
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanoresponsive luminescence and liquid-crystalline behaviour of a cyclophane featuring two 1,6-bis(phenylethynyl)pyrene groups†
Yoshimitsu Sagara* and
Nobuyuki Tamaoki *
*
Research Institute for Electronic Science, Hokkaido University, N20, W10, Kita-Ku, Sapporo 001-0020, Japan. E-mail: sagara@es.hokudai.ac.jp; tamaoki@es.hokudai.ac.jp
First published on 5th October 2017
Abstract
Cyclophanes have been attractive targets over recent decades because of their fascinating molecular structures and inherent ability to act as supramolecular hosts. However, cyclophanes that exhibit stimuli-responsive luminescence in their condensed states are still very rare, although luminescent cyclophanes could be suitable platforms for sophisticated photofunctional molecular assembled materials. We here report that a pyrenophane featuring two π-extended pyrene groups exhibits mechanoresponsive luminescence in the solid states and shows a nematic liquid-crystalline phase at elevated temperature. Two 1,6-bis(phenylethynyl)pyrene moieties form intramolecular excimers in a diluted chloroform solution, whereas no clear intra- and/or intermolecular excimer formation was observed for the solid state accessed through slow cooling from the nematic phase. Photoluminescence spectroscopic measurements, emission lifetime measurements, and X-ray diffraction patterns confirmed that mechanical stimuli result in conversion to much less ordered molecular assembled states in which some luminophores form excimers, leading to changes in photoluminescence colours.
Introduction
A variety of cyclophanes, which are cyclic compounds composed of aromatic groups and aliphatic linkers, have been developed from the viewpoints of synthetic chemistry and supramolecular chemistry due to their unusual molecular structures and usefulness as host cavities.1–3 Photoluminescent cyclophanes have attracted much attention because their photophysical properties can change when guest ions or molecules are incorporated into the cavities.4–8 The fascinating characteristics make them promising candidates for ion or molecular sensors.3d In contrast to an increasing number of studies on luminescent cyclophanes in solution, little attention has been paid to the photophysical properties or phase transition behaviour of luminescent cyclophanes in the condensed states. In 2013, Ramaiah's group reported that an anthracene-based cyclophane exhibits a photoluminescent colour change in the solid state when exposed to methanol vapour.9 Our group also demonstrated that the photophysical properties of anthracene-based luminescent cyclophanes are controlled by thermal and mechanical stimuli in the solid states.10 Our cyclophanes have two 9,10-bis(phenylethynyl)anthracene groups that are bridged by tetraethyleneglycol10a or hexaethyleneglycol10b linkers. Furthermore, the latter was found to show a nematic liquid-crystalline phase.10b These results unambiguously indicate that luminescent cyclophanes could be an intriguing platform to develop various photofunctional condensed materials.Here, we report the mechanoresponsive luminescence and liquid-crystalline behaviour of a luminescent cyclophane having two π-extended pyrene groups. Pyrene is a well-known aromatic hydrocarbon due to its highly emissive properties and excimer formation.11 Since several pyrene-based cyclophanes, pyrenophanes, were prepared by Misumi's group in the 1970s,12 various pyrenophanes have been developed to examine their strained molecular structures and curious photophysical properties.2d,6 For example, Inouye's group demonstrated that a series of water-soluble cyclophanes having two π-extended pyrene groups show intramolecular excimer formation and that some aromatic guests can form inclusive complexes with the cyclophanes due to the neutral cavities.6 However, little attention has been given to the phase transition behaviour of pyrenophanes, and no pyrenophanes have been reported to show external stimuli-responsive luminescence in the solid states, except for one.13
In this decade, we have seen an increasing number of organic or organometallic compounds that show mechanoresponsive luminescence.13–23 A variety of molecular structures have been found to show mechanical stimuli-responsive behaviour.13–23 However, cyclic compounds that exhibit mechanoresponsive luminescence are very rare, although the introduction of a cyclic structure is a reliable approach to developing mechano- and/or thermoresponsive luminescent materials.10,13 Very recently, we reported the first pyrenophane that exhibits mechano- and thermochromic luminescence in the condensed states.13 The asymmetric pyrenophane has one π-extended pyrene group in its cyclic structure and exhibits a nematic phase above ∼110 °C. Rapid cooling of the sample in the nematic phase to room temperature leads to the formation of a supercooled nematic liquid-crystalline phase. The kinetically trapped state is immediately converted to a thermodynamically stable crystalline phase on thermal treatment, which is accompanied by a photoluminescent colour change. Furthermore, the obtained crystalline state exhibits mechanochromic luminescence. However, the correlation between the molecular structure and the stimuli-responsive luminescence remains unclear. Further investigation of varying molecular structures is required to obtain more insight into the stimuli-responsive luminescence characteristics and phase transition behaviour.
