
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleobase-modified antisense oligonucleotides containing 5-(phenyltriazol)-2′-deoxyuridine nucleotides induce exon-skipping in vitro†
Bao T.
Le
ab,
Mick
Hornum
 c,
Pawan K.
Sharma
d,
Poul
Nielsen
c and
Rakesh N.
Veedu
*ab
c,
Pawan K.
Sharma
d,
Poul
Nielsen
c and
Rakesh N.
Veedu
*ab
aCentre for Comparative Genomics, Murdoch University, Perth, Australia-6150. E-mail: R.Veedu@murdoch.edu.au
bPerron Institute for Neurological and Translational Science, Perth, Australia-6009
cDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark
dDepartment of Chemistry, Kurukshetra University, Kurukshetra-113-119, India
First published on 28th November 2017
Abstract
Chemically-modified antisense oligonucleotide-mediated exon-skipping has been validated as a therapeutic strategy for tackling several disease pathologies, particularly duchenne muscular dystrophy. To date, only sugar-modified and internucleotide linkage-modified oligonucleotide chemistries have been explored for exon-skipping applications. Herein, for the first time, we have investigated the potential of nucleobase-modified antisense oligonucleotides to induce exon-skipping. For this purpose, we have synthesised 5-(phenyltriazol)-2′-deoxyuridine-modified 2′-O-methyl mixmer antisense oligonucleotides, and evaluated their efficacy to induce exon-23 skipping in H-2Kb-tsA58 (H2K) mdx mouse myotubes as a model system. Our results showed that the phenyltriazol base-modified AOs successfully induced efficient exon-skipping in a DMD transcript. Our findings open up the exploration of novel base-modified antisense oligonucleotides for exon-skipping applications.
Introduction
Antisense oligonucleotide (AO)-based therapy has now been established as one of the most promising approaches for the treatment of various diseases. The use of chemically-modified nucleotides is paramount to improve the pharmacokinetics of AO-based therapeutic molecules. Last year, two AO drug candidates, Exondys 51 for the treatment of duchenne muscular dystrophy (DMD) and nusinersen for the treatment of spinal muscular atrophy (SMA), have been approved by the US Food and Drug Administration (FDA), bringing the total number of FDA approved antisense oligonucleotide drugs to four.1,2 With the approval of Exondys 51 for the treatment of DMD, AO-mediated splice modulation has now emerged and been validated as a viable treatment alternative. DMD is a serious X-linked muscle wasting genetic disorder.3,4 Patients with DMD lack dystrophin protein due to mutations in one or more exons of the dystrophin gene required for muscle contraction and protection. Exondys 51 is a 30-nucleotide long phosphorodiamidate morpholino oligomer (PMO) AO targeting exon-51 in the dystrophin gene transcript. In addition, another AO drug candidate drisapersen composed of 2′-O-methyl (2′-O-Me) nucleotides on a phosphorothioate (PS) backbone entered phase-3 clinical trials, and was later rejected by the FDA based on poor efficacy and toxicity issues.So far, several antisense oligonucleotide chemistries have been explored for their potential to modulate splicing or induce exon-skipping.5–16 However, only sugar moiety and phosphate backbone modified-AOs have been investigated for exon-skipping applications. We envisioned the scope of nucleobase-modified AOs to induce exon-skipping. Towards this, Nielsen and colleagues reported the synthesis of 5-(phenyltriazol)-2′-deoxyuridines that showed improved duplex stability when targeting a complementary RNA.17–20 Improved duplex stability upon incorporation of 5-(phenyltriazol)-2′-deoxyuridine nucleotides is believed to be due to the π–π stacking of the aromatic substituents. Herein, we describe for the first time that the design and synthesis of 5-(phenyltriazol)-2′-deoxyuridine/2′-O-Me (Fig. 1) mixmer RNA AOs and evaluate its efficacy to induce exon-skipping in vitro in H-2Kb-tsA58 (H2K) mdx myotubes.
