
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the regioselectivity of Michael addition reactions to asymmetric divinylic compounds†
Rodrigo Navarro *a,
Cristina Monterdebc,
Serena Molinad,
Mónica Pérez-Perrinoa,
Felipe Reviriegoa,
Anselmo del Prado‡
a,
Alberto Gallardoa and
Helmut Reineckea
*a,
Cristina Monterdebc,
Serena Molinad,
Mónica Pérez-Perrinoa,
Felipe Reviriegoa,
Anselmo del Prado‡
a,
Alberto Gallardoa and
Helmut Reineckea
aInstitute of Polymer Science and Technology, Juan de la Cierva 3, 28006, Madrid, Spain. E-mail: rnavarro@ictp.csic.es; Fax: +34 91 56 44 853; Tel: +34 91 56 22 900
bInstitute of Organic Chemistry, Juan de la Cierva 3, 28006, Madrid, Spain
cInstitute of Material Science, Sor Juana Inés de la Cruz 3, 28049, Cantoblanco, Madrid, Spain
dImdea Water Institute, Av. Punto Com 2, 28805, Alcalá de Henares, Madrid, Spain
First published on 13th December 2017
Abstract
In the present paper, we describe the synthesis of novel monomers prepared by regioselective Michael addition to asymmetric divinylic compounds. This chemoselectivity was experimentally studied employing different reaction conditions and theoretically calculated using chemical global and local descriptors. The global reactivity data show that incoming nucleophilic secondary amines preferentially attack the acrylic derived units acrylate and acrylamide, while deactivated methacrylate and N-vinyl-pyrrolidone require harder reaction conditions, which leads to the formation of by-products. Moreover, it is demonstrated that the presence of two vinyl units within a studied divinylic agent leads to a significant increase in its global reactivity parameters. Besides, the local reactivity parameters of asymmetric divinyl compounds show a preference for an attack at the Cβ of activated units compared to the Cβ center of deactivated units. Based on these results, asymmetric divinyl compounds are very interesting starting materials for the preparation of new functionalized monomers.
1 Introduction
The synthesis of monomers with tailored performance and desirable properties, by simple and high yield procedures, is a challenging problem in modern materials science. A procedure that fulfils the requirements of simplicity and high yields is the Michael addition reaction, which is one of the most useful and applied synthetic strategies and is often an important icon to provide a range of biologically active β-functionalized systems. In particular, β-thioether esters or β-amino esters are found in a large number of alkaloids and polyketides and are also versatile building blocks in organic synthesis.Since the pioneering work of Mülhaupt et al.1 on the selective Michael addition of diamines to the acrylate group of ethyleneglycol diacrylate methacrylate for the preparation of dimethacrylates, some other examples can be found in the recent literature that use this selectivity towards different acrylic groups in asymmetric divinylic compounds to obtain novel methacrylates. Thus, this synthetic strategy has allowed for the easy preparation of polymethacrylates bearing pendant special groups such as phosphonate,2,3 hydroxyl,4 stearyl,5 or prodendronic amine moieties.6 The latest example, described by us, reported the successful use of this strategy with an asymmetric acrylamide/methacrylate divinylic compound. Some of these monomers could be prepared in bulk. In addition, the formed β-aminoesters exhibit an enhanced sensitivity to hydrolysis, compared to standard aliphatic esters.6 Very recently, Armes et al. have described for the first time the usefulness of this facile strategy using thiol-type nucleophiles to obtain cysteine- or glutathione-containing methacrylates7 in water and on a multi-gram scale. Besides, our group has also shown the effectiveness of this facile strategy using thiol-type nucleophiles to obtain cysteine- or glutathione-containing methacrylates7 in water and on a multi-gram scale. Besides, our group has also shown the chemoselectivity of this reaction on polymerizable vinylamide groups like vinylpyrrolidone,8 which did not interfere in the Michael addition of amines to acrylates.
The selectivity of the Michael addition (carbonyl or Cβ carbon addition) towards Michael acceptors is strongly dependent on the type and nature of the substituent linked to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. In principle, the reaction can take two different reaction pathways, addition to a carbonyl carbon or conjugated addition (addition to Cβ). By varying the nature of the substituent of the Michael acceptor, the ratio of Ccarbonyl/Cβ that describes the preference of an addition to one carbon atom or to the other can significantly be altered. Therefore, the electronic features of α-β unsaturated Michael acceptors play a central role in determining the selectivity of the Michael addition. A number of global and local reactivity descriptors, namely, global electrophilicity, global hardness, global softness, local hardness, local softness, the Fukui function (f), and the nucleophilicity index (N) etc., have been proposed and derived from density functional theory (DFT). These descriptors have received great importance in order to determine the fate of a given reaction. Understanding the reactivity f of the total chemical process is quite important to study the changes in the global and local reactivity profiles. In this context, Domingo’s9 and Contreras’10,11 groups have been involved for a long time in the study of α-β unsaturated compounds using these parameters to establish the polar character of chemical reactions.
C double bond. In principle, the reaction can take two different reaction pathways, addition to a carbonyl carbon or conjugated addition (addition to Cβ). By varying the nature of the substituent of the Michael acceptor, the ratio of Ccarbonyl/Cβ that describes the preference of an addition to one carbon atom or to the other can significantly be altered. Therefore, the electronic features of α-β unsaturated Michael acceptors play a central role in determining the selectivity of the Michael addition. A number of global and local reactivity descriptors, namely, global electrophilicity, global hardness, global softness, local hardness, local softness, the Fukui function (f), and the nucleophilicity index (N) etc., have been proposed and derived from density functional theory (DFT). These descriptors have received great importance in order to determine the fate of a given reaction. Understanding the reactivity f of the total chemical process is quite important to study the changes in the global and local reactivity profiles. In this context, Domingo’s9 and Contreras’10,11 groups have been involved for a long time in the study of α-β unsaturated compounds using these parameters to establish the polar character of chemical reactions.
In the present paper, we explain the regioselectivity experimentally observed in the Michael addition reaction of nucleophiles to non-symmetric divinyl agents using different descriptors. Based on these results we present a versatile route for the preparation of novel functionalized monomers with high yields and it is applicable in some cases for the preparation of hydrolysable conjugates.
