O4S3, m/z calcd: 771.35150 [M + Na]+; found: 771.34955 [M + Na]+.
Compound 13. HRMS (ESI+): C8H17D1O1S1, m/z calcd: 164.12139 [M + H]+, found: 164.12170 [M + H]+.
Compound 14. HRMS (ESI+): C8H16D2O1S1, m/z calcd: 165.12767 [M + H]+, found: 165.12774 [M + H]+.
General procedure for cleavage/substitution of thiacalixarene monosulfoxide
Thiacalixarene 5 (50 mg, 0.063 mmol) was added to a 25 mL Schlenk flask and put under high vacuum (oil pump). After 20 minutes, the flask was filled several times with argon and anhydrous THF (5 mL) was added through septum. The solution was cooled down to −78 °C and n-BuLi (10 eq., 2 M in THF) was added in one portion to the flask. The reaction mixture was stirred for 30 seconds at −78 °C, quenched by the addition of the corresponding electrophile, and stirred for 10 min at −78 °C. The cooling bath was then removed and the content of the flask was allowed to warm to RT. After the addition of distilled water (3 mL), the mixture was extracted with CH2Cl2 (3 × 15 mL) and brine. Collected organic layers were washed with water (50 mL) and dried over MgSO4. The solvent was removed under reduced pressure to yield crude product which was further purified using preparative thin layer chromatography.Data for 19. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.41 (1H, d, J = 2.3 Hz, Ar-H), 7.39 (2H, m, Ar-H), 7.33 (2H, t, J = 7.3 Hz, Ar-H), 7.30 (1H, m, Ar-H), 7.26 (2H, m, Ar-H), 7.22 (1H, d, J = 2.3 Hz, Ar-H), 6.96 (1H, d, J = 2.3 Hz, Ar-H), 6.95 (1H, d, J = 2.3 Hz, Ar-H), 6.87 (2H, m, Ar-H), 6.74 (1H, d, J = 2.3 Hz, Ar-H), 6.03 (1H, s, CH–OH), 3.92 (3H, s, –OCH3), 3.91 (3H, s, –OCH3), 3.83 (3H, s, –OCH3), 3.57 (3H, s, –OCH3), 2.94 (1H, br.s, OH), 1.24 (9H, s, t-Bu), 1.23 (9H, s, t-Bu), 1.06 (9H, s, t-Bu), 1.04 (9H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 156.25, 155.14, 154.58, 153.94, 148.00, 147.93, 147.80, 144.16, 143.88, 137.20, 131.06, 130.84, 129.99, 128.97, 128.38, 128.33, 127.77, 127.46, 127.42, 127.18, 126.60, 126.42, 126.38, 125.91, 125.32, 121.21, 110.61, 72.95, 61.33, 60.85, 60.77, 55.97, 34.60, 34.49 (2×), 34.17, 31.40, 31.32, 31.10, 31.08; IR (KBr) ν 2960, 2868, 1725, 1549, 1459, 1417, 1362, 1262, 1163, 1070, 999, 734, 700 cm−1. HRMS (ESI+): (C51H64O5S3) m/z calcd: 875.38081 [M + Na]+, 891.35475 [M + K]+, found: 875.38116 [M + Na]+, 891.35321 [M + K]+.
Data for 20. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.42 (2H, d, J = 2.2 Hz, Ar-H), 7.38 (4H, m, Ar-H), 7.33 (4H, t, J = 7.5 Hz, Ar-H), 7.25 (2H, t, J = 7.5 Hz, Ar-H), 7.23 (2H, d, J = 2.2 Hz, Ar-H), 6.92 (2H, d, J = 2.2 Hz, Ar-H), 6.74 (2H, d, J = 2.2 Hz, Ar-H), 6.03 (2H, s, CH–OH), 3.91 (6H, s, –OCH3), 3.57 (6H, s, –OCH3), 2.92 (2H, br.s, –OH), 1.24 (18H, s, t-Bu), 1.03 (18H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 155.19, 153.99, 148.04, 147.95, 143.86, 137.22, 131.14, 130.32, 128.58, 128.38, 127.55, 127.42, 126.48, 126.44, 126.41, 125.38, 72.95, 61.34, 60.78, 34.60, 34.48, 31.32, 31.05; IR (KBr) ν 2961, 2931, 2868, 1681, 1549, 1459, 1418, 1393, 1362, 1264, 1239, 1163, 1090, 999, 735, 699 cm−1. HRMS (ESI+): (C58H70O6S3) m/z calcd: 981.42267 [M + Na]+, 997.39661 [M + K]+, found: 981.42185 [M + Na]+, 997.39548 [M + K]+.