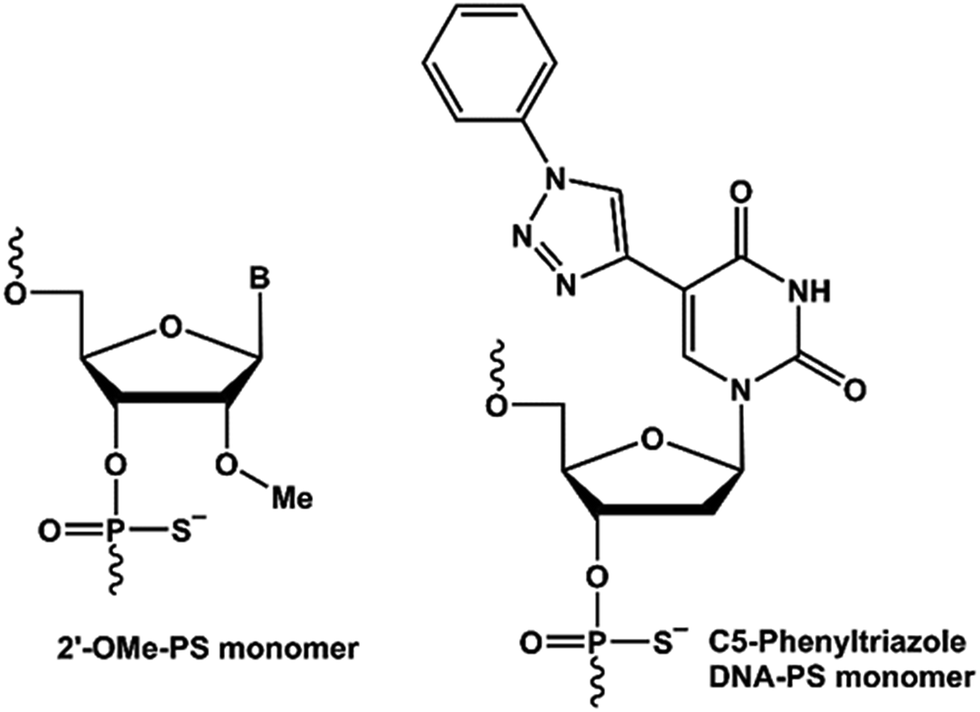 | ||
| Fig. 1 Structural presentation of the nucleic acid analogues used in this study. | ||
Experimental
Design and synthesis of antisense oligonucleotides used in this study
All AOs (Table 1) were prepared in-house on an AKTA Oligopilot synthesizer (GE Healthcare Life Sciences) via standard phosphoramidite chemistry in 1 μmol scale. Synthesized oligonucleotides were deprotected and cleaved from the solid support by treatment with NH4OH at 55 °C overnight. The crude oligonucleotides were then purified, desalted and verified by HPLC and MALDI-ToF MS analysis (see ESI† for detailed procedure and analysed data).| AOs names | Sequence, 5′ → 3′ direction | T m (°C) |
|---|---|---|
| a Complementary synthetic RNA target: 5′-r(AG GUA AGC CGA GGU UUG GCC)-3′. 5-(Phenyltriazol)-2′-deoxyuridine monomers are represented in bold underlined letters. Melting temperature curves are shown in Fig. S1 (ESI). | ||
| 2′-O-MePS | GGCCAAACCUCGGCUUACCU | 62.9 |
| ON1 | GGCCAAACC![[U with combining low line]](https://www.rsc.org/images/entities/i_char_0055_0332.gif) CGGCUACCU CGGCUACCU |
58.8 |
| ON2 |
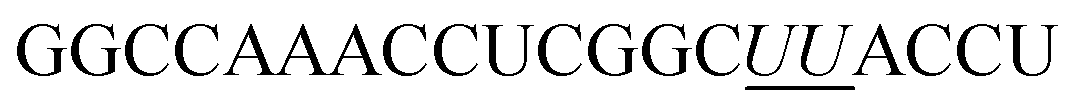
|
62.8 |
Melting temperature analysis of the antisense oligonucleotides
Three antisense oligonucleotides: control 2′-O-MePS, ON1 and ON2 were prepared at 2 μM concentrations in a buffer solution containing 10 mM NaCl, 0.01 mM EDTA adjusted to pH 7.0 by 10 mM sodium phosphate buffer. The AOs were then mixed with the synthetic complementary RNA sequence (2 μM) at equal volume and denatured at 95 °C for 10 minutes followed by cooling down to room temperature and loaded onto a quartz cuvette of 1 mm path-length. The melting process was monitored by Shimadzu UV-1800 with the temperature controller over the range of 20–90 °C at a ramp rate of 1.0 °C min−1. Tm values were then calculated by the first derivative.Cell culture and transfection
H-2K b -tsA58 (H2K) mdx mouse myoblast (provided by Prof. Sue Fletcher and Prof. Steve Wilton's laboratory, Murdoch University, Australia) were cultured as described previously.21,22 Briefly, when 60–80% confluent, primary mdx myoblast cultures were treated with trypsin (Life Technologies) and seeded at a density of 2 × 104 cells per well into 24 well plates. The plates were pre-treated with 50 μg ml−1 poly-D-lysine (Sigma) and 100 μg ml−1 Matrigel (Corning). Cultures were then allowed to differentiate into myotubes in Dulbecco's Modified Eagle Medium (DMEM) containing 5% horse serum by incubating at 37 °C, 5% CO2 for 24 hours. Antisense oligonucleotides were complexed with Lipofectin (Life Technologies) at the ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Lipofectin:AO) and used in a final transfection volume of 500 μl per well in a 24-well plate as per the manufacturer's instructions, except that the solution was not removed after 3 hours.