2 Experimental
2.1 Materials
Unless otherwise noted, materials and solvents were obtained from commercially available sources and used without further purification steps. N-Vinylpyrrolidone (VP), methacryloyl chloride, acryloyl chloride, 2-hydroxyethyl methacrylate (HEMA) and 2-hydroxyethyl acrylamide were distilled and freshly used. The asymmetric crosslinkers 2-(acryloyloxy)ethyl methacrylate (AOEMA) and 2-(acrylamidoethyl)methacrylate (AAEMA) were prepared following the synthetic protocols described elsewhere.6,12,132.2 Synthesis
1H-NMR (CDCl3, 300 MHz) δ: 6.40 (dd, 1H, J = 17.3, 1.5 Hz), 6.30–6.22 (m, 2H, NH), 6.13 (dd, 1H, J = 10.6, 1.5 Hz), 6.07 (dd, 1H, J = 10.1, 1.7 Hz), 5.84 (dd, 1H, 10.6, 1.5 Hz), 5.63 (dd, 1H, J = 10.1, 1.7 Hz), 4.27 (t, 2H, J = 5.5 Hz), 3.62 (q, 2H, J = 5.5 Hz).
1H-NMR (CDCl3, 300 MHz) δ: 6.12 (s, 1H), 5.50 (s, 1H), 4.18 (t, 2H, J = 5.8 Hz), 3.51 (m, 2H), 2.72–2.65 (m, 2H), 2.59–2.46 (m, 4H), 2.35–2.32 (m, 2H), 1.93 (s, 3H), 1.00–0.96 (m, 6H).
2.3 Computational details
Full geometry optimizations for the whole series of the vinyl and divinyl compounds have been performed at the B3LYP/6-31G(d,p) level of theory, which was implemented using Gaussian 03W. All of the optimizations were implemented under vacuum. The local Fukui functions for the electrophilic (fk−) and nucleophilic (fk+) attacks were obtained from a single point calculation at the optimized structures of the GS of the molecules using a method described elsewhere.14 Within this approach, the electrophilic and nucleophilic Fukui functions that were condensed to groups of atoms are evaluated in terms of the coefficients of the frontier molecular orbitals involved and the overlap matrix. With the nucleophilic Fukui function at hand, the regional (site) electrophilicity power is readily obtained by (ωk− = ωfk−). The values of the global and local maximum charge transfer towards the electrophile are evaluated using the following equations (ΔNmax = (−μ/η); (ΔNmax(k) = ΔNmaxfk+)).3 Results and discussion
As mentioned above, asymmetric divinylic acrylate/methacrylate compounds, such as AOEMA (structure shown in Fig. 1), have been used to obtain novel methacrylates by profiting from the different selectivities of both amine and mercapto groups in Michael addition.1,3 Our group has recently extended this synthetic strategy to the molecular system acrylamide/methacrylate (AAEMA, structure shown in Fig. 1).6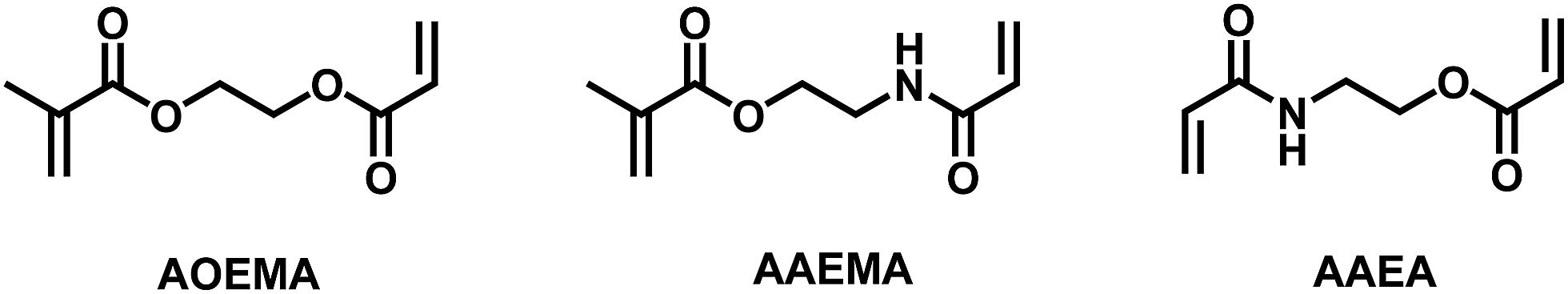 | ||
| Fig. 1 The chemical structure of asymmetric divinylic compounds based on acrylic units. | ||
Thus, a generic synthetic pathway to obtain tailor-made methacrylates may be drawn as indicated in Scheme 1, where X may be O or NH (for acrylate and acrylamide, respectively) and Y may be an amine or mercaptane. These asymmetric vinyl-agents contain two different polymerizable units which are preferred for the reaction with incoming nucleophiles such as amines or thiols.
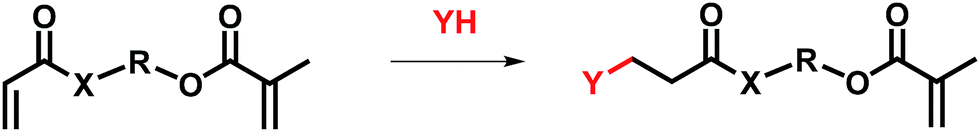 | ||
| Scheme 1 The synthetic strategy to obtain functionalized methacrylates based on Michael addition. | ||
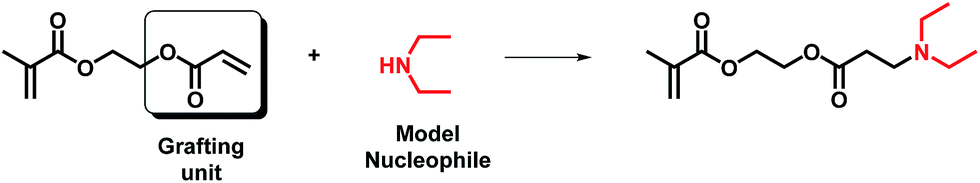
The feasibility of this route using either AOEMA or AAEMA is shown in this work with secondary amines such as diethylamine or dibutylamine. The result of this conjugate-addition reaction under optimal experimental conditions was a regioselective attack of the acrylic unit, leading to the selective formation of a β-aminoester or β-aminoamide bearing an unreacted methacrylic unit. These compounds are interesting in the biomedical field as β-aminoester groups have been demonstrated to be hydrolytically unstable and may be used as key elements for controlled drug delivery because of their attractive pH-sensitive hydrolysis.15,16
3.1 Michael addition to AOEMA
The structural characterization and monitoring of the reaction mixture were performed using 1H-NMR spectroscopy. In the case of AOEMA, the products obtained under certain experimental conditions showed high selectivity towards β-aminoester. Fig. 2 shows the 1H-NMR spectrum of the reaction between AOEMA and diethylamine.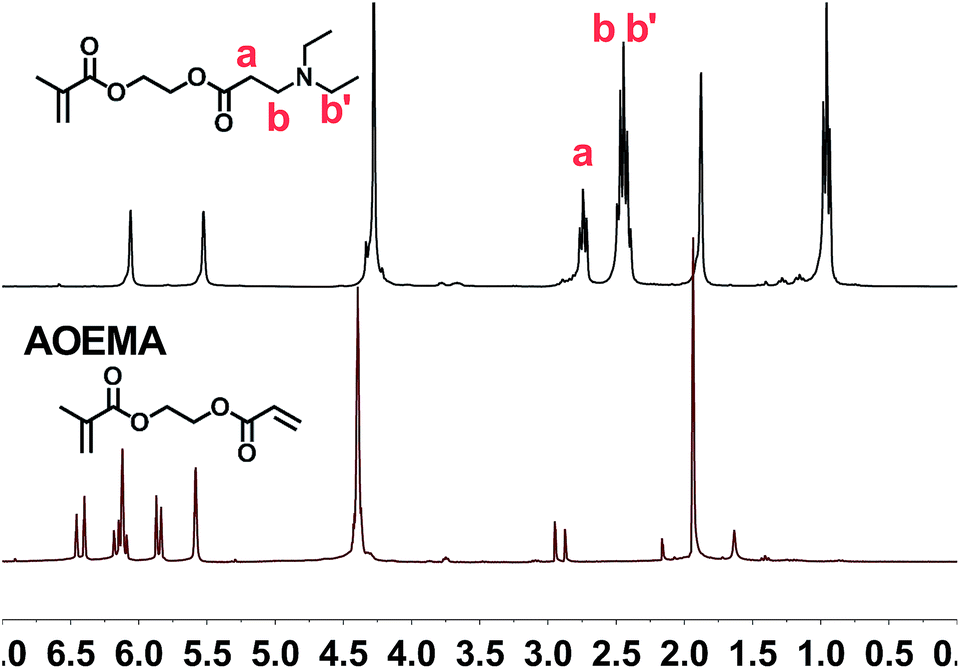 | ||
| Fig. 2 1H-NMR-spectra of AOEMA (bottom) and the reaction between AOEMA and diethylamine (top). | ||