Results and discussion
A new symmetric cyclophane 1 was designed to have two 1,6-bis(phenylethynyl)pyrene groups as luminophores (Fig. 1). Hexaethyleneglycol was chosen as the linker between two luminophores since a previously reported anthracene-based cyclophane having hexaethyleneglycol linkers exhibits a nematic liquid-crystalline phase.10b Compound 1 was synthesised through Williamson ether synthesis from 1,6-bis(4-hydroxyphenylethynyl)pyrene and 1,6-bis(phenylethynyl)pyrene with two bromo-terminated hexaethyleneglycol groups. The molecular structures of the reported asymmetric cyclophane 2 and linear reference analogue 3 are also shown in Fig. 1.13 A comparison of the 1H NMR spectra of 1 and 3 in CDCl3 (Fig. 2) reveals upfield shifts of peaks corresponding to the luminophores. The inherent cyclic structure of 1 brings one luminophore to proximity of the other luminophore in CDCl3 and thus affects the circular current of the luminophores.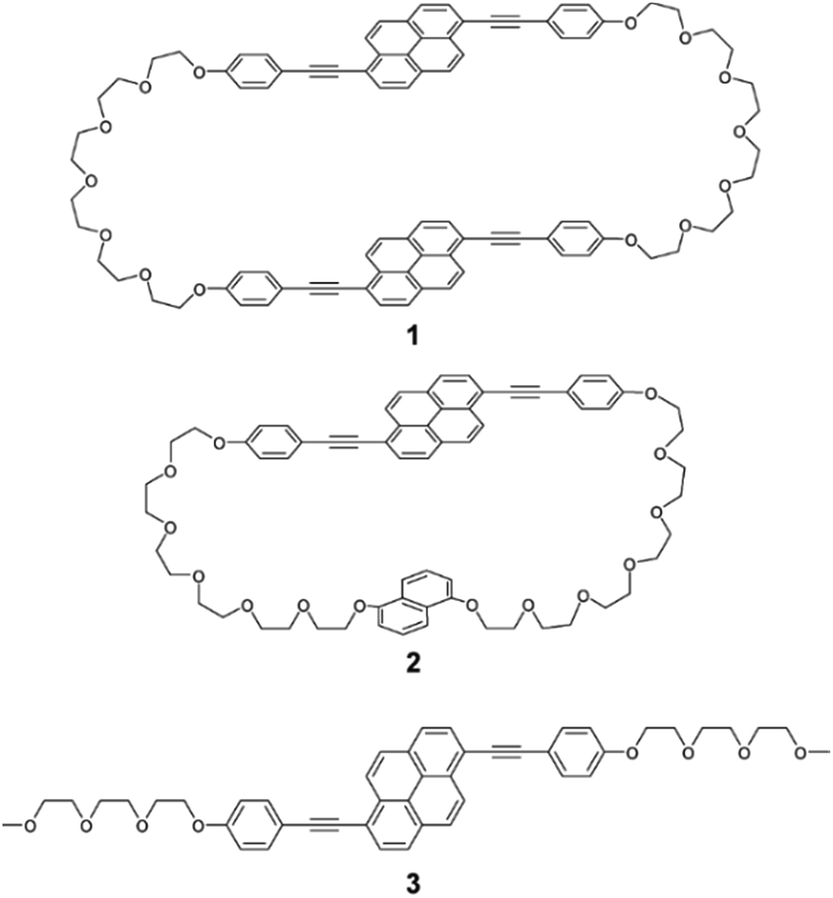 | ||
| Fig. 1 Molecular structures of symmetric pyrenophane 1, asymmetric pyrenophane 2, and linear reference compound 3. | ||
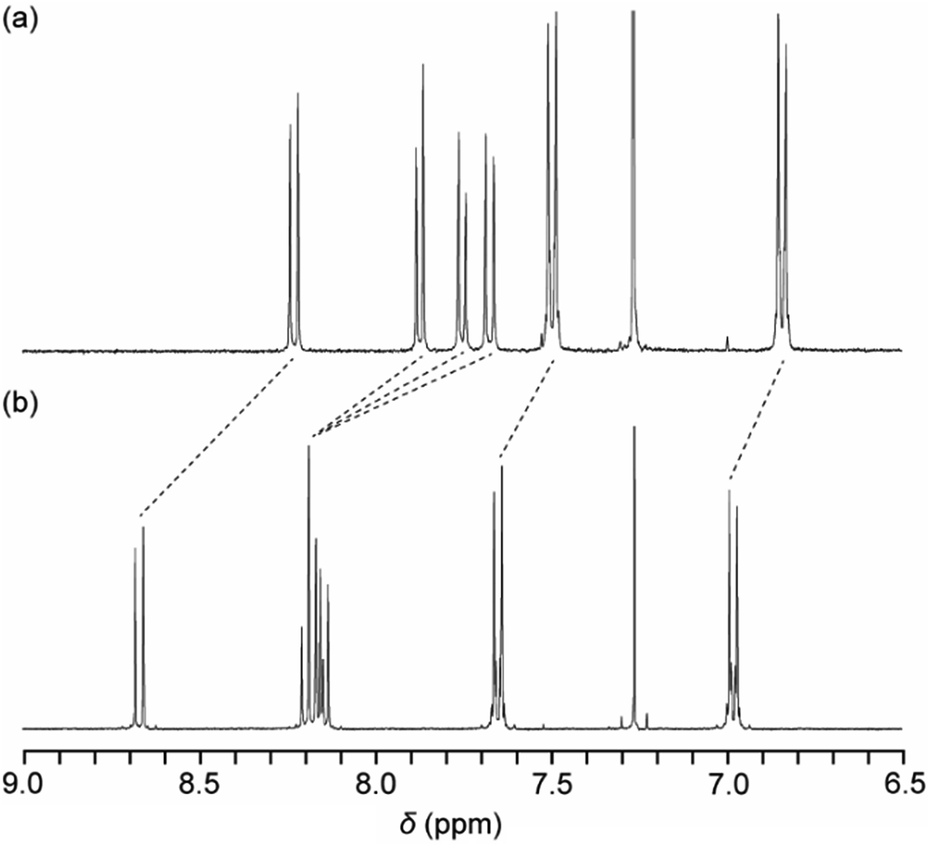 | ||
| Fig. 2 Partial 1H NMR spectra of (a) pyrenophane 1 and (b) linear reference compound 3 in CDCl3 at room temperature. | ||
Absorption and photoluminescence spectral measurements were first carried out to confirm that introduction of another luminophore into the cyclic structure results in significant changes in the photophysical properties. Fig. 3a shows absorption spectra of the symmetric pyrenophane 1, asymmetric pyrenophane 2, and linear reference compound 3 in chloroform solution (c = 1.0 × 10−5 M). A chloroform solution of 1 gives an absorption band between 350 and 450 nm. One peak was observed at 404 nm with a molar extinction coefficient of 1.0 × 10−5 L mol−1 cm−1 and one shoulder was detected at 426 nm (ε = 0.85 × 10−5 L mol−1 cm−1). The fact that the absorption spectral feature of 1 is clearly different from those of compounds 2 and 3, which have one pyrene moiety, indicated