Data for 21. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.41 (1H, d, J = 2.3 Hz, Ar-H), 7.39 (2H, m, Ar-H), 7.33 (2H, t, J = 7.3 Hz, Ar-H), 7.30 (1H, m, Ar-H), 7.26 (2H, m, Ar-H), 7.22 (1H, d, J = 2.3 Hz, Ar-H), 6.96 (1H, d, J = 2.3 Hz, Ar-H), 6.95 (1H, d, J = 2.3 Hz, Ar-H), 6.87 (2H, m, Ar-H), 6.74 (1H, d, J = 2.3 Hz, Ar-H), 6.03 (1H, s, CH–OH), 3.92 (3H, s, –OCH3), 3.91 (3H, s, –OCH3), 3.83 (3H, s, –OCH3), 3.57 (3H, s, –OCH3), 2.94 (1H, br.s, OH), 1.24 (9H, s, t-Bu), 1.23 (9H, s, t-Bu), 1.06 (9H, s, t-Bu), 1.04 (9H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 156.32, 156.25, 154.54, 153.41, 148.02, 147.79, 146.79, 146.47, 144.16, 141.84, 132.08, 130.85, 130.75, 129.99, 128.77, 128.52, 128.34, 127.93, 127.85, 127.69, 127.36, 127.16, 126.55, 126.00, 125.91, 125.32, 121.20, 110.61, 82.43, 60.85 (2×), 60.71, 55.97, 34.54, 34.49, 34.39, 34.17, 31.41, 31.09 (2×), 31.07; IR (KBr) ν 3488, 2960, 2867, 1595, 1549, 1459, 1393, 1362, 1262, 1245, 1070, 1023, 997, 908, 733, 699 cm−1. HRMS (ESI+): (C57H68O5S3) m/z calcd: 951.41211 [M + Na]+, 967.38605 [M + K]+, found: 951.41287 [M + Na]+, 967.38480 [M + K]+.
Data for 22. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.32 (16H, m, Ar-H), 7.29 (4H, m, Ar-H), 7.28 (2H, d, J = 2.5 Hz, Ar-H), 6.89 (2H, d, J = 2.3 Hz, Ar-H), 6.66 (2H, d, J = 2.3 Hz, Ar-H), 6.59 (2H, d, J = 2.5 Hz, Ar-H), 5.62 (2H, s, –OH), 3.92 (6H, s, –OCH3), 3.15 (6H, s, –OCH3), 1.08 (18H, s, t-Bu), 1.07 (18H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 156.38, 153.41, 148.05, 146.81, 146.44, 141.88, 132.16, 130.90, 128.38, 128.27, 127.92, 127.85, 127.37, 127.16, 125.86, 125.35, 82.43, 60.86, 60.74, 34.52, 34.39, 31.06, 31.04; IR (KBr) ν 3493, 2961, 2867, 1549, 1460, 1419, 1393, 1362, 1265, 1232, 1159, 1091, 1032, 996, 908, 733, 700 cm−1. HRMS (ESI+): (C70H78O6S3) m/z calcd: 1133.48527 [M + Na]+, 1149.45921 [M + K]+, found: 1133.48591 [M + Na]+, 1149.45857 [M + K]+.
Data for 23. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.67 (1H, d, J = 2.4 Hz, Ar-H), 7.30 (1H, dd, J = 8.5, 2.5 Hz, Ar-H), 7.28 (1H, d, J = 2.4 Hz, Ar-H), 7.24 (1H, d, J = 2.5 Hz, Ar-H), 7.01 (3H, m, Ar-H), 6.89 (1H, d, J = 2.4 Hz, Ar-H), 6.87 (1H, d, J = 8.5 Hz, Ar-H), 4.40 (2H, J = 7.2 Hz, –OCH2CH3), 3.92 (6H, s, –OCH3), 3.90 (3H, s, –OCH3), 3.83 (3H, s, –OCH3), 1.41 (3H, J = 7.2 Hz, –OCH2CH3), 1.24 (9H, s, t-Bu), 1.21 (9H, s, t-Bu), 1.10 (9H, s, t-Bu), 1.08 (9H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 166.6, 156.3, 155.8, 155.2, 154.8, 148.2, 147.9, 147.3, 144.2, 132.7, 130.9, 130.6, 130.2, 129.9, 128.7, 128.4, 128.3, 128.1, 128.0, 127.3, 127.0, 126.0, 125.2, 121.1, 110.6, 62.1, 61.3, 61.0, 60.9, 56.0, 34.6, 34.5, 34.4, 34.2, 31.4, 31.2 (2×), 31.1, 14.3; IR (KBr) ν 2960, 1727, 1549, 1460, 1418, 1393, 1364, 1259, 1092, 1000, 875, 791 cm−1. HRMS (ESI+): (C47H62O6S3) m/z calcd: 841.36007 [M + Na]+, 857.33401 [M + K]+, found: 841.36038 [M + Na]+, 857.33298 [M + K]+.