In this figure, the 1H signals from the acrylic unit have vanished, however the singlets of the methacrylate subunit remained constant. Moreover, in the top-spectrum two relevant new signals labelled a and b have appeared. The methylene group signal of a of the α-position to the aminoester function appears at 2.74 ppm, while signal b corresponds to the methylene group in the β-position. As the environment of this b group is similar to that of the methylenes of diethylamine (b′), all of these methylene signals are located at the same position (2.45 ppm). Both signals were observed for the addition of acrylates and were employed to monitor the progress of the addition reaction. The regioselective conjugate addition to the acrylate unit at the expense of other reactive moieties suggests that the acrylate motif is much more favored against nucleophilic attack. To better understand how the formation of this product depends on the reaction conditions, a screening of different experimental conditions was carried out. Table 1 shows the solvents used in the reaction and the yields reached after 3 hours at room temperature.
Comparing the entries concerning the catalyst and yield, one can easily see that the use of catalytic amounts of acetic acid leads to an increase in the reaction yield. In all of the studied cases, higher yields are reached when a catalyst is used. The role of the Brønsted acid is critical because it is capable for activating the carbonyl group through the formation of a complex. Several studies have been devoted to study the influence of this complex on the activation of the Michael reaction.17
On the other hand, rapid conversions were observed when the catalyzed reaction was carried out in chloroform or acetonitrile. However, solvents with weakly basic oxygen functionalities such as THF (ether) or acetone (carbonyl) interfered with the protonation equilibria and gave lower conversions, which has also been found by Wabnitz et al.18 Hence, the following tests were performed using chloroform as the preferred solvent.
Interestingly, the purity of the isolated products was high enough to allow for their subsequent polymerization without further purification steps. These monomers exhibited a good polymerizability under conventional radical polymerization conditions. The obtained polymers were completely soluble in conventional solvents and no crosslinking was detected, demonstrating the complete conversion of all acrylic groups. Therefore, the present synthetic protocol for functionalized monomers opens up new perspectives for potential industrial applications because expensive purification procedures are completely avoided.
3.2 Michael addition to AAEMA
It is well-known that the conjugate addition of an incoming amine to an acrylamide unit leads to a β-aminoamide group. These functional groups are more stable towards hydrolysis than their ester counterparts and require strongly basic conditions and a far longer reaction time.6 This is observed in the case of AAEMA, which leads to functionalized methacrylates bearing β-aminoamides according to the reaction scheme shown in Scheme 2. In this case, the molecule consists of a methacrylate and an acrylamide which may participate in the Michael addition reaction.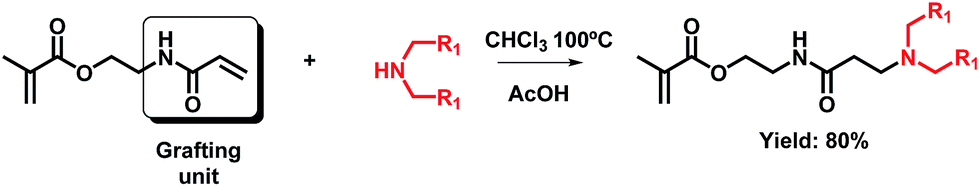 | ||
| Scheme 2 General scheme of the Michael addition of a model amine to AAEMA. | ||
The reaction conditions previously used in the case of AOEMA (diethylamine, AcOH as catalyst in chloroform) were employed as the starting point for the preparation of this second class of functionalized monomer. Applying those conditions, the obtained yield was 23%, determined by NMR analysis. To increase the yield of this reaction, in the next set of experiments, the reaction temperature was gradually increased from 50 to 100 °C. It was also necessary to change the incoming amine, due to the low boiling point of diethylamine (55 °C). We decided to use a secondary pro-dendronic amine, which has also proven useful for applications in different sectors such as gene therapy.6 Moreover, to avoid side reactions like undesired polymerization, hydroquinone was added as a potent free radical scavenger. The reaction yield gradually increased and reached a maximum value of 80% at 100 °C. Under these harsh conditions, some conjugate addition to the methacrylate unit was also detected by NMR analysis; however, the fraction of this by-product was too low and could not be isolated by column chromatography.
As described above, the formation of the β-aminoamide product can easily be monitored by 1H-NMR spectroscopy, because two new signals are detected. As shown in Fig. 3, the positions of these new peaks are located at 2.56 and 3.43 ppm, corresponding to α-methylene and β-methylene protons, respectively.