that some electronic ground-state interactions occur between the two luminophores of symmetric pyrenophane 1. Similar alterations in absorption spectra were also seen for the anthracene-based cyclophanes that we had reported10 and some perylene-based cyclophanes.24 These cyclophanes have two luminophores in each cyclic structure and the luminophores form H-type geometry in solution.10,24
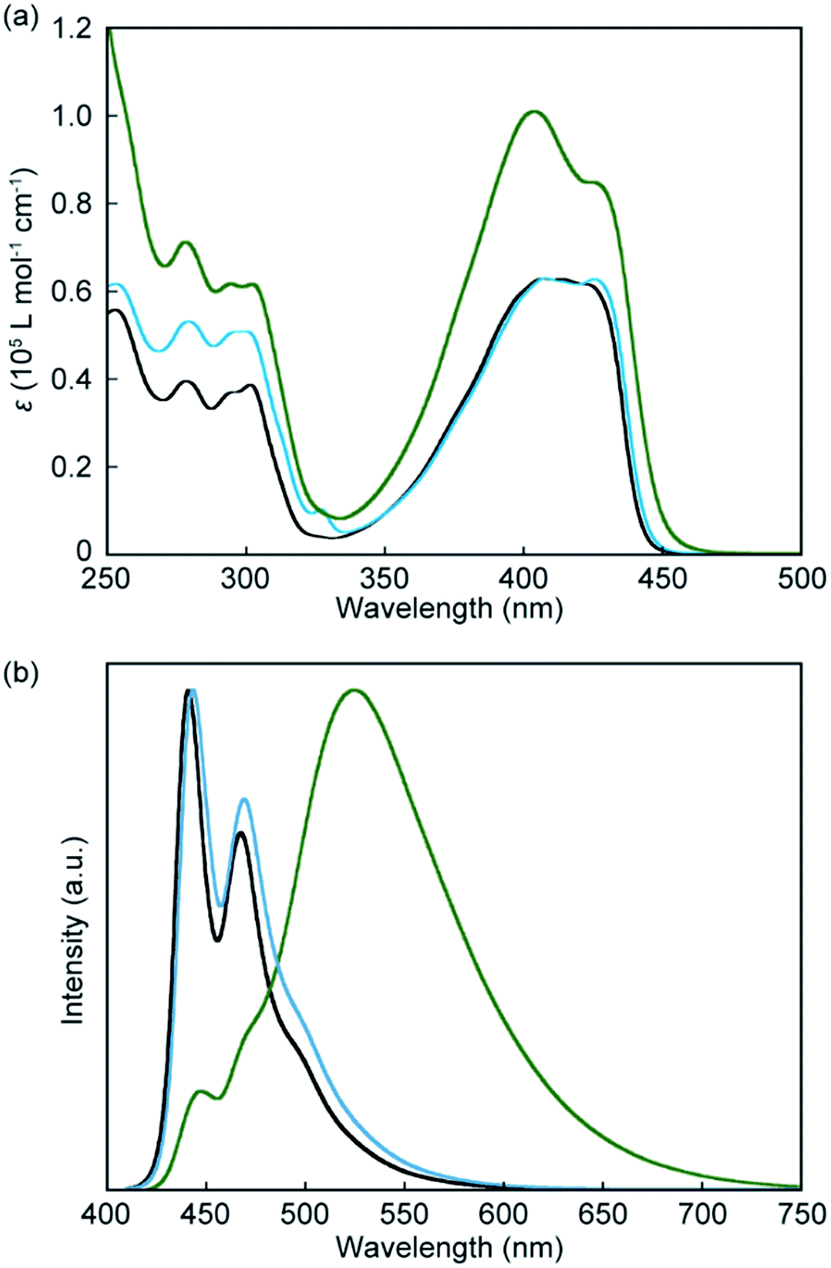 | ||
| Fig. 3 (a) Absorption and (b) normalised photoluminescence spectra of chloroform solutions (c = 1.0 × 10−5 M) of symmetric pyrenophane 1 (green line), asymmetric pyrenophane 2 (blue line), and linear reference compound 3 (black line). All spectra were recorded at room temperature. Photoluminescence spectra were obtained with excitation light of 400 nm. | ||
Introduction of two luminophores into one cyclic structure also induces significant changes in photoluminescence spectra. As shown in Fig. 3b, well-resolved vibronic structures were seen for a chloroform solution of compound 3 (c = 1.0 × 10−5 M), suggesting that compound 3 is well-individualised in chloroform (Fig. 3b, black line). The emission spectrum of asymmetric pyrenophane 2 in chloroform also shows a monomer-like emission spectral feature (Fig. 3b, blue line). Conversely, a chloroform solution of symmetric pyrenophane 1 displays a broad and structureless emission band with a peak at 525 nm (Fig. 3b, green line). One additional peak and a shoulder also appear at 448 and 470 nm, respectively. The former broad emission band is ascribed to intramolecular excimers between the luminophores, whereas the latter corresponds to monomer emission. The concentration dependency of emission spectra was also examined to confirm intramolecular excimer formation for 1 in chloroform (Fig. S1, ESI†). The spectral feature does not change as the concentration is varied from 1.0 × 10−5 to 1.0 × 10−7 M except for the relative intensity of the peak due to monomer emission. The decrease in the emission intensity of the monomer peak results from self-absorption.
