
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly active and thermally stable 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex as catalyst for norbornene polymerization
Ping Huo*a,
Jingbo Lia,
Wanyun Liu *a,
Guangquan Meia and
Xiaohui Heb
*a,
Guangquan Meia and
Xiaohui Heb
aKey Laboratory of Jiangxi University for Applied Chemistry and Chemical Biology, Yichun University, Yichun 336000, China. E-mail: ycxyhuoping@163.com; liuwanyun2006@163.com
bDepartment of Chemistry/Institute of Polymers, Nanchang University, 999 Xuefu Avenue, Nanchang 330031, China
First published on 9th November 2017
Abstract
The 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex was synthesized and characterized. The nickel complex exhibited high activity toward the homopolymerization of norbornene and copolymerization of norbornene with 5-norbornene-2-carboxylic acid methyl ester in the presence of B(C6F5)3. It was observed that the nickel catalyst with large steric hindrance from polycyclic aromatic systems on the backbone was found to exhibit good thermal stability and afford high incorporation of polar monomers in norbornene copolymerization.
Introduction
The norbornene (NB) (bicyclo[2.2.1]hept-2-ene) addition polymer exhibits excellent dielectric properties, optical transparency and rather high thermal stability, which has been of great interest over the past decade.1–3 It is well-known that the NB polymerization reaction can be efficiently carried out using conventional Ziegler–Natta catalysts,4,5 metallocene catalysts6,7 and late-transition metal catalysts.8–14 Recently, the late-transition metal catalysts have shown lower oxophilicity and resistance toward polar functionalities, which resulted in renewed interest. The common late transition metal catalysts are nickel(II) and palladium(II) complexes. Since α-diimine nickel, palladium complexes have been successfully developed by Brookhart and his collaborators and show high catalytic activity for α-olefin polymerization.15–17 A series of breakthroughs have been made in this field, mainly involving other late transition metal α-diimine catalysts.18–21A key insight from these early studies is that the incorporation of bulky axial substituents in the α-diimine ligand is crucial in the enhancement of thermally stable of catalysts, because the bulky substituents can hinder the rotation of aniline moieties and protect the metal center effectively. In addition, these catalysts can produce polyolefins with high molecular weight and have good tolerance toward polar groups. Since its discovery, this class of bulky catalysts has attracted a tremendous amount of interest. Much of the work is focused on changing the N-bound aryl group22–25 or modifying the backbone,26–28 including the use of axial donating diimine ligands. A macro cyclic ligand by connecting the aryl substituents of 2,6-diarylphenylimino moieties using ethylene tether was synthesized by Guan and co-workers.29 Both the cyclophane-based Ni(II) and Pd(II) catalysts showed significantly higher thermal stability than the acyclic analogs. Wu have synthesized α-diimine nickel catalyst with a bulky camphyl backbone, which show good thermal stability toward olefin polymerizations and obtain special branched polyolefin.30,31 A series of Ni(II) dihalide complexes bearing unsymmetrical α-diimine ligands were synthesized and exhibited high activity and thermal stability in olefin polymerization.32,33 Recently, we have successfully explored a type of thermostable and high activity three-dimensional geometry 9,10-dihydro-9,10-ethanoanthracene-11,12-diimine Ni(II) and Pd(II) catalysts toward norbornene polymerization.34,35 The three-dimensional geometry α-diimine metal catalysts with big steric hindrance on the backbone were capable of norbornene polymerization at 120 °C only in the presence of B(C6F5)3 and afford high incorporation of polar monomer.
Based on the above results, we herein continue to increase the hindrance on the backbone of α-diimine late transition metal catalyst and synthesized the 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex with large steric hindrance of polycyclic aromatic systems. The catalytic activity of complex with only B(C6F5)3 as cocatalyst in NB polymerization were also investigated. The nickel(II) complex appeared to be thermostable and highly active catalyst on the NB homopolymerization and copolymerization of NB and 5-norbornene-2-carboxylic acid methyl ester (NB-COOCH3).