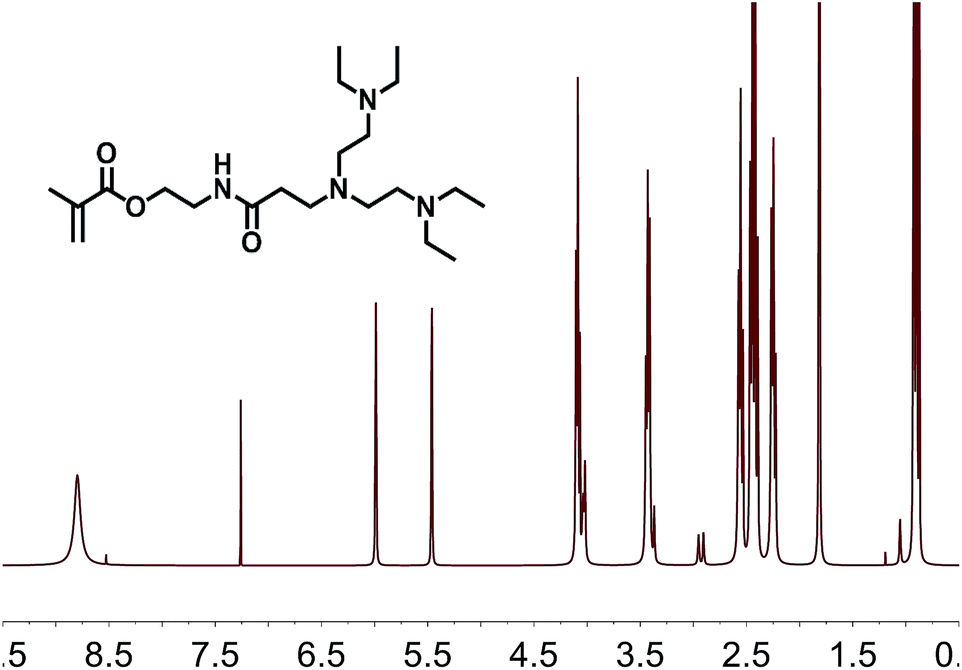 | ||
| Fig. 3 1H-NMR-spectrum of the adduct from AAEMA and a pro-dendronic secondary amine. | ||
This study shows that the addition to an acrylamide AAEMA is less favored than to an acrylate, although a high selectivity with respect to acrylamide addition is still maintained. Therefore, conjugate addition of this type of divinyl agent generates functionalized methacrylates with pendant groups that are more stable and readily polymerizable. However, the monomer formed in good yield has to be purified by column chromatography in order to eliminate small amounts of the methacrylate adduct, which would otherwise lead to crosslinking of the system.
After purification, the monomer could be transformed into a polymer by conventional radical polymerization. The chemical and physical behavior of the homopolymer has previously been analyzed and its usefulness as a gene vector has been studied.6,19
3.3 Michael addition to AAEA
In light of the observed differences in the reactivities between the acrylates and acrylamides (AOEMA and AAEMA), we were interested in the study of a new asymmetric structure bearing both acrylic groups. For this reason, AAEA (Fig. 1) has been synthesized and evaluated for its use in Michael addition. In principle, both acrylic centers are susceptible to Michael addition. If the results would show a regioselectivity towards the acrylate, this difunctional compound could be used to obtain novel acrylamides with hydrolysable side groups. To our knowledge, this divinyl agent has not been used previously for conjugate addition reactions and opens up new perspectives because polyacrylamide products are very attractive in a wide range of biomedical applications.20,21 In this case, we have used dibutylamine as a model secondary amine, following the reaction conditions described for AOEMA (AcOH as the catalyst in chloroform at RT). The reaction is shown in Scheme 3.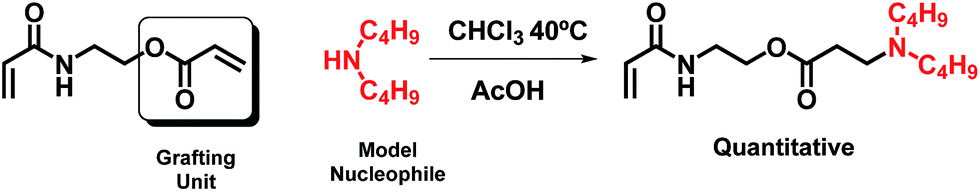 | ||
| Scheme 3 Reaction scheme of AAEA with a model nucleophile. | ||
As shown in Fig. 4, under the experimental conditions described above, the addition of the model amine takes place selectively on the acrylic ester side, remaining unaltered at the acrylamide terminal. This is shown by NMR analysis which shows that the acrylic system with proton signals at higher ppm has disappeared. In addition, the triplet at 4.26 ppm (–CH2CH2O–) is shifted to 3.78 ppm after the reaction, while the signal at 3.61 ppm (–NCH2CH2–) has hardly moved. All these results indicate that under identical experimental conditions the reactivity of the acrylate is considerably higher than that of the acrylamide. The reaction yield was quantitative and the β-aminoester acrylamide could be used for the polymerization step without further purification, yielding a soluble polymer by conventional free radical polymerization.