Emission lifetime measurements confirmed the conclusions drawn from the steady-state absorption and photoluminescence spectroscopic experiments. A chloroform solution of linear reference compound 3 gave a simple emission decay profile that was fitted with a single exponential decay function (Fig. 4a), and the calculated lifetime is 1.4 ns, which is indicative of monomer emission (Table 1).13 In contrast, the emission decay curve obtained from pyrenophane 1 in chloroform could be fitted with a triexponential decay function (Fig. 4b), and a much longer emission lifetime of 21 ns was observed compared to that of the monomer. A similar emission lifetime (19 ns) was observed for asymmetric pyrenophane 2 in the supercooled nematic phase where the luminophores form intermolecular excimers.13
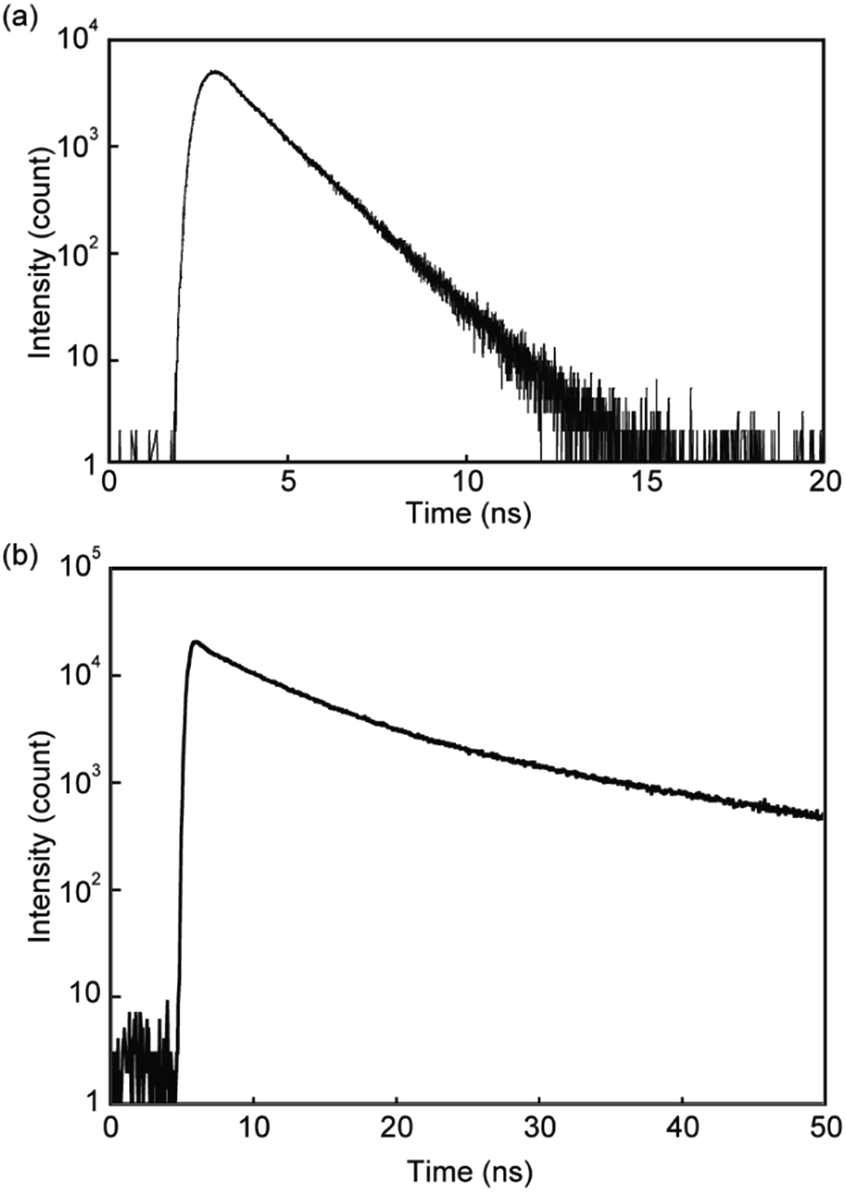 | ||
| Fig. 4 Emission decay profiles of (a) linear reference compound 3 and (b) pyrenophane 1 in chloroform. All profiles were collected at room temperature with excitation light of 405 nm. | ||
| τi (ns) | χ2 | ΦPL | |
|---|---|---|---|
| a All measurements were carried out at room temperature.b Monitored at 530 nm.c Monitored at 490 nm.d Monitored at 520 nm.e Monitored at 450 nm. | |||
| 1 in chloroformb | 0.1, 5.4, 21 | 1.10 | 0.81 |
| 1 in crystalline statec | 1.0, 2.5, 4.9 | 1.07 | 0.11 |
| 1 after grindingd | 1.2, 5.0, 18 | 1.08 | 0.23 |
| 2 in chloroforme | 0.5, 2.3 | 1.00 | 0.95 |
| 3 in chloroforme | 1.4 | 1.09 | 0.96 |
Photoluminescence quantum yields were also measured for compound 1 (Table 1). Compound 1 shows a relatively high quantum yield of 0.81 in chloroform. This means that the intramolecular excimer formation of 1 does not significantly decrease the photoluminescence quantum yield. In the case of a symmetric cyclophane featuring two 9,10-bis(phenylethynyl)anthracene moieties bridged by hexaethyleneglycol groups, the quantum yield significantly decreased compared to the monomer analogue because of intramolecular excimer formation (0.90 → 0.14).10b This reflects the fact that the anthracene-based cyclophane exhibits large peaks ascribed to monomer emission in its emission spectra,10b whereas the broad excimer band was dominant for pyrenophane 1 in the emission spectrum (Fig. 3b, green line).