Results and discussion
Synthesis of complexes NiLBr2
The stoichiometric condensation reaction of 6,13-dihydro-6,13-ethanopentacene-15,16-dione and 2,6-dimethyl aniline produced α-diimine ligand (Scheme 1). Novel synthesized α-diimine ligand formed NiLBr2 complex in good yields (Scheme 1) on reaction with Ni(DME)Br2 (DME = dimethoxyethane) in dichloromethane. Due to low solubility in dichloromethane, the crystal structure of NiLBr2 complex could not be obtained and its structure was determined by elemental analysis and NMR spectroscopy. | ||
| Scheme 1 Synthesis of (α-diimine ligand) nickel dibromide complex. | ||
Homopolymerization of NB
First of all, NB polymerization activities of the synthesized complex NiLBr2 were investigated with B(C6F5)3 as cocatalyst and toluene as the reaction solvent. It is well known that catalytic property was depended on the structure of catalyst. To verify that the polycyclic aromatic backbone can improve stability of α-diimine nickel complex NiLBr2, a series of norbornene homopolymerization were carried out at different temperature. Table 1 displayed the results of norbornene homopolymerization with NiLBr2 in the temperature range 0–110 °C. The observed basic trend was that the activity of NiLBr2 reached a maximum value at 100 °C and then decreased with temperature increasing. Even at 110 °C, a high activity 3.75 × 107 gpolymer molNi−1 h−1 can be achieved. NiLBr2 exhibited good thermal stability could be attributed to its rigid and bulky backbone.| No. | T (°C) | Reaction time (min) | PNB yield (%) | Activityb (gpolymer molNi−1 h−1) |
|---|---|---|---|---|
a Conditions: n[NiLBr2] = 5 × 10−6 mol, cocatalyst is B(C6F5)3, B/complex/monomer (n/n/n) is 5/1/15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, the solvent is toluene, the total volume is 22 mL.b In units of 106 gpolymer molNi−1 h−1. 000, the solvent is toluene, the total volume is 22 mL.b In units of 106 gpolymer molNi−1 h−1. |
||||
| 1 | 0 | 30 | 6.38 | 0.18 |
| 2 | 30 | 30 | 51.21 | 1.44 |
| 3 | 60 | 2 | 80.30 | 33.97 |
| 4 | 90 | 2 | 97.93 | 41.43 |
| 5 | 100 | 2 | 98.95 | 41.84 |
| 6 | 110 | 2 | 88.26 | 37.50 |
Copolymerization of NB and NB-COOCH3
Since NiLBr2/B(C6F5)3 system had high activity (4.18 × 107 gpolymer molNi−1 h−1) toward NB homopolymerization, it was selected as catalyst to investigate whether it also had high activity toward copolymerization of NB with NB-COOCH3. A series of copolymerizations of NB with NB-COOCH3 were investigated with the catalyst system of NiLBr2/B(C6F5)3 in toluene at 60 °C for 1 h under nitrogen atmosphere (Table 2). From the Table 2, we could find that the NiLBr2/B(C6F5)3 system exhibited high activity toward copolymerizations of NB with NB-COOCH3, which was far higher than the activities of the other α-diimine catalysts.34 However, NB-COOCH3 could not be homopolymerized in the same conditions. The reason was that oxygen atom of functionalized NB derivatives competed with the double-bond for the coordination, which led to high concentration of oxygen atom would impede double-bond coordination.35| NB/NB-COOCH3 (mol/mol) | Reaction time (min) | Yield (%) | Activityb | Mwc (g mol−1) | Mw/Mnc | NB-COOCH3d (mol%) |
|---|---|---|---|---|---|---|
| a Conditions: c[Cat.] = 5.0 × 10−6 mol; cocatalyst is B(C6F5)3, B/complex/monomer (n/n/n) is 10/1/15000, polymerization temperature is 60 °C, the solvent is toluene, the total volume is 22 mL.b In units of gpolymer molNi−1 h−1.c Determined by GPC vs. polystyrene standards in THF.d Determined by 1H NMR spectroscopy in CDCl3.e Not determined. |
||||||
| 100/0 | 2 | 80.30 | 3.40 × 107 | —e | —e | —e |
| 80/20 | 60 | 47.89 | 7.59 × 105 | 3.6 × 105 | 1.1 | 10.2 |
| 60/40 | 60 | 38.90 | 6.83 × 105 | 6.4 × 104 | 1.2 | 19.4 |
| 50/50 | 60 | 16.87 | 3.11 × 105 | 2.7 × 104 | 1.1 | 28.3 |
| 0/100 | 60 | Trace | —e | —e | —e | —e |
Although the obtained polynorbornene (PNB) were hardly soluble in common organic solvents such as CHCl3, CH2Cl2, THF and so on, the copolymers of NB and NB-COOCH3 were soluble in these common organic solvents, which exhibited better solubility than PNB. Compared with NB homopolymerization, the yield and activities of copolymerization of NB and NB-COOCH3 dramatically decreased with the content of NB-COOCH3 increasing.