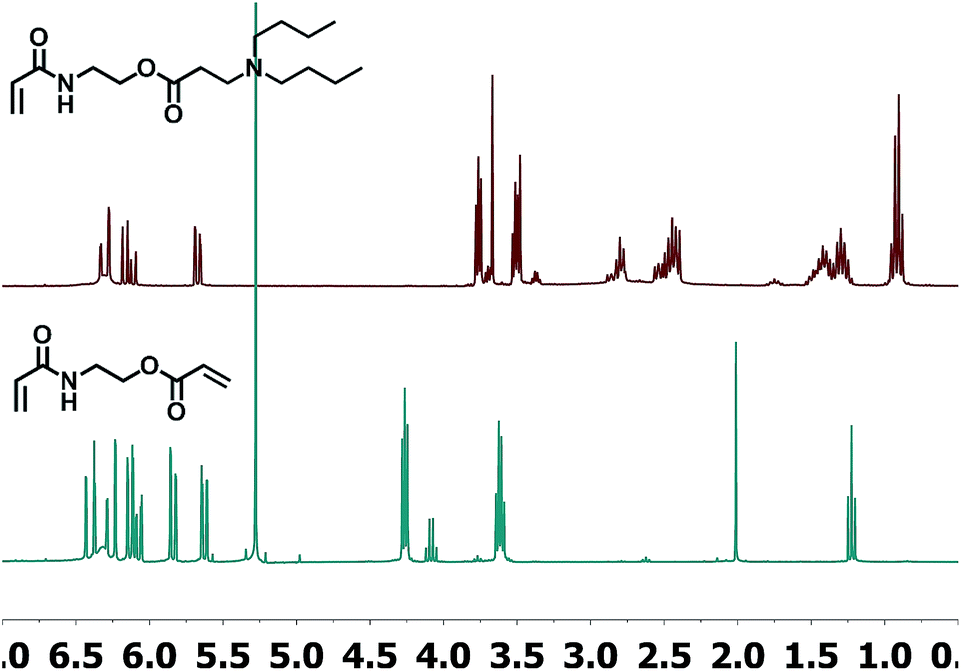 | ||
| Fig. 4 1H-NMR-spectra of the divinylic compound AAEA (bottom) and adduct of AAEA and dibutylamine (top). | ||
4 Theoretical study of the Michael addition reaction to asymmetric divinyl agents
To find a plausible explanation for the reactivities shown by the asymmetric compounds under study, a theoretical computational study was carried out in the framework of Density Functional Theory (DFT). The results of this study have shown an agreement with our experimental results, where the preferential formation of one product over other possible products depends on several factors that will be explained throughout this work. Besides, the computational study has been extended to other polymerizable units such as methacrylamides and vinyl-X (X = O or N) compounds with the aim to predict theoretically the reactivity of other virtual asymmetric divinyl agents. Our group8 has previously shown the selective reactivity of nucleophiles towards difunctional agents carrying a vinylamide unit in their structure, like the N-vinylpyrrolidone moiety. The chemical structures of all of the studied divinylic compounds are depicted in Fig. 5.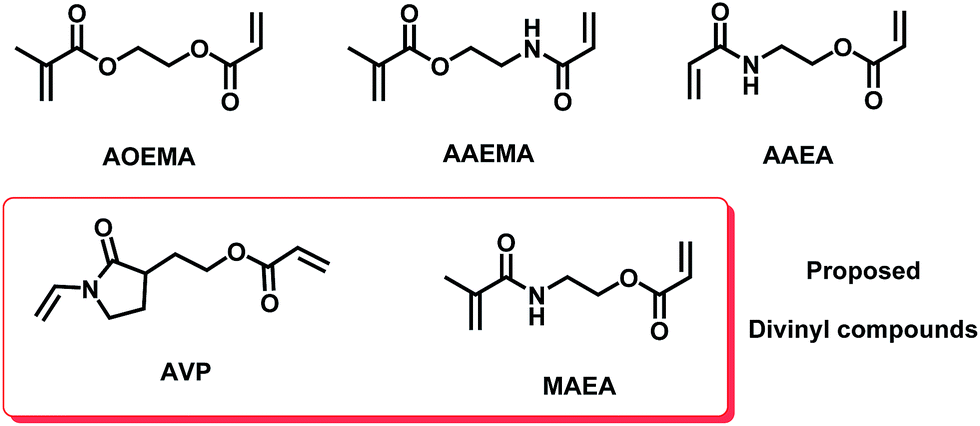 | ||
| Fig. 5 The structures of the asymmetric divinyl compounds. Theoretical asymmetric compounds bearing other polymerizable units (vinylpyrrolidone and methacrylamide) are highlighted in the red box. | ||
The compound called AVP contains an acrylate and vinylpyrrolidone subunit, and the expected reactivity of this compound should lead to functionalized polyvinylpyrrolidones, which are very interesting in biomedical fields.8,13 The other proposed asymmetric divinyl compound (MAEA) contains an acrylate and a methacrylamide moiety. Initially, this theoretical work was focused on finding the most stable form in which each studied molecule occurs, that is, the most stable conformer (geometrical features). Then, the chemical reactivity behavior of this conformer was determined using global and local parameters. The overall work could explain the observed reactivity of each molecule.
4.1 Geometrical features of model vinyl compounds and asymmetric divinylic compounds
On the basis of free rotation around the C–C and C–X bonds, a conformational search was firstly carried out to find out the most stable conformer of the studied species. Moreover, to understand the reactivity of the asymmetric divinyl compounds, we have used the following model units.Acrylic moieties can basically adopt two different conformations based on the relationship of the alkene motif and the carbonyl group. As shown in Fig. 6, they can be arranged in an s-cis and s-trans conformation. Different authors22–24 have studied the conformers of model monomers and their findings suggest that all acrylic monomers, except MMA, are more stable in their cis conformation. For the particular case of the MMA monomer, long-range stabilizing interactions have been proposed as the origin of its preferential trans-conformation.
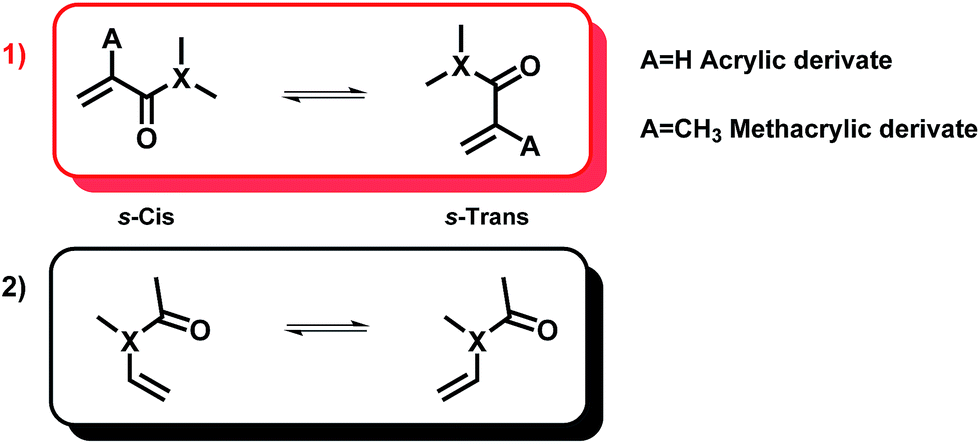 | ||
| Fig. 6 Relevant conformers for (1) an acrylic unit and (2) a vinyl-X moiety. | ||
With respect to vinyl-X monomers, there exist mainly two conformers called s-cis and s-trans. For this type of system, in the trans-conformation the oxygen atom and the hydrogen connected to the first carbon of the vinyl group (HCC) are at the same side of the C–N bond. Different studies25 have previously demonstrated that these moieties basically adopt an s-trans conformation. The interaction with the carbonyl oxygen stabilizes this predominant conformer. Furthermore, the torsional energy profiles show a remarkable barrier, which leads to an unfavorable interconversion.
On the other hand, in the case of asymmetric divinyl-compounds bearing two acrylic units, it is possible to define mainly four possible conformers.
The relative energy values of the studied conformers are included in Table 1 of the ESI.† For all of the asymmetric agents containing a methacrylate subunit, the s-trans conformer is 0.35 kcal mol−1 more stable than its cis-partner. Our finding is consistent with the result obtained by Degirmenci et al.22 These authors have attributed this preferential arrangement to the existence of long-distance stabilizing interactions between the α-methyl and the carbonyl group. However, for other acrylate and acrylamide subunits, the cis-arrangement is presented as the most stable conformation. In the case of acrylate, the energy difference between the conformers was calculated to be around 0.29 kcal mol−1; whereas for the acrylamide derivatives (acrylamide, methacrylamide) the torsional energy profiles for the cis/trans configurational transitions show remarkably high barriers, which is the reason why interconversion between the conformers is not favorable. The most predominant conformations for the acrylamide and methacrylamide units are s-cis and s-trans, respectively.