:1 (Lipofectin:AO) and used in a final transfection volume of 500 μl per well in a 24-well plate as per the manufacturer's instructions, except that the solution was not removed after 3 hours.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR)
RNA was extracted from transfected cells using Direct-zol™ RNA MiniPrep Plus with TRI Reagent® (Zymo Research) as per the manufacturer's instructions. The dystrophin transcripts were then analysed by nested RT-PCR across exons 20–26 as described previously.22,23 PCR products were separated on 2% agarose gels in Tris–acetate–EDTA buffer and the images were captured on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). Densitometry analyses was performed by Image J software.24 To quantify the actual exon-skipping efficiency, the amount of exon-23 skipped RT-PCR product is expressed as a percentage of total dystrophin transcript products.Cell viability assay
Cells were seeded and transfected with the AOs at 50 and 200 nM as described previously. After 24 h, cell viability assay was performed using a colorimetric assay (WST-1, Sigma). Briefly, WST-1 solution was added at ratio 1:10 (v/v) per well and incubated for 4 h at 37 °C, 5% CO2. The absorbance was then measured with a microplate reader (FLUOstar Omega, BMG Labtech, Germany) at 450 nm.
Results and discussion
First, we designed and synthesised two 20-mer 2′-O-MePS AOs containing 5-(phenyltriazol)-2′-deoxyuridine nucleotides in parallel to a previously reported 2′-O-Me phosphorothioate (PS) AO.22,23 The first AO sequence (ON1) contained two 5-(phenyltriazol)-2′-deoxyuridine nucleotides at positions 10 and 16, and the second AO (ON2) had two consecutive 5-(phenyltriazol)-2′-deoxyuridine nucleotides at positions 15 and 16 respectively (Table 1). To assess the binding affinity of the modified AOs against its RNA target, we initially performed the thermal stability analysis using a synthetic complementary RNA sequence (Table 1) in parallel to the control 20-mer 2′-O-MePS AO. In line with previous reports,17–20ON1 containing two 5-(phenyltriazol)-2′-deoxyuridine nucleotides positioned five nucleotides apart from each other showed slight destabilization of the heteroduplex with lower Tm of 58.8 °C compared to the 20-mer 2′-O-MePS control AO sequence (62.9 °C). On the other hand, the AO sequence containing two consecutive 5-(phenyltriazol)-2′-deoxyuridine nucleotides showed similar Tm (62.8 °C) to the unmodified control 2′-O-MePS AO sequence, probably due to stronger π–π stacking interaction (Table 1).We then evaluated the exon-skipping efficiency of the AOs in vitro using H-2Kb-tsA58 (H2K) mdx mouse myotubes. Briefly, H2K mdx myoblasts were plated for differentiation into myotubes 24 h prior to transfection with the AOs using lipid-based delivery agent Lipofectin (2:1 ratio of lipo:AO) at 25, 50, 100 and 200 nM. The cells were then collected after 24 h of treatment and the RNA was extracted followed by reverse-transcriptase polymerase chain amplification (RT-PCR) analysis by amplifying the product across exon 20–26 as previously reported.22,23 The results clearly demonstrated that all three AOs efficiently induced exon-23 skipping in DMD transcript