Data for 24. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 7.68 (2H, d, J = 2.5 Hz, Ar-H), 7.25 (2H, d, J = 2.5 Hz, Ar-H), 7.06 (2H, d, J = 2.4 Hz, Ar-H), 7.02 (2H, d, J = 2.4 Hz, Ar-H), 4.40 (2H, J = 7.2 Hz, –OCH2CH3), 3.92 (6H, s, –OCH3), 3.89 (6H, s, –OCH3), 1.41 (6H, J = 7.2 Hz, –OCH2CH3), 1.22 (18H, s, t-Bu), 1.11 (18H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 166.5, 155.9, 155.4, 148.3, 147.3, 133.2, 133.0, 132.9, 130.4, 129.1, 128.9, 128.8, 128.7, 128.6, 127.4, 127.1, 125.3, 62.2, 61.3, 61.0, 34.6, 34.5, 31.2 (2×), 14.3; IR (KBr) ν 2962, 1728, 1550, 1464, 1419, 1392, 1366, 1261, 1093, 1003, 876, 791 cm−1. HRMS (ESI+): (C50H66O8S3) m/z calcd: 913.38120 [M + Na]+, 929.35514 [M + K]+, found: 913.38123 [M + Na]+, 929.35513 [M + K]+.
:1, v/v) mixture as an eluent. The presence of derivative 26 in the crude reaction mixture was confirmed by MS, nevertheless, it has never been isolated in a pure form.
Data for 25. 1H NMR (400 MHz, CDCl3, 296 K) δ (ppm): 10.79 (1H, br s, –COOH), 8.07 (1H, d, J = 2.4 Hz, Ar-H), 7.41 (1H, d, J = 2.4 Hz, Ar-H), 7.31 (1H, dd, J = 8.5, 2.4 Hz, Ar-H), 7.29 (1H, d, J = 2.4 Hz, Ar-H), 7.02 (1H, d, J = 2.4 Hz, Ar-H), 7.00 (1H, d, J = 2.4 Hz, Ar-H), 6.91 (1H, d, J = 2.4 Hz, Ar-H), 6.89 (1H, d, J = 2.4 Hz, Ar-H), 6.88 (1H, d, J = 8.5 Hz, Ar-H), 4.09 (3H, s, –OCH3), 3.92 (3H, s, –OCH3), 3.91 (3H, s, –OCH3), 3.83 (3H, s, –OCH3), 1.24 (18H, s, t-Bu), 1.09 (9H, s, t-Bu), 1.08 (9H, s, t-Bu); 13C NMR (100 MHz, CDCl3, 296 K) δ (ppm): 165.2, 156.4, 155.5, 154.8, 154.6, 149.2, 148.4, 147.9 (2×), 144.2, 135.7, 131.1, 130.5, 130.4, 129.2, 128.9, 128.3 (2×), 127.6, 127.5, 127.4, 126.1, 122.0, 120.9, 110.7, 63.0, 61.0, 60.9, 55.9, 34.8, 34.6, 34.5, 34.2, 31.4, 31.2, 31.1, 31.0; IR (KBr) ν 2960, 2868, 1692, 1549, 1459, 1417, 1392, 1362, 1260, 1070, 999, 907, 808, 730 cm−1. HRMS (ESI+): (C45H58O6S3) m/z calcd: 813.32877 [M + Na]+, found: 813.32880 [M + Na]+.
Data for 26. HRMS (ESI+): (C46H58O8S3) m/z calcd: 857.31860 [M + Na]+, found: 857.31805 [M + Na]+.
X-ray measurement
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation (Grant 17-18108S). Financial support from specific university research (MSMT 20/2017) is also acknowledged.Notes and references
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971 CrossRef CAS.
- For selected books on calixarenes and their applications, see: (a) Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M. X. Wang, Springer Int. Publishing, Switzerland, 2016 Search PubMed; (b) C. D. Gutsche, Calixarenes An introduction, The Royal Society of Chemistry, Thomas Graham House, Cambridge, 2nd edn, 2008 Search PubMed; (c) Calixarenes in the Nanoworld, ed. J. Vicens, J. Harrowfield and L. Backlouti, Springer, Dordrecht, 2007 Search PubMed; (d) Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed; (e) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 Search PubMed.
- For selected reviews on thiacalixarenes, see: (a) M. Zhao, J. Lv and D.-S. Guo, RSC Adv., 2017, 7, 10021 RSC; (b) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824 RSC; (c) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed; (d) P. Lhotak, Eur. J. Org. Chem., 2004, 1675 CrossRef CAS.