To obtain the reactivity ratios of NB and NB-COOCH3 monomers, we changed the polymerization time and the feed content of catalysts to control the conversion rate to be under 10%. The results were shown in Table 3. The reactivity ratios were determined by the Kelen–TüdÕs method (Fig. 1).36 For 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex/B(C6F5)3, the reactivity ratio of NB-COOCH3 was 0.146, and the reactivity ratio of NB was 2.282.
| Entry | NB/NB-COOCH3 (mol/mol) | Time (h) | Yield (%) | mol% of NB-COOCH3 in copolymerb |
|---|---|---|---|---|
| a Conditions: n[NiLBr2] = 5 × 10−6 mol, B/complex/monomer (n/n/n) is 5/1/15000, the solvent is toluene, the total volume is 22 mL.b Determined by 1H NMR spectroscopy in CDCl3. |
||||
| 1 | 60/40 | 0.30 | 6.34 | 18.9 |
| 2 | 50/50 | 1 | 5.78 | 26.3 |
| 3 | 40/60 | 2 | 1.46 | 33.4 |
| 4 | 30/70 | 12 | 3.32 | 42.3 |
| 5 | 20/80 | 24 | 0.91 | 48.2 |
 | ||
| Fig. 1 Linear fitting of the NB/NB-COOCH3 copolymers catalyzed by NiLBr2/B(C6F5)3 systems according to Kelen–TÜdÕs method. | ||
Characterization of copolymers
The obtained copolymers were characterized by GPC and the corresponding data were shown in Table 2. The molecular weights of the obtained copolymers were up to 105, and the molecular weight distributions MWD of the obtained polymers were all relative narrow (MWD < 1.5).These obtained copolymers were characterized by spectra (IR, 1H NMR) and thermal analysis (DSC, TGA). In Fig. 2, the signal of double-bond at 1710 cm−1, assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the ROMP structure of PNB, was not observed. The signals at about 941 cm−1 could be attributed to the ring of bicyclo[2.2.1]heptanes further illustrating the monomers via vinyl-type addition polymerization. The sharp signals at 1740 cm−1 from the vibration of the carbonyl group were characteristic of ester moieties. This confirmed that the NB-COOCH3 had been inserted into the chains of the copolymers and to be true vinyl-type addition copolymer rather than blend polymer.
C bond of the ROMP structure of PNB, was not observed. The signals at about 941 cm−1 could be attributed to the ring of bicyclo[2.2.1]heptanes further illustrating the monomers via vinyl-type addition polymerization. The sharp signals at 1740 cm−1 from the vibration of the carbonyl group were characteristic of ester moieties. This confirmed that the NB-COOCH3 had been inserted into the chains of the copolymers and to be true vinyl-type addition copolymer rather than blend polymer.
 | ||
| Fig. 2 FTIR spectra of poly(NB-co-NB-COOCH3) with (a) 10.2%, (b) 19.4%, and (c) 28.3% of NB-COOCH3 molar ratios separate obtained by NiLBr2/B(C6F5)3 systems. | ||
Fig. 3 proved that the polymers are vinyl-addition type by the absence of the resonance of the proton hydrogen connected to the double bond at 5.3–5.9 ppm. The peak at 0.7–1.6 ppm could be assigned to methene hydrogen corresponding to H5/H6/H7/H6′/H7′/H9′; the peak at 1.6–2.5 ppm could be assigned to the methine hydrogen corresponding to H1/H2/H3/H4/H1′/H2′/H3′/H4′; the peak at 2.5–2.8 ppm could be attributed to the methine hydrogen corresponding to H5′; and the characteristic peak at 3.4–3.9 ppm was assigned to the methyl hydrogen corresponding to H8′. The content of NB-COOCH3 in copolymers was calculated by the following way.37
| NB-COOCH3 (mol%) = 2IH8′/3(IH2/H3 + IH2′/H3′) | (1) |
 | ||
| Fig. 3 1H NMR spectra of poly(NB-co-NB-COOCH3) with (a) 10.2%, (b) 19.4%, and (c) 28.3% of NB-COOCH3 molar ratios separate obtained by NiLBr2/B(C6F5)3 system. | ||
The results of calculated NB-COOCH3 content were presented in Table 2. It could be observed that the content of NB-COOCH3 in copolymers was calculated from 10.2 to 28.3% by varying the comonomer feed ratios (NB-COOCH3/NB) from 10 to 50%.