at all concentrations from 25 nM to 200 nM (Fig. 2A) in a dose-dependent manner by yielding the skipped product of 688 bp from the full-length 901 bp product. The actual exon-skipping efficiencies of the AOs were analysed by densitometry (semiquantitative) and the percentage of exon-23 skipping was determined by analysing the amount of exon-23 RT-PCR product over the full-length dystrophin product band. Densitometry results revealed that the ON1 containing two distantly positioned 5-(phenyltriazol)-2′-deoxyuridines achieved slightly higher exon-23 skipping efficiency at 25 nM (57%) and 50 nM (61%) concentrations compared to the control 2′-O-MePS AO (51% and 60% respectively at 25 and 50 nM) (Fig. 2A and B). However, at higher concentrations, the full 2′-O-MePS AO showed higher exon-23 skipping efficacy (69% at 100 and 70% 200 nM; Fig. 2A and B). Notably, ON2 containing two consecutive 5-(phenyltriazol)-2′-deoxyuridines showed slightly reduced exon-skipping efficacy at both lower (47 and 54% at 25 and 50 nM, respectively) and higher concentrations (57 and 62% at 100 and 200 nM, respectively; Fig. 2A and B). This observation does not correlate with the melting temperature analysis data. We speculate that this may be due to the structural features of the AO in the cellular environment which may distort the AO-target complex because of the positioning of the 5-(phenyltriazol)-2′-deoxyuridine monomers, and this needs to be further investigated.
 | ||
| Fig. 2 (A) RT-PCR analysis of exon-23 skipping in H2K mdx mouse myotubes; (B) densitometry analysis of exon-23 skipping in H2K mdx mouse myotubes. Concentration range: 25, 50, 100 and 200 nM; gray: full-length exon 23 product; green: exon-23 skipping product; blue: dual exon-22/23 skipping product; UT: untreated; −ve: negative control. | ||
We also observed the presence of another weak product band at 542 bp which is believed to be due to the unfavourable dual exon-22/23 skipping (Fig. 2A). All three AOs induced the dual exon-22/23 skipping at all concentrations and the intensity increases with increasing concentrations, however, the yields were generally very low in all cases, ranging from 15–28%, compared to the major expected exon-23 skipped product of 688 bp (51–70%). ON1 induced slightly more dual exon-22/23 skipping compared to other AOs at 100 nM (28%), but the percentage reduced to 25% at 200 nM, which is comparable to the control AO (26%) and ON2 (24%) (Fig. 2B).
We then performed the cell viability assay to assess the toxicity of the base-modified AOs in parallel to 2′-O-MePS AO. Overall, the AOs (2′-O-MePS, ON1 and ON2, respectively) were found to be relatively non-toxic to cells at 50 nM dose with 95, 96 and 98% cell viable compared to the untreated (Fig. 3). In line with our previous report,6 the 2′-O-MePS AO was found to be slightly toxic at 200 nM with the cell vitality reduced to 84%, while the nucleobase modified ON1 and ON2 maintained 93 and 91% of cell viability respectively at 200 nM (Fig. 3). These results may indicate the benefit of incorporating the nucleobase-modified nucleotides such as 5-(phenyltriazole)-2′-deoxyuridine in reducing the cytotoxic effect of the 2′-O-MePS AO.