- (a) O. Kundrat, I. Cisarova, S. Böhm, M. Pojarova and P. Lhotak, J. Org. Chem., 2009, 74, 4592 CrossRef CAS PubMed; (b) O. Kundrat, H. Dvorakova, I. Cisarova, M. Pojarova and P. Lhotak, Org. Lett., 2009, 11, 4188 CrossRef CAS PubMed; (c) O. Kundrat, H. Dvorakova, V. Eigner and P. Lhotak, J. Org. Chem., 2010, 75, 407 CrossRef CAS PubMed.
- (a) O. Kundrat, J. Kroupa, S. Böhm, J. Budka, V. Eigner and P. Lhotak, J. Org. Chem., 2010, 75, 8372 CrossRef CAS PubMed; (b) O. Kundrat, H. Dvorakova, S. Böhm, V. Eigner and P. Lhotak, J. Org. Chem., 2012, 77, 2272 CrossRef CAS PubMed.
- (a) O. Kundrat, V. Eigner, H. Dvorakova and P. Lhotak, Org. Lett., 2011, 13, 4032 CrossRef CAS PubMed; (b) O. Kundrat, H. Dvorakova, S. Böhm, V. Eigner and P. Lhotak, J. Org. Chem., 2012, 77, 2272 CrossRef CAS PubMed.
- (a) H. Katagiri, T. Hattori, N. Morohashi, N. Iki and S. Miyano, J. Org. Chem., 2007, 72, 8327 CrossRef CAS PubMed; (b) G. Mislin, E. Graf, M. W. Hosseini, A. De Cian and J. Fischer, Tetrahedron Lett., 1999, 40, 1129 CrossRef CAS; (c) H. Katagiri, N. Iki, T. Hattori, C. Kabuto and S. Miyano, J. Am. Chem. Soc., 2001, 123, 779 CrossRef CAS PubMed; (d) N. Morohashi, H. Katagiri, N. Iki, Y. Yamane, C. Kabuto, T. Hattori and S. Miyano, J. Org. Chem., 2003, 68, 2324 CrossRef CAS PubMed; (e) N. Morohashi, N. Iki, C. Kabuto and S. Miyano, Tetrahedron Lett., 2000, 41, 2933 CrossRef CAS; (f) N. Morohashi, N. Iki, T. Onodera, C. Kabuto and S. Miyano, Tetrahedron Lett., 2000, 41, 5093 CrossRef CAS; (g) P. Lhotak, Tetrahedron, 2001, 57, 4775 CrossRef CAS.
- J. Miksatko, V. Eigner, H. Dvorakova and P. Lhotak, Tetrahedron Lett., 2016, 57, 3781 CrossRef CAS.
- J. Miksatko, V. Eigner, M. Kohout and P. Lhotak, Tetrahedron Lett., 2017, 58, 1687 CrossRef CAS.
- J. Clayden, Directed Metallation of Aromatic Compounds, in Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2009 Search PubMed.
- L. Melzig, C. B. Rauhut, N. Naredi-Rainer and P. Knochel, Chem.–Eur. J., 2011, 17, 5362 CrossRef CAS PubMed.
- (a) N. Furukawa, S. Ogawa, K. Matsumura and H. Fujihara, J. Org. Chem., 1991, 56, 6341 CrossRef CAS; (b) S. Oae, T. Kawai and N. Furukawa, Phosphorus Sulfur Relat. Elem., 1987, 34, 123 CrossRef CAS; (c) T. Shibutani, H. Fujihara and N. Furukawa, Heteroat. Chem., 1991, 2, 521 CrossRef CAS.
- N. Furukawa and S. Sato, Top. Curr. Chem., 1999, 205, 90 CrossRef.
- J. Clayden, C. C. Stimson and M. Keenan, Chem. Commun., 2006, 1393 RSC.
- For the formation of dianions from dialkyl sulfoxides, see e.g.: (a) E. M. Kaiser, R. D. Beard and C. R. Hauser, J. Organomet. Chem., 1973, 59, 53 CrossRef CAS; (b) C. M. Thompson, Dianion Chemistry in Organic Synthesis, CRC Press, Boca Raton, 1994 Search PubMed.
- (a) T. Sone, K. Moriya and Y. Ohba, Bull. Chem. Soc. Jpn., 1991, 64, 576 CrossRef; (b) T. Sone, Y. Ohba, K. Moriya, H. Kumada and K. Ito, Tetrahedron, 1997, 53, 10689 CrossRef CAS.
- Bruker, APEX3, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2015 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1554286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11218a |
| This journal is © The Royal Society of Chemistry 2017 |