The TGA curves of the copolymers with different NB-COOCH3 content prepared by NiLBr2/B(C6F5)3 were shown in Fig. 4. The decomposition temperatures (Td)s of the polymers were as high as 339.6–377.7 °C and decreased with the increasing of NB-COOCH3 content in copolymers, which was similar to the transition temperatures.
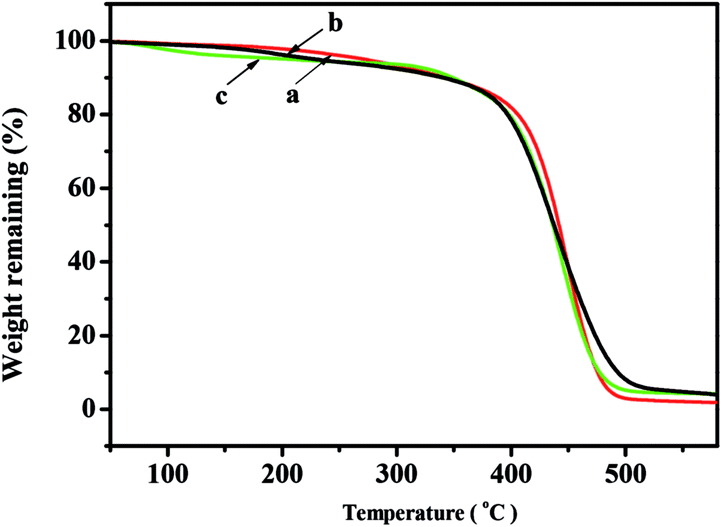 | ||
| Fig. 4 TGA curves of poly(NB-co-NB-COOCH3) with(a) 10.2%, (b) 19.4%, and (c) 28.3% of NB-COOCH3 molar ratios separate obtained by NiLBr2/B(C6F5)3 systems. The (Td)s of the polymers are: (a) 377.7 °C, (b) 364.5 °C, (c) 339.6 °C. | ||
The DSC curves and glass transition temperatures (Tg)s of the copolymers with different NB-COOCH3 ratio prepared by NiLBr2/B(C6F5)3 were shown in Fig. 5. The (Tg)s of the polymers were reduced to 265.7–279.6 °C when the content of NB-COOCH3 increased in the copolymers.