For the vinyl-X subunits, one could also define two main conformers, based on the relative orientation of the vinyl moiety and the carbonyl group. Considering this second type of vinylic unit, we have also analyzed the behavior of the vinyl-pyrrolidone subunit as an example of a vinyl-X monomer. In this case, our results are similar to the reported data by Martinez-Aparicio et al.,25 who calculated the energy difference between both conformers (3.20 kcal mol−1) of these subunits. These authors claimed the existence of a long-range interaction stabilization, which is able to minimize the energy of the trans-conformer. Our results have estimated this barrier energy to be around 3.17 kcal mol−1.
The most stable optimized structures of all of the studied compounds are depicted in Fig. 7. All optimized geometries are recognized as true minima due to the lack of imaginary harmonic frequencies. Analyzing their structure, one can extract valuable information; for instance, in the case of AOEMA, both acrylic units are coplanar, that is, the main atoms of its backbone are located in the same plane. However, in the AAEMA, AAEA and MAEA compounds, no coplanarity is observed for the acrylic units. To get a deeper understanding concerning the degree of deviation between both planes, the dihedral angel defined between the carbonyl groups of both monomeric units was measured. For AAEMA, AAEA, and MAEA the dihedral angle was −104.3, 104.9 and −109.0°, respectively. The length of the spacer between both acrylic units could have an influence on the orientation of both monomers in the optimized structures. Furthermore, although the subunit VP of the AVP crosslinker is not flat, since this type of ring adopts an envelope conformation, the dihedral angle between both carbonyl groups exhibits a value of 61.8°. This finding shows that this difunctional agent is not flat and both monomer units are twisted.
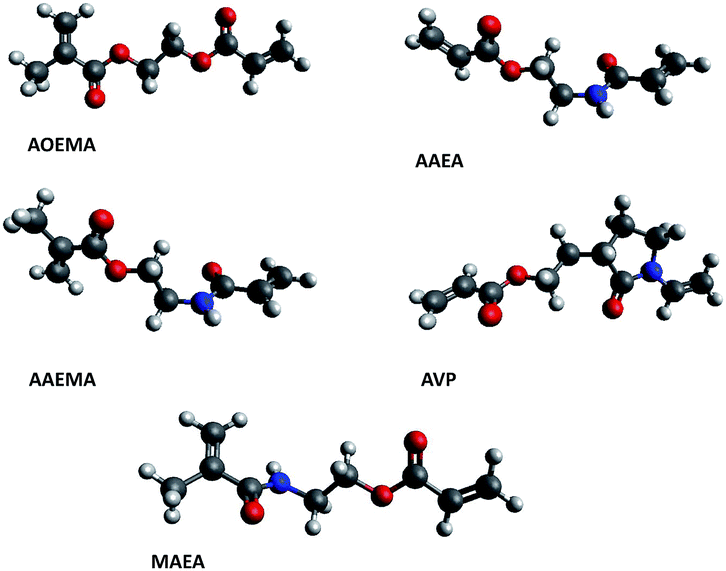 | ||
| Fig. 7 Representation of the optimized conformers of the asymmetric divinylic compounds. | ||
After determining the optimized structure of the divinyl-agents, in the next set of calculations we have calculated the global and local reactivity parameters, which govern the chemical behavior of the studied compounds. Starting with these optimized geometries, the calculations were done at the same level of theory for determining the energy in a single point.
4.2 Global reactivity parameters
The reaction conditions employed for the addition of an amine to an acrylate, acrylamide or a methacrylate are very different. To clarify the difference in the chemical reactivity, the global parameters of the optimized structures of these agents were calculated. In addition, it was also necessary to analyze the isolated vinyl units that were used as a reference. The concepts of global electrophilicity parameters have been rigorously employed to interpret the reactivity patterns.26,27 Table 2 summarizes the global reactivity parameters calculated for the vinyl model and divinylic agents.| Compound | EHOMO (eV) | ELUMO (eV) |
|
η = εL − εH |
|
ΔNmax |
|---|---|---|---|---|---|---|
| Model monomeric units | ||||||
| Methyl acrylate | −7.399 | −1.231 | −4.32 | 6.17 | 1.51 | 0.70 |
| Methyl methacrylate | −7.284 | −1.041 | −4.16 | 6.24 | 1.39 | 0.67 |
| Dimethyl acrylamide | −6.399 | −0.568 | −3.48 | 5.83 | 1.04 | 0.60 |
| Dimethyl methacrylamide | −6.367 | −0.245 | −3.31 | 6.12 | 0.89 | 0.54 |
| Vinyl pyrrolidone | −5.977 | −0.062 | −3.02 | 5.92 | 0.77 | 0.51 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Asymmetric divinylic compound | ||||||
| AOEMA | −7.403 | −1.366 | −4.38 | 6.03 | 1.59 | 0.73 |
| AAEA | −6.886 | −1.229 | −4.06 | 5.66 | 1.45 | 0.71 |
| AAEMA | −6.893 | −1.077 | −3.98 | 5.81 | 1.36 | 0.68 |
| AVP | −6.886 | −1.220 | −4.05 | 5.66 | 1.45 | 0.71 |
| MAEA | −6.898 | −1.256 | −4.08 | 5.64 | 1.47 | 0.72 |
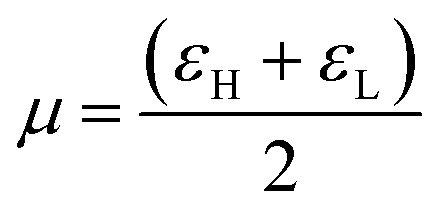
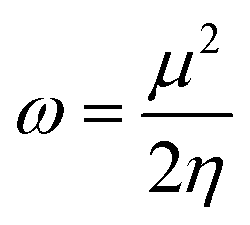
According to the electrophilic scale proposed by Mayr et al.,28 α,β-unsaturated compounds are characterized as strong electrophiles with global electrophilicity values ω ≥ 1.50 eV. The substituted α,β-unsaturated compounds bearing electro-withdrawing groups are moderate electrophiles, with ω ranging between 1.50 and 1.10 eV. The introduction of electron-donating groups linked to the double bond induces a strong deactivation (ω ≤ 1.10 eV), therefore these compounds can be considered as marginal electrophiles (or nucleophiles). The order of electrophilicity is consistent with the substitution pattern at the CC double bond.