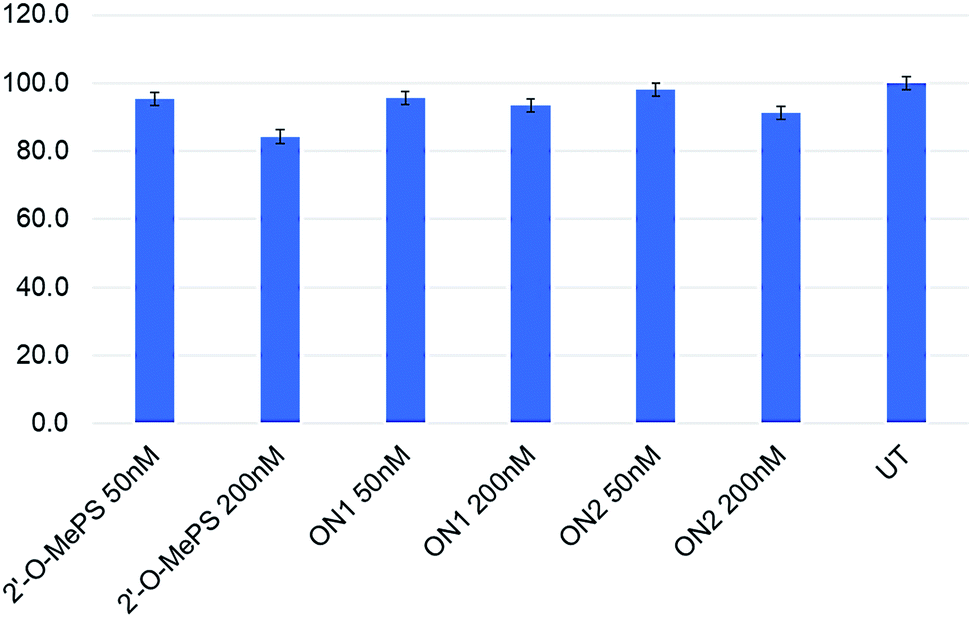 | ||
| Fig. 3 Cell viability assay of the AOs in H2K mdx mouse myotubes. UT: untreated. | ||
It is worth mentioning that the 5-(phenyltriazole)-2′-deoxyuridine building block is a logical choice since it is one of the nucleobase-modifications that has demonstrated the most positive effects on the thermal duplex stability to date,17 but this property requires at least two of these modifications are placed consecutively. Not only is this work the first time this building block is incorporated along with 2′-O-Me in phosphorothioated oligonucleotides, but it is also the first time that this building block is examined in vitro in cells for antisense applications. Notably, the current work unintuitively demonstrates that the exon-skipping potential of the oligonucleotides is slightly better when the 5-(phenyltriazole)-2′-deoxyuridine building blocks are placed distantly compared to consecutively, and that the melting temperature (i.e. hybridization strength) is therefore may not be necessarily a good indicator for skipping efficiency in the context of the 5-(phenyltriazol)-2′-deoxyuridine building block. As these are only our preliminary investigation towards the application of base-modified AOs in exon-skipping, this finding can help to improvise the AO design in future exon-skipping experiments containing base-modified nucleotides with multiple chemistries.
Conclusions
In summary, we have designed and synthesized two 2′-O-MePS mixmer RNA AOs containing two 5-(phenyltriazol)-2′-deoxyuridine monomers placed at distant positions and consecutively, and evaluated their potential to induce exon-skipping in DMD transcript using H2K mdx mouse myotubes. We found that a 2′-O-MePS AO containing two distantly placed 5-(phenyltriazol)-2′-deoxyuridines induced higher exon-23 skipping at low concentrations, compared to the previously reported control 2′-O-MePS AO. Contrary to the melting temperature analysis data, the AO containing two consecutive 5-(phenyltriazol)-2′-deoxyuridines were not as effective, highlighting the impact of the modified monomer positions. Although we report the first demonstration of exon-skipping using nucleobase-modified AOs, further investigations are necessary to gain more insights about the positioning and also in combination with other sugar-modified nucleotides such as locked nucleic acid (LNA) nucleotides.25,26 Our results open the scope of exploring novel base-modified nucleic acid analogues in combination with other chemistries for their potential in constructing efficient exon-skipping AOs.Conflicts of interest
Authors declare no conflicts of interests.Acknowledgements
We thank Prof. Steve Wilton and Prof. Sue Fletcher and their research group for providing H-2Kb-tsA58 (H2K) mdx mouse cells and guidance in cell culture protocols. RNV acknowledges funding from the McCusker Foundation and the Perron Institute for Neurological and Translational Science. R. N. V. also acknowledges the funding support from the Department of Health Western Australia Merit Award Scheme. B. T. L. thanks the MIPS funding scheme of Murdoch University.References
- Y. Y. Syed, Drugs, 2016, 76, 1699–1704 CrossRef CAS.
- D. R. Corey, Nat. Neurosci., 2017, 20, 497–499 CrossRef CAS.