We next examined the phase transition behaviour of pyrenophane 1 in the condensed states. The differential scanning calorimetry (DSC) curve reveals that compound 1 exhibits a phase transition from a low-ordered solid state to a nematic liquid-crystalline phase at 227 °C on heating (Fig. 5). Although some cyclophanes have been reported to show liquid-crystalline phases to date,25,26 highly emissive luminophores have not been incorporated in liquid-crystalline cyclic compounds except in our research.10b,13 Because pyrenophane 1 has two π-extended pyrene groups and thus the molecular structure is symmetric, the phase transition temperature of the nematic phase becomes much higher than that of asymmetric pyrenophane 2 (107 °C).13 Furthermore, although rapid cooling to compound 2 in the nematic phase leads to a supercooled nematic liquid-crystalline phase,13 compound 1 does not form kinetically trapped states even at the highest attainable cooling rates. This is presumably due to the strong tendency of pyrenophane 1 to form the solid state. As shown in Fig. 6, a schlieren texture indicative of nematic liquid-crystalline phases was observed above ∼230 °C in the absence of a coverslip. In contrast to pyrenophane 2, which shows homeotropic alignment in the presence of a coverslip when in the nematic phase,13 homeotropic alignment was not observed for pyrenophane 1 in the presence of a coverslip. Symmetrisation of the molecular structures also affects the molecular alignment in liquid-crystalline phases.
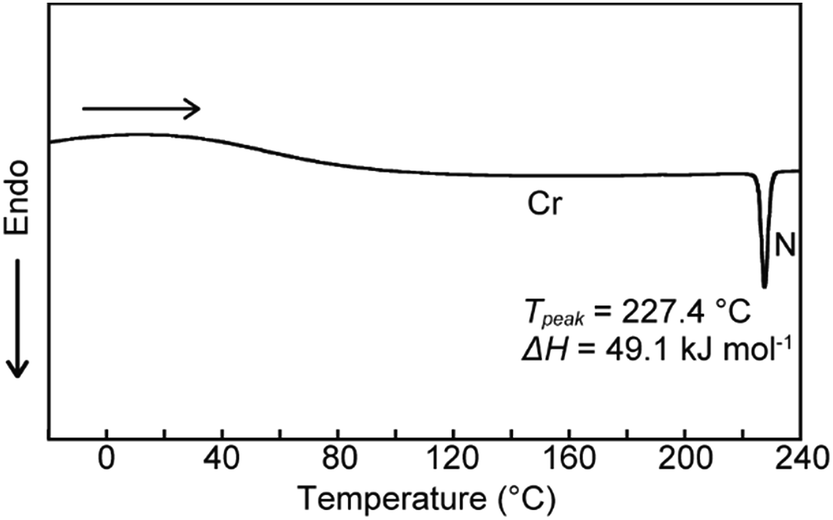 | ||
| Fig. 5 DSC trace of pyrenophane 1 on heating. Scanning rate was 10 °C min−1. | ||
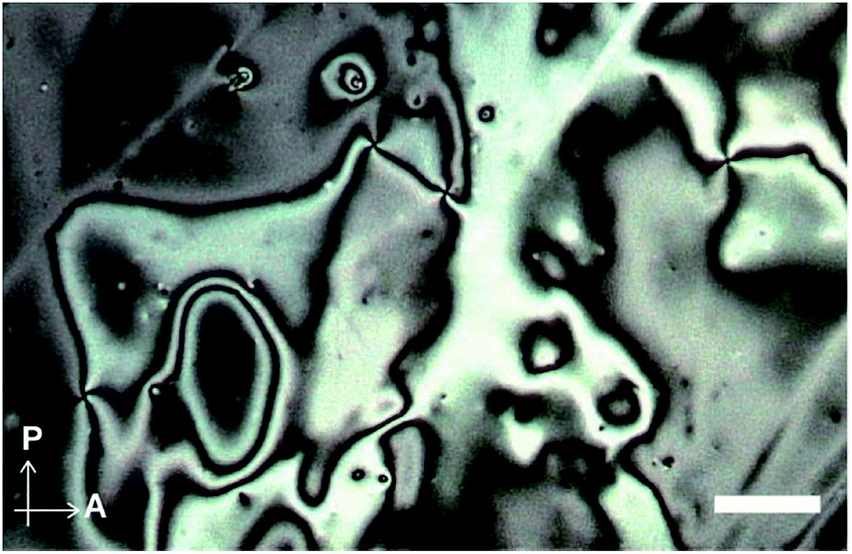 | ||
| Fig. 6 Polarised optical microscopic image of pyrenophane 1 at 240 °C in the absence of a coverslip. Scale bar: 40 μm. | ||
Pyrenophane 1 was found to exhibit mechanochromic luminescence in the condensed state at room temperature. Cyclophanes showing thermo- or mechanoresponsive luminescence in the solid states are very rare.10,13 The solid state of 1, which was obtained by slow cooling from the nematic phase, exhibits sky-blue photoluminescence under excitation light (365 nm). The emission colour turns yellow-green when the solid state is mechanically ground at room temperature. Recovery of the initial sky-blue emissive state requires heating of the ground sample until pyrenophane 1 forms the nematic phase and subsequent cooling to room temperature.
The photoluminescence spectral measurements (Fig. 7) clearly confirmed the change in the photoluminescence colour in response to mechanical stimuli. The photoluminescence spectrum of pyrenophane 1 in the initial solid state displays a sharp peak at 490 nm before mechanical grinding (Fig. 7, blue line). No clear broad emission bands were observed, indicating that few luminophores form intra- or intermolecular excimers before grinding in contrast to in the chloroform solution. Mechanical grinding to the solid phase results in a red shift of the photoluminescence spectrum (Fig. 7, green line). The broad and structureless emission band shows a peak at 523 nm, which is a similar wavelength to the chloroform solution of 1, which forms intramolecular excimers. These results indicate that, after mechanical grinding, some luminophores form inter- and/or intramolecular excimers in the condensed states. Emission lifetime measurements confirmed excimer formation after grinding for the solid states of pyrenophane 1. The emission decay profile recorded for the ground sample (Fig. 8, green line) was fitted with a triexponential decay function and a long lifetime of 18 ns was detected (Table 1). The initial solid phase of 1 before mechanical grinding gave a different emission decay profile (Fig. 8, blue line), and longer emission lifetimes ascribed to excimers were not observed (Table 1).