Considering methyl acrylate as the reference system, its global electrophilicity index (ω = 1.51 eV) is located at the limit between a strong and a moderate electrophile. Previous studies10 have proposed the methyl acrylate as a strong electrophile, which therefore acts as an active electrophile in the Michael addition of nucleophiles. For a methacrylate unit, due to the formal linkage of a methyl group to the double bond, an electrophilic deactivation has been detected by significantly reducing ω to 1.39 eV. On the other hand, by changing the nature of the carboxyl group, from ester to amide, the monomeric unit becomes a marginal electrophile because its global electrophilicity is significantly reduced to 1.04 eV. Finally, the lowest global electrophilicity index within this model monomer series corresponds to N-vinyl-2-pyrrolidone (ω = 0.77 eV), which consequently has to be considered as a marginal electrophile.
For divinyl agents, the presence of two polymerizable units significantly increases their global electrophilicity with respect to that of both isolated vinyl moieties. Therefore, all of the studied divinylic compounds are considered as strong or moderate electrophiles. For instance, AOEMA is composed of two monomeric units (methacrylate/acrylate) with strong and moderate electrophilic behavior, respectively. However, their combination yields an asymmetric divinyl agent with strong character, suggesting the existence of a synergistic effect between both vinyl units. Similarly, AVP presents the same tendency beside this divinylic compound to act as a moderate electrophile, with one of its monomeric units (VP) characterized by its nucleophilic character (marginal electrophile).
Another important global electronic parameter is called the chemical potential (μ), which is usually associated with the charge transfer ability of the system in its ground state geometry, that is, the direction of a charge transfer (CT) between the Michael donor to Michael acceptor. The asymmetric crosslinker AOEMA is the most electron deficient system, which is evidenced by the highest value of Δω (ωdiethylamine − ωAOEMA = 0.80 − 1.59 = −0.79 eV) for the interaction between AOEMA and N,N-diethylamine. This is accountable for a charge transfer from the incoming amine to the electron-deficient electrophile.
Furthermore, besides these global indexes it is also possible to define the maximum amount of electronic charge (ΔNmax) that the electrophilic system may acquire additionally from the environment. This can also give an estimation about the relative electrophilicity and nucleophilicity of a particular species toward Michael addition. For all of the studied compounds, the acrylate derivatives (AOEMA, AAEA, AVP, and MAEA) present the highest values, while the model compound VP gives the smallest one of the series.
4.3 Regioselective studies (local reactivity parameters)
The interaction between a non-symmetrical difunctional agent with an incoming nucleophile (i.e. secondary amine) can provide four isomeric adducts, depending on the carbon-attack of the monomeric unit. For each acrylic moiety one can define two possible carbon-attack centers (Ccarbonyl and Cβ). The selective formation of one condensed product over the other is called regioselectivity, and the obtained isomer is termed the regioisomer. Recently, several studies, devoted to regioselective Michael addition using DFT, have shown that a combined analysis of the local electrophilicity ωk and the local nucleophilicity Nk of the electrophile and nucleophile allows for the prediction of the obtained regioisomer that is to be expected.29 To explain this regioselectivity of the asymmetric divinyl agents, the condensed Fukui functions (nucleophilic and electrophilic) were calculated. These functions are very useful for the discrimination between the different reactive sites on the same molecule. These condensed Fukui functions were calculated using UCA-Fukui software.30Table 3 summarizes the condensed Fukui functions of the most relevant atoms of these asymmetric crosslinkers (Ccarbonyl and Cβ).
| fk− | fk+ | ||||
|---|---|---|---|---|---|
| CCO |
Cβ | CCO |
Cβ | ||
| AOEMA | Methacrylate | 0.0005 | 0.4087 | 0.0029 | 0.0046 |
| Acrylate | 0.0000 | 0.0000 | 0.1671 | 0.4024 | |
| AAEMA | Methacrylate | 0.0003 | 0.0004 | 0.1893 | 0.3678 |
| Acrylamide | 0.0168 | 0.0092 | 0.0003 | 0.0003 | |
| AAEA | Acrylate | 0.0002 | 0.0001 | 0.1644 | 0.4118 |
| Acrylamide | 0.0174 | 0.0096 | 0.0002 | 0.0001 | |
| AVP | Acrylate | 0.0000 | 0.0000 | 0.1614 | 0.4203 |
| VP | 0.0075 | 0.3353 | 0.0003 | 0.0000 | |
| MAEA | Acrylate | 0.0000 | 0.0001 | 0.1676 | 0.4174 |
| Methacrylamide | 0.0025 | 0.0335 | 0.0000 | 0.0000 | |
Analyzing the results of this table one may draw several conclusions:
(a) For all of the asymmetric divinyl agents studied, the electrophilic Fukui function (fk+) is strongly localized on an acrylate subunit. In fact, this function is strongly assembled in both reactive centers (Ccarbonyl and Cβ) of the acrylate group; moreover, considering the complete function, it can be assessed that around 80% of this function is located on the reactive subunit (data not given).
(b) By contrast, the nucleophilic Fukui function (fk−) is located on the other subunit of the studied compounds. Moreover, around 80–85% of this function is focused on this polymerizable moiety.
(c) For AOEMA, AAEA, AVP and MAEA, the electrophilic functions can easily explain the nucleophilic attack of the Cβ of the acrylates. Therefore, this confirms the preferential formation of the experimentally obtained adduct. In addition, these results may provide a theoretical framework for the possible addition of a nucleophile to acrylate in the case of asymmetric AVP and MAEA.
(d) Finally, in the case of AAEMA, the condensed Fukui function failed. The poor description of the local reactivity by these functions may be due to the activation of inner orbitals at the early stages of the reaction or the fact that this reaction takes place along a different pathway. This hypothesis is based on the role played by a Brønsted acid catalyst, which could be coordinated to the acrylamide subunit and, consequently, facilitate the Michael addition to this subunit. Previously, other authors have proposed the activation of the carbonyl group using (Lewis or Brønsted) catalysts. It has been postulated that the proton or metal center could be coordinated to the carbonyl oxygen atom, causing a redistribution of the electron density. Consequently, the conjugate addition process is triggered.18,31 This result is in full accordance with our experimental results and shows that the Cβ of the acrylamide subunit presents more susceptibility.