- S. D. Wilton, R. N. Veedu and S. Fletcher, Trends Mol. Med., 2015, 21, 417–426 CrossRef CAS.
- T. L. Bao, R. N. Veedu, S. Fletcher and S. D. Wilton, Expert Opin. Orphan Drugs, 2015, 4, 139–152 CrossRef.
- P. Sazani, A. Astriab-Fischer and R. Kole, Antisense Nucleic Acid Drug Dev., 2003, 13, 119–128 CrossRef CAS.
- B. T. Le, V. V. Filichev and R. N. Veedu, RSC Adv., 2016, 6, 95169–95172 RSC.
- B. T. Le, S. Chen, M. Abramov, P. Herdewijn and R. N. Veedu, Chem. Commun., 2016, 52, 13467–13470 RSC.
- S. Chen, B. T. Le, K. Rahimizadeh, K. Shaikh, N. Mohal and R. N. Veedu, Molecules, 2016, 21, 1582 CrossRef.
- B. T. Le, K. Murayama, F. Shabanpoor, H. Asanumac and R. N. Veedu, RSC Adv., 2017, 7, 34049–34052 RSC.
- B. T. Le, A. M. Adams, S. Fletcher, S. D. Wilton and R. N. Veedu, Mol. Ther.--Nucleic Acids, 2017, 9, 155–161 CrossRef CAS.
- A. Goyenvalle, G. Griffith, A. Babbs, S. El Andaloussi, K. Ezzat and A. Avril, et al. , Nat. Med., 2015, 21, 270–275 CrossRef CAS.
- T. Shimo, K. Tachibana, K. Saito, T. Yoshida, E. Tomita and R. Waki, et al. , Nucleic Acids Res., 2014, 42, 8174–8187 CrossRef CAS.
- A. Aartsma-Rus, W. E. Kaman, M. Bremmer-Bout, A. A. Janson, J. T. den Dunnen, G. J. van Ommen and J. C. van Deutekom, Gene Ther., 2004, 11, 1391–1398 CrossRef CAS.
- X. Gao, X. Shen, X. Dong, N. Ran, G. Han, L. Cao, B. Gu and H. Yin, Mol. Ther.--Nucleic Acids, 2015, 4, e255 CrossRef CAS.
- S. M. Jirka, C. L. Tanganyika-de Winter, J. W. Boertje-van der Meulen, M. van Putten, M. Hiller, R. Vermue, P. C. de Visser and A. Aartsma-Rus, Mol. Ther.--Nucleic Acids, 2015, 4, e265 CrossRef CAS.
- M. Takagi, M. Yagi, K. Ishibashi, Y. Takeshima, A. Surono, M. Matsuo and M. Koizumi, Nucleic Acids Symp. Ser., 2004, 48, 297–298 CrossRef.
- P. Kocalka, N. K. Andersen, F. Jensen and P. Nielsen, ChemBioChem, 2007, 8, 2106–2116 CrossRef CAS.
- N. K. Andersen, H. Døssing, F. Jensen, B. Vester and P. Nielsen, J. Org. Chem., 2011, 76, 6177–6187 CrossRef CAS.
- M. Hornum, P. Kumar, P. Podsiadly and P. Nielsen, J. Org. Chem., 2015, 80, 9592–9602 CrossRef CAS.
- M. Hornum, A. Djukina, A.-K. Sassnau and P. Nielsen, Org. Biomol. Chem., 2016, 14, 4436–4447 RSC.
- T. A. Rando and H. M. Blau, J. Cell Biol., 1994, 125, 1275–1287 CrossRef CAS.
- C. J. Mann, K. Honeyman, A. J. Cheng, T. Ly, F. Lloyd, S. Fletcher, J. E. Morgan, T. A. Partridge and S. D. Wilton, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 42–47 CrossRef CAS.
- S. D. Wilton, F. Lloyd, K. Carville, S. Fletcher, K. Honeyman, S. Agrawal and R. Kole, Neuromuscul. Disord., 1999, 9, 330–338 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- R. N. Veedu and J. Wengel, Chem. Biodivers., 2010, 7, 536 CrossRef CAS.
- R. N. Veedu and J. Wengel, RNA Biol., 2009, 6, 321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10964d |
| This journal is © The Royal Society of Chemistry 2017 |