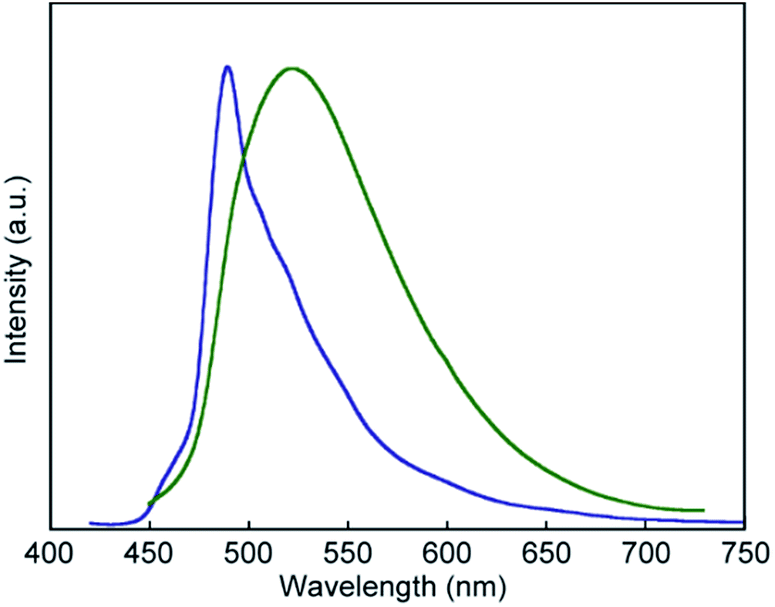 | ||
| Fig. 7 Normalised photoluminescence spectra of pyrenophane 1 in its solid states before (blue line) and after (green line) mechanical grinding at room temperature. Emission spectra were recorded with excitation light of 400 nm. | ||
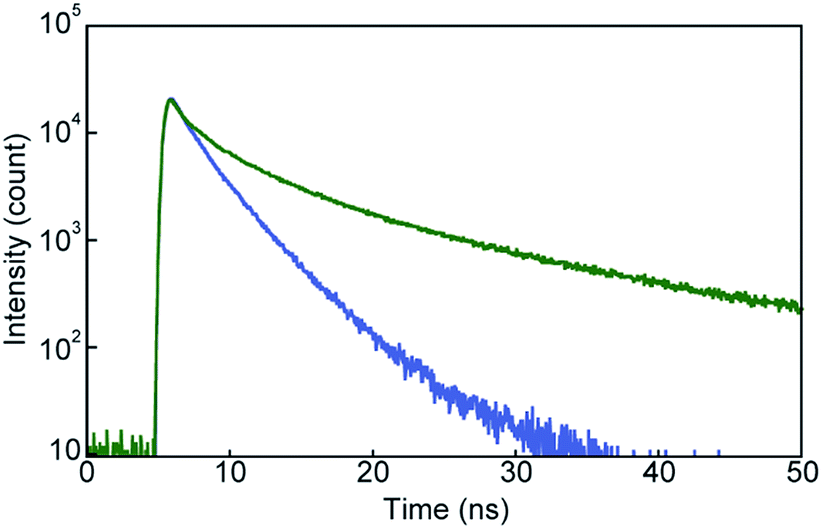 | ||
| Fig. 8 Emission decay profiles of pyrenophane 1 in the initial solid state (blue line) and after grinding (green line). All curves were recorded at room temperature with excitation light of 405 nm. | ||
X-ray diffraction (XRD) measurements were performed to obtain an insight into the change in molecular order in the solid states. The XRD pattern before grinding shows four sharp peaks at 30.4, 9.9, 7.5, and 3.9 Å and four broad peaks at 5.6, 5.0, 4.3, and 3.4 Å (Fig. 9a), indicating that pyrenophane 1 forms low-ordered molecular assembled states after cooling from the nematic phase rather than a well-ordered crystalline state. The mechanical grinding results in broadening of all peaks and a decrease in the peak intensities as shown in Fig. 9b. The results suggested that mechanical stimuli considerably reduce the molecular order compared to before grinding because the peak positions do not change. The observed change in the XRD patterns supports the conclusion drawn from the photophysical experiments conducted for 1 in the solid states before and after mechanical grinding. In the mechanical stimuli-induced much less ordered structures, the luminophores can form inter- and/or intramolecular excimers, leading to a change in the photoluminescence colour.