 | ||
| Fig. 5 DSC curves of poly(NB-co-NB-COOCH3) with (a) 10.2%, (b) 19.4%, and (c) 28.3% of NB-COOCH3 molar ratios separate obtained by NiLBr2/B(C6F5)3 systems. | ||
All the copolymers, irrespective of the polymerization conditions employed showed identical powder WXRD pattern. Fig. 6 showed the WXRD curves of the copolymers with different NB-COOCH3 contents prepared by NiLBr2/B(C6F5)3. There was no trace of Bragg reflection in the characteristic of crystalline regions, so the polymers were noncrystalline. The occurrence of two halos at 2θ values of 9.72–10.95° (peak 1) and 18.07–19.80° (peak 2) were characteristic for PNB.38
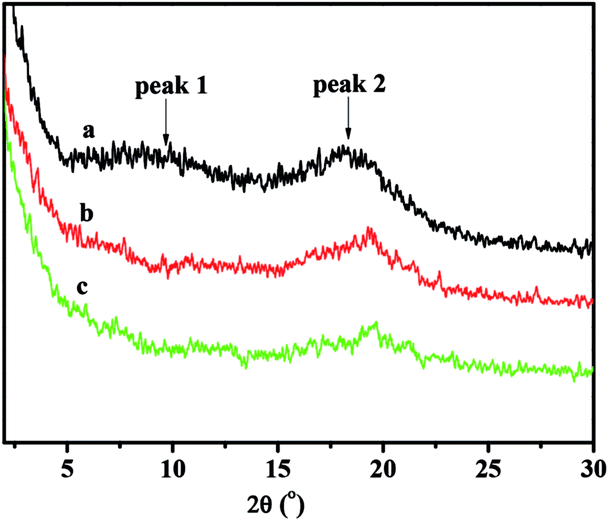 | ||
| Fig. 6 WXRD curves of poly(NB-co-NB-COOCH3) with(a) 10.2%, (b) 19.4%, and (c) 28.3% of NB-COOCH3 molar ratios separate obtained by NiLBr2/B(C6F5)3 systems. | ||
Experimental
Materials
All the reactions were performed under an atmosphere of dry and oxygen-free argon using standard vacuum, Schlenk, or under nitrogen atmosphere in glove box (M Braun). Toluene and hexane were refluxed over metallic sodium for 24 hours before being used. Dichloromethane was dried over CaH2 for 8 hours and distilled under a nitrogen atmosphere. NB was purchased from Alfa Aesar and purified through drying by sodium and distilled under dry nitrogen, then made a solution in toluene. NB-COOCH3 was purchased from Aldrich and purified by CaH2. Other commercially available reagents were purchased and used without purification.General
The element analysis was performed on elementar-vario EL cube elemental analyzer. The nuclear magnetic resonance (NMR) was recorded on a Bruker ARX 400 NMR spectrometer at ambient temperature, with CDCl3 as the solvent. Chemical shifts for the 1H NMR and 13C NMR spectrum were given in parts per million from the peak for internal tetramethylsilane. The gel permeation chromatography (GPC) was conducted with a Breeze Waters system using polystyrenes as the standard and tetrahydrofuran as the eluent at a flow rate of 1.0 mL min−1. The IR spectra were recorded by a Shimadzu IR Prestige-21 FTIR spectrophotometer. The wide-angle X-ray diffraction (WXRD) curves were recorded on a Bruker D8 Focus X-ray diffractometer, operating at 40 kV and 40 mA with a copper target (λ = 1.54 Å) and at a scanning rate of 2° min−1. The thermal gravimetric analysis (TGA) measurements were performed on a Perkin-Elmer instrument TGA 7 from room temperature to 700 °C at a rate of 10 °C min−1 under nitrogen atmosphere. The differential scanning calorimetry (DSC) measurements were obtained on a Shimadzu DSC-60 with a heating/cooling rate of 10 °C min−1 under nitrogen atmosphere.Synthesis of α-diimine ligand (L)
Under a nitrogen atmosphere, to a solution of 3.0 mmol of the 2,6-dimethyl aniline in 150 mL toluene, 1.0 mmol of 6,13-dihydro-6,13-ethanopentacene-15,16-dione and a catalytic amount of p-toluenesulfonic acid were added. The mixture was heated under reflux in the dark. The resulting water was removed as azeotropic mixture using a Dean–Stark apparatus. After 24 hours, the reaction mixture was cooled to room temperature and evaporated at reduced pressure. The residual solids were further purified by silica column chromatography (v/v, 10/1, petroleum ether/ethyl acetate) and were crystallized from the mixture of petroleum ether and ethyl acetate to get the product as yellow crystals in 42.5% yield. The α-diimine ligand was characterized by elemental analysis and 1H and 13C NMR spectroscopy. Mp: 228–229 °C. Elemental analysis (%), found: C, 88.61; H, 6.10; N, 5.29; calcd: C, 88.85; H, 5.97; N, 5.18. 1H NMR (CDCl3, δ, ppm): 1.90 (s, 12H), 4.88 (s, 2H), 7.05–7.28 (m, 6H), 7.54 (m, 4H), 7.88 (m, 4H), 7.96 (s, 4H). 13C NMR (CDCl3, δ, ppm): 17.82, 51.43, 124.51, 125.61, 126.08, 127.71, 128.39, 131.52, 133.74, 136.05, 148.10, 160.24.Synthesis of (α-diimine ligand) nickel dibromide complex (NiLBr2)