5 Conclusions
From the results obtained by experimental and theoretical methods, we can conclude that the synthesis of new functionalized vinyl compounds bearing β-aminoester and β-aminoamide groups is based on a selective addition of a nucleophile to an acrylic unit. As starting products, asymmetric divinylic agents were used, which contain two vinyl moieties with a different nature. The dependence of the nature of the acrylic derivative was examined by studying the addition of a model amine to an acrylate, methacrylate and acrylamide unit. We have also performed a conformation study of these divinylic agents. The two main conformers, cis and trans, are very close in energy; however, the s-cis conformer is slightly more stable than the s-trans counterpart, except for the methacrylate subunits.It has also been found that the plane of the vinyl units is twisted by around −104°. The global parameters (global electrophilicity, chemical potential and ΔNmax) suggest the existence of a synergistic effect between the two vinyl units, which is responsible for the significant increase in the chemical reactivity of the divinylic compounds compared to the isolated vinyl units. Furthermore, the local parameters (such as the Fukui functions) disclose selective and preferential formation of a regioisomer adduct. However, these theoretical results can not explain the reactivity of AAEMA making it necessary to consider additional aspects such as the activation of internal orbitals or catalyst participation in the activation of the acrylamide unit.
The results of this work seem to allow us to make some predictions about other similar systems. If we assume that secondary amines preferentially attack acrylic subunits (acrylate and acrylamide), then other types of nucleophile such as thiols or oximes should also preferentially attack these moieties, leading to attractive vinyl compounds bearing other functional groups. The addition of oxime leads to oxime ethers, which presents an interesting hydrolysis process in acidic media. Experimental studies along these lines are currently being performed in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Consejo Superior de Investigaciones Científicas (CSIC). Equally, this work was financially supported by the Ministerio de Economía y Competitividad (MINECO) through MAT2013-42957-R. R. N. is grateful for a contract of the Postdoctoral Formation program from the Ministerio de Economía y Competitividad.References
- E. Müh, J. Marquardt, J. E. Klee, H. Frey and R. Mülhaupt, Macromolecules, 2001, 34, 5778–5785 CrossRef.
- Z. S. Bilgici, O. D. Ordu, M. Isik and D. Avci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5042–5048 CrossRef CAS.
- M. Schmider, E. Müh, J. E. Klee and R. Mülhaupt, Macromolecules, 2005, 38, 9548–9555 CrossRef CAS.
- L. J. Mathias, B. S. Shemper, M. Alirol and J.-F. Morizur, Macromolecules, 2004, 37, 3231–3238 CrossRef CAS.
- H. Weickmann, M. Gurr, O. Meincke, R. Thomann and R. Mülhaupt, Adv. Funct. Mater., 2010, 20, 1778–1786 CrossRef CAS.
- J. A. Redondo, R. Navarro, E. Martínez-Campos, M. Pérez-Perrino, R. París, J. L. López-Lacomba, C. Elvira, H. Reinecke and A. Gallardo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2297–2305 CrossRef CAS.
- V. Ladmiral, A. Charlot, M. Semsarilar and S. P. Armes, Polym. Chem., 2015, 6, 1805–1816 RSC.
- A. del Prado, R. Navarro, A. Gallardo, C. Elvira and H. Reinecke, RSC Adv., 2014, 4, 35950–35958 RSC.
- L. R. Domingo, J. A. Sáez and M. Arnó, Org. Biomol. Chem., 2014, 12, 895–904 CAS.
- L. R. Domingo, P. Pérez and R. Contreras, Tetrahedron, 2004, 60, 6585–6591 CrossRef CAS.
- P. Pérez, L. R. Domingo, A. Aizman and R. Contreras, Theor. Comput. Chem., 2007, 19, 139–201 Search PubMed.
- D. Demirgöz, R. Navarro, M. Pérez, H. Reinecke and A. Gallardo, J. Appl. Polym. Sci., 2010, 115, 896–900 CrossRef.
- M. Pérez Perrino, R. Navarro, M. Gómez Tardajos, A. Gallardo and H. Reinecke, Macromol. Chem. Phys., 2009, 210, 1973–1978 CrossRef.
- R. R. Contreras, P. Fuentealba, M. Galván and P. Pérez, Chem. Phys. Lett., 1999, 304, 405–413 CrossRef CAS.
- Y.-b. Lim, Y. H. Choi and J.-s. Park, J. Am. Chem. Soc., 1999, 121, 5633–5639 CrossRef CAS.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- T. C. Wabnitz and J. B. Spencer, Org. Lett., 2003, 5, 2141–2144 CrossRef CAS PubMed.
- T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem.–Eur. J., 2004, 10, 484–493 CrossRef CAS PubMed.
- T. Acunha, C. Ibáñez, P. Reguera, M. Isabel, M. Sarò, R. Navarro, J. Alfonso Redondo, H. Reinecke, A. Gallardo and C. Simó, et al., Electrophoresis, 2015, 36, 1564–1571 CrossRef CAS PubMed.
- J. Kopeček and P. Kopečková, Adv. Drug Delivery Rev., 2010, 62, 122–149 CrossRef PubMed.
- Z. Qin, W. Liu, L. Li, L. Guo, C. Yao and X. Li, Bioconjugate Chem., 2011, 22, 1503–1512 CrossRef CAS PubMed.
- I. Degirmenci, V. Aviyente, V. Van Speybroeck and M. Waroquier, Macromolecules, 2009, 42, 3033–3041 CrossRef CAS.
- X. Yu, J. Pfaendtner and L. J. Broadbelt, J. Phys. Chem. A, 2008, 112, 6772–6782 CrossRef CAS PubMed.
- G. Zhang, I. A. Konstantinov, S. G. Arturo, D. Yu and L. J. Broadbelt, J. Chem. Theory Comput., 2014, 10, 5668–5676 CrossRef CAS PubMed.
- S. Martinez-Aparicio, K. Hall and P. Balbuena, J. Phys. Chem. A, 2006, 110, 9183–9193 CrossRef PubMed.
- P. K. Chattaraj, B. Maiti and U. Sarkar, J. Phys. Chem. A, 2003, 107, 4973–4975 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- T. Lemek and H. Mayr, J. Org. Chem., 2003, 68, 6880–6886 CrossRef CAS PubMed.
- G. Madjarova, A. Tadjer, T. P. Cholakova, A. Dobrev and T. Mineva, J. Phys. Chem. A, 2005, 109, 387–393 CrossRef CAS PubMed.
- J. Sánchez-Márquez, D. Zorrilla, A. Sánchez-Coronilla, M. Desireé, J. Navas, C. Fernández-Lorenzo, R. Alcántara and J. Martín-Calleja, J. Mol. Model., 2014, 20, 2492 CrossRef PubMed.
- H. Suga, H. Takemoto and A. Kakehi, Heterocycles, 2007, 71, 361–371 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Reference Cartesian coordinates for all the studied molecules and the relative energy of each conformer. See DOI: 10.1039/c7ra11005g |
| ‡ A. del Prado present address: Adolphe Merkle Institute, Chemin des Verdiers 4, Fribourg, Switzerland. |
| This journal is © The Royal Society of Chemistry 2017 |