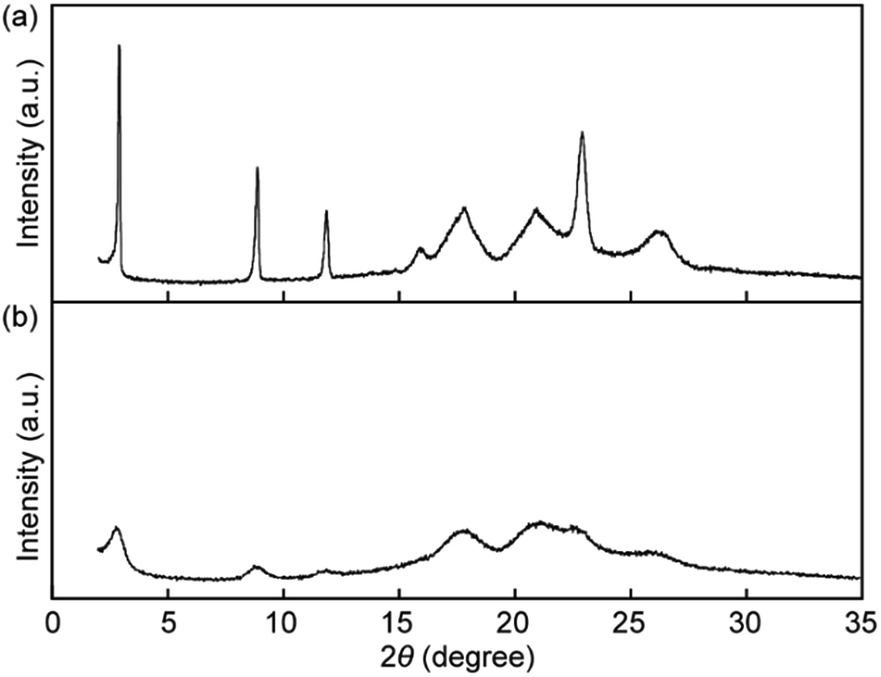 | ||
| Fig. 9 XRD patterns of 1 in the solid states (a) before and (b) after grinding at room temperature. | ||
Conclusions
We demonstrated that pyrenophane 1 featuring two 1,6-bis(phenylethynyl)pyrene groups exhibits mechanoresponsive luminescence at room temperature in the solid state and shows a nematic liquid-crystalline state above 227 °C on heating. Introduction of another 1,6-bis(phenylethynyl)pyrene moiety induced unambiguous changes in the phase transition behaviour and stimuli-responsive luminescence properties as well as the photophysical properties of the diluted solution compared to those of asymmetric pyrenophane 2.13 However, the fact that both symmetric pyrenophane 1 and asymmetric pyrenophane 2 show mechanoresponsive luminescence confirms that introduction of cyclic structures into luminophores that show assembled structure-dependent photoluminescence colours is a reliable strategy to develop stimuli-responsive luminescent solid materials. As luminescent cyclophanes have proved to be promising platforms to achieve stimuli-responsive luminescent molecular materials, work to develop other types of stimuli-responsive luminescent cyclophanes is under way.Experimental
All reagents and solvents were purchased from Aldrich, Tokyo Kasei, and Wako. Unless otherwise noted, all reactions were carried out under nitrogen atmosphere. Silica gel column chromatography was carried out with a Biotage Isolera Flash system. 1H NMR spectra were recorded on a JEOL JNM-ECX 400 spectrometer and all chemical shifts are quoted on the δ-scale in ppm relative to the signal of tetramethylsilane (at 0.00) as an internal standard. Proton-decoupled 13C NMR spectra were recorded on a JEOL JNM-ECX 400 spectrometer and all chemical shifts (δ) are reported in ppm using residual solvent as the internal standard (CDCl3 at 77.16). Coupling constants (J) are reported in Hz and relative intensities are also shown. Matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectra were obtained on an AB SCIEX TOF/TOF 5800. High resolution electrospray ionization (ESI) mass spectrum was obtained on a Thermo Scientific Exactive. The DSC measurements were conducted using a Rigaku Thermo Plus EVO DSC8230 with a heating/cooling rate of 10 °C min−1 under nitrogen atmosphere. Powder XRD measurements were carried out with a Rigaku SmartLab. UV-vis absorption spectra were measured with a JASCO V-550. Steady-state fluorescence spectra were recorded on a JASCO FP-6500. Time-resolved fluorescence measurements were carried out with a Hamamatsu Photonics Quantaurus-Tau. Quantum efficiencies were measured with a Hamamatsu Photonics Quantaurus-QY. Polarised optical microscopic observation was conducted with an Olympus BX-60 optical microscope equipped with a Moticam 1080 camera.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Y. Urano for photoluminescence quantum yield measurements and Prof. T. Nakamura and Prof. S. Noro for DSC measurements. Y. S. acknowledges support from JSPS KAKENHI Grant JP16H00818, the Asahi Glass Foundation, and the Iketani Science and Technology Foundation.Notes and references
- Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) D. J. Cram and J. M. Cram, Acc. Chem. Res., 1971, 4, 204–213 CrossRef CAS; (b) C. Seel and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1992, 31, 528–549 CrossRef; (c) J. O. Jeppesen, M. B. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115–5131 CrossRef CAS PubMed; (d) P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS PubMed; (b) A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99, 931–967 CrossRef CAS PubMed; (c) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (d) D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158–4168 RSC; (e) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- (a) P. Čudić, M. Žinić, V. Tomišić, V. Simeon, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1995, 1073–1075 RSC; (b) M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1996, 2169–2175 RSC; (c) O. Baudoin, F. Gonnet, M.-P. Teulade-Fichou, J.-P. Vigneron, J.-C. Tabet and J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2762–2771 CrossRef CAS.
- (a) P. P. Neelakandan and D. Ramaiah, Angew. Chem., Int. Ed., 2008, 47, 8407–8411 CrossRef CAS PubMed; (b) P. P. Neelakandan, K. S. Sanju and D. Ramaiah, Photochem. Photobiol., 2010, 86, 282–289 CrossRef CAS PubMed.
- (a) M. Inouye, K. Fujimoto, M. Furusyo and H. Nakazumi, J. Am. Chem. Soc., 1999, 121, 1452–1458 CrossRef CAS; (b) H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495–504 CrossRef CAS PubMed.
- L. Qiu, C. Zhu, H. Chen, M. Hu, W. He and Z. Guo, Chem. Commun., 2014, 50, 4631–4634 RSC.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- P. C. Nandajan, P. P. Neelakandan and D. Ramaiah, RSC Adv., 2013, 3, 5624–5630 RSC.
- (a) Y. Sagara, Y. C. Simon, N. Tamaoki and C. Weder, Chem. Commun., 2016, 52, 5694–5697 RSC; (b) Y. Sagara, C. Weder and N. Tamaoki, RSC Adv., 2016, 6, 80408–80414 RSC.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- (a) T. Umemoto, S. Satani, Y. Sakata and S. Misumi, Tetrahedron Lett., 1975, 16, 3159–3162 CrossRef; (b) T. Kawashima, T. Otsubo, Y. Sakata and S. Misumi, Tetrahedron Lett., 1978, 19, 5115–5118 CrossRef.