0.2 mmol α-diimine ligand (L) and 0.2 mmol dimethoxymethane nickel dibromide were added to a Schlenk tube together with 10 mL dried dichloromethane, the reaction mixture was then stirred for 8 h at room temperature and 10 mL absolute hexane was added. The dark red crystal (α-diimine ligand) nickel dibromide complex (NiLBr2) was crystallized from the mixture of dichloromethane and hexane in 82% yield. Complex NiLBr2 was characterized by elemental analysis and 1H NMR spectroscopy. Mp: 342–344 °C. Elemental analysis (%), found: C, 62.96; H, 4.36; N, 3.84; calcd: C, 63.28; H, 4.25; N, 3.69. 1H NMR (CDCl3, δ, ppm): 1.92 (s, 12H), 4.95 (s, 2H), 7.04–7.30 (m, 6H), 7.56 (m, 4H), 7.91 (m, 4H), 7.97 (s, 4H).Homopolymerization of NB
In a typical procedure, the appropriate B(C6F5)3 solid was introduced into the round-bottom glass flask, and then a certain amount of a toluene solution of NB and quantitative nickel complex NiLBr2 solution were syringed into the well-stirred solution in order, and the reaction mixture was continuously stirred for an designed period at the polymerization temperature. Polymerizations were terminated by addition of the acidified ethanol (v/v, 10/1, ethanol/HCl) and stayed overnight. The polymers were then obtained through filtration or centrifugation and washed by ethanol several times and were dried at 40 °C to a constant weight.Polymerization of norbornene (NB) and 5-norbornene-2-carboxylic acid methyl ester (NB-COOCH3)
A typical procedure was as follows: the appropriate B(C6F5)3 solid was introduced into the 100 mL round-bottom glass flask, then a certain amount of toluene solution of NB, NB-COOCH3, and appropriate amount of catalyst were syringed into the well stirred solution in order, and the reaction was continuously stirred for an appropriate period at the polymerization temperature. The polymerization was stopped by addition of acidic ethanol (v/v, 10/1, ethanol/HCl) and stayed overnight. The polymers were then obtained through filtration or centrifugation and washed by ethanol several times and were dried at 40 °C to a constant weight.Conclusions
The 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex with large steric hindrance of polycyclic aromatic systems on the backbone was synthesized and characterized. The new 6,13-dihydro-6,13-ethanopentacene-15,16-diimine nickel(II) complex on NB polymerization showed high activities even up to 4.18 × 107 gpolymer molNi−1 h−1. Meanwhile, the complex also displayed high activity toward the copolymerization of NB and NB-COOCH3. The complex with big hindrance on backbone was found to show good thermal stability for NB polymerization and afford high-molecular-weight copolymers with high incorporation of polar monomer. These findings identify a promising strategy to explore new catalysts with stability and good tolerance to polar comonomer by designing bulkier backbone α-diimine catalytic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21664014), the Natural Science Foundation of Jiangxi Province (20151BAB206021 and 20161BAB215197).Notes and references
- C. Janiak and P. G. Lassahn, J. Mol. Catal. A: Chem., 2001, 166, 193 CrossRef CAS.
- N. R. Grove, P. A. Kohl, S. A. B. Allen, S. Jayaraman and R. Shick, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3003 CrossRef CAS.
- S. Ahmed, S. A. Bidstrup, P. A. Kohl and P. J. Ludovice, J. Phys. Chem. B, 1998, 102, 9783 CrossRef CAS.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541 CrossRef CAS.
- G. Natta, P. Pino, P. Corradini, F. Dannusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708 CrossRef CAS.
- A. El-Batta, A. W. Waltman and R. H. Grubbs, J. Organomet. Chem., 2011, 696, 2477 CrossRef CAS.
- N. Diteepeng, X. Y. Tang, X. H. Hou, Y. S. Li, K. Phomphrai and K. Nomuraa, Dalton Trans., 2015, 44, 12273 RSC.
- L. Piche, J. C. Daigle, G. Rehse and J. P. Claverie, Chem.–Eur. J., 2012, 18, 3277 CrossRef CAS PubMed.
- Q. Xing, T. Zhao, Y. Qiao, L. Wang, C. Redshaw and W. H. Sun, RSC Adv., 2013, 3, 26184 RSC.