- Y. Sagara, C. Weder and N. Tamaoki, Chem. Mater., 2017, 29, 6145–6152 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- (a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed; (b) K. Araki and T. Mutai, Photochemistry, 2016, 43, 191–225 CAS; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (d) A. P. Haehnel, Y. Sagara, Y. C. Simon and C. Weder, Top. Curr. Chem., 2015, 369, 345–375 CrossRef CAS PubMed; (e) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed.
- (a) Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki and C. Weder, Chem. Mater., 2017, 29, 1273–1278 CrossRef CAS; (b) Y. Sagara, A. Lavrenova, A. Crochet, Y. C. Simon, K. M. Fromm and C. Weder, Chem.–Eur. J., 2016, 22, 4374–4378 CrossRef CAS PubMed; (c) J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577–582 RSC; (d) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC; (e) S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J.-H. Park, B.-K. An and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 203–207 CrossRef CAS PubMed; (f) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed; (g) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed; (h) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CAS; (i) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed; (j) Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed; (k) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed; (l) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (m) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS; (n) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- (a) Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, J. Am. Chem. Soc., 2014, 136, 4273–4280 CrossRef CAS PubMed; (b) Y. Sagara, T. Komatsu, T. Terai, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, Chem.–Eur. J., 2014, 20, 10397–10403 CrossRef CAS PubMed; (c) Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, Adv. Funct. Mater., 2013, 23, 5277–5284 CrossRef CAS.
- (a) M. Mitani, S. Ogata, S. Yamane, M. Yoshio, M. Hasegawa and T. Kato, J. Mater. Chem. C, 2016, 4, 2752–2760 RSC; (b) M. Mitani, S. Yamane, M. Yoshio, M. Funahashi and T. Kato, Mol. Cryst. Liq. Cryst., 2014, 594, 112–121 CrossRef CAS; (c) S. Yamane, Y. Sagara, T. Mutai, K. Araki and T. Kato, J. Mater. Chem. C, 2013, 1, 2648–2656 RSC; (d) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed; (e) Y. Sagara, S. Yamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869–1875 CrossRef CAS; (f) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS PubMed.
- (a) M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS PubMed; (b) T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS PubMed; (c) T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed; (d) D. Genovese, A. Aliprandi, E. A. Prasetyanto, M. Mauro, M. Hirtz, H. Fuchs, Y. Fujita, H. Uji-I, S. Lebedkin, M. Kappes and L. De Cola, Adv. Funct. Mater., 2016, 26, 5271–5278 CrossRef CAS; (e) Q. Benito, X. F. Le Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J. P. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311–11320 CrossRef CAS PubMed; (f) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 Search PubMed; (g) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef CAS PubMed; (h) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed; (i) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed.
- (a) B. Huitorel, Q. Benito, A. Fargues, A. Garcia, T. Gacoin, J.-P. Boilot, S. Perruchas and F. Camerel, Chem. Mater., 2016, 28, 8190–8200 CrossRef CAS; (b) V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286–6289 CrossRef CAS PubMed.
- (a) B. R. Crenshaw, M. Burnworth, D. Khariwala, A. Hiltner, P. T. Mather, R. Simha and C. Weder, Macromolecules, 2007, 40, 2400–2408 CrossRef CAS; (b) A. Pucci, F. Di Cuia, F. Signori and G. Ruggeri, J. Mater. Chem., 2007, 17, 783–790 RSC; (c) M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946–955 CrossRef CAS; (d) B. R. Crenshaw and C. Weder, Macromolecules, 2006, 39, 9581–9589 CrossRef CAS; (e) A. Pucci, M. Bertoldo and S. Bronco, Macromol. Rapid Commun., 2005, 26, 1043–1048 CrossRef CAS; (f) B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717–4724 CrossRef CAS; (g) C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef.
- D. W. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS PubMed.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- (a) W. Wang, L. Wang, B. J. Palmer, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 11150–11159 CrossRef CAS PubMed; (b) J. Feng, Y. Zhang, C. Zhao, R. Li, W. Xu, X. Li and J. Jiang, Chem.–Eur. J., 2008, 14, 7000–7010 CrossRef CAS PubMed; (c) F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed; (d) K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588–2593 CrossRef CAS PubMed.
- (a) V. Percec, A. D. Asandei and G. Ungar, Chem. Mater., 1996, 8, 1550–1557 CrossRef CAS; (b) V. Percec, A. D. Asandei and P. Chu, Macromolecules, 1996, 29, 3736–3750 CrossRef CAS; (c) V. Percec, M. Kawasumi, P. L. Rinaldi and V. E. Litman, Macromolecules, 1992, 25, 3851–3861 CrossRef CAS; (d) V. Percec, P. J. Turkaly and A. D. Asandei, Macromolecules, 1997, 30, 943–952 CrossRef CAS.
- (a) B. Neumann, D. Joachimi and C. Tschierske, Adv. Mater., 1997, 9, 241–244 CrossRef CAS; (b) B. Neumann, T. Hegmann, R. Wolf and C. Tschierske, Chem. Commun., 1998, 105–106 RSC; (c) B. Neumann, T. Hegmann, C. Wagner, P. R. Ashton, R. Wolf and C. Tschierske, J. Mater. Chem., 2003, 13, 778–784 RSC; (d) T. Hegmann, B. Neumann, R. Wolf and C. Tschierske, J. Mater. Chem., 2005, 15, 1025–1034 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic schemes and additional fluorescence spectra. See DOI: 10.1039/c7ra10776e |
| This journal is © The Royal Society of Chemistry 2017 |