- M. P. Weberski Jr, L. Chen, M. Delferro and T. J. Marks, Chem.–Eur. J., 2012, 18, 10715 CrossRef PubMed.
- A. Sachse, S. Demeshko, S. Dechert, V. Daebel, A. Lange and F. Meyer, Dalton Trans., 2010, 39, 3903 RSC.
- M. P. Weberski, C. Chen, M. Delferro, C. Zuccaccia, A. Macchioni and T. J. Marks, Organometallics, 2012, 31, 3773 CrossRef CAS.
- L. H. Guo, H. Y. Gao, Q. R. Guan, H. B. Hu, F. S. Liu and Q. Wu, Organometallics, 2012, 31, 6054 CrossRef CAS.
- H. B. Hu, H. Y. Gao, D. R. Chen, G. L. Li, Y. X. Tan, G. D. Liang, F. M. Zhu and Q. Wu, ACS Catal., 2015, 5, 122 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664 CrossRef CAS.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS.
- H. D. Mkoyi, S. O. Ojwach, I. A. Guzei and J. Darkwa, J. Organomet. Chem., 2013, 724, 95 CrossRef CAS.
- L. H. Guo and C. L. Chen, Sci. China: Chem., 2015, 58, 1663 CrossRef CAS.
- H. Liu, W. Z. Zhao, X. Hao, C. Redshaw, W. Huang and W. H. Sun, Organometallics, 2011, 30, 2418 CrossRef CAS.
- F. Zhai and R. F. Jordan, Organometallics, 2017, 36, 2784 CrossRef CAS.
- B. Liu, M. Z. Fang, S. Y. Jie, Z. Y. Bu and B. G. Li, Chin. J. Polym. Sci., 2016, 34, 221 CrossRef CAS.
- A. Meduri, T. Montini, F. Ragaini, P. Fornasiero, E. Zangrando and B. Milani, ChemCatChem, 2013, 5, 1170 CrossRef CAS.
- J. L. Rhinehart, L. A. Broen and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316 CrossRef CAS PubMed.
- S. Y. Dai, X. L. Sui and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948 CrossRef CAS PubMed.
- C. S. Popeney, C. M. Levins and Z. B. Guan, Organometallics, 2011, 30, 2432 CrossRef CAS.
- F. S. Liu, H. Y. Gao, Z. L. Hu, H. B. Hu, F. M. Zhu and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3859 CrossRef CAS.
- D. H. Leung, J. W. Ziller and Z. B. Guan, J. Am. Chem. Soc., 2008, 130, 7538 CrossRef CAS PubMed.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. B. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821 CrossRef CAS PubMed.
- L. H. Guo, H. Y. Gao, Q. R. Guan, H. B. Hu, J. A. Deng, J. Liu, F. S. Liu and Q. Wu, Organometallics, 2012, 31, 6054 CrossRef CAS.
- J. Liu, D. R. Chen, H. Wu, Z. F. Xiao, H. Y. Gao, F. M. Zhu and Q. Wu, Macromolecules, 2014, 47, 3325 CrossRef CAS.
- S. Z. Du, S. L. Kong, Q. S. Shi, J. Mao, C. Y. Guo, J. J. Yi, T. L. Liang and W. H. Sun, Organometallics, 2015, 34, 582 CrossRef CAS.
- D. D. Jia, W. J. Zhang, W. L. Liu, L. Wang, C. Redshaw and W. H. Sun, Catal. Sci. Technol., 2013, 3, 2737 CAS.
- P. Huo, W. Y. Liu, X. H. He, Z. H. Wei and Y. W. Chen, Polym. Chem., 2014, 5, 1210 RSC.
- P. Huo, W. Y. Liu, X. H. He, H. M. Wang and Y. W. Chen, Organometallics, 2013, 32, 2291 CrossRef CAS.
- T. Kelen and F. J. TüdÕs, J. Macromol. Sci., Part A, 1975, 9, 1 CrossRef.
- Y. P. Xing, Y. W. Chen and X. H. He, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4425 CrossRef CAS.
- X. Mi, L. Wang, Y. C. Ke and Y. L. Hu, Macromol. Chem. Phys., 2003, 204, 868 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |