
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of 2,3-disubstituted indolines via an organocatalytic intramolecular Michael addition†
Jusung Lee,
Kwang Min Ko and
Sung-Gon Kim *
*
Department of Chemistry, Kyonggi University, 154-42, Gwanggyosan-ro, Yeongtong-gu, Suwon 16227, Republic of Korea. E-mail: sgkim123@kyonggi.ac.kr
First published on 15th December 2017
Abstract
An asymmetric synthesis of 2,3-disubstituted indolines has been developed via an organocatalytic intramolecular Michael addition. When a primary amine derived from cinchona alkaloid was used as the catalyst, the intramolecular cyclization reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-arylprop-2-en-1-ones afforded the corresponding cis-2,3-disubstituted indoline derivatives with high yields, moderate diastereoselectivities, and excellent enantioselectivities (up to 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99% ee). Moreover, the catalytic reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-alkylprop-2-en-1-ones afforded trans-2,3-disubstituted indolines in high yields and with good-to-excellent diastereo- and enantioselectivities (up to 20:1 dr and 99% ee).
:1 dr and 99% ee). Moreover, the catalytic reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-alkylprop-2-en-1-ones afforded trans-2,3-disubstituted indolines in high yields and with good-to-excellent diastereo- and enantioselectivities (up to 20:1 dr and 99% ee).
Optically active indolines are structurally essential elements in biologically active natural alkaloids and chiral pharmaceuticals.1 Among various indolines, chiral 2,3-disubstituted indolines are significant building blocks in both synthetic and medicinal chemistry because they are present in numerous natural products and pharmaceuticals including (−)-strychnine, aspidospermidine, WAY-163909, and (−)-physostigmine, as shown in Fig. 1.2 They have two chiral centers leading to four possible diastereomers, each of which can exhibit significantly different chemical and biological activities. Therefore, it is challenging and interesting to develop novel synthetic methodologies for constructing enantioenriched 2,3-disubstituted indoline derivatives in synthetic organic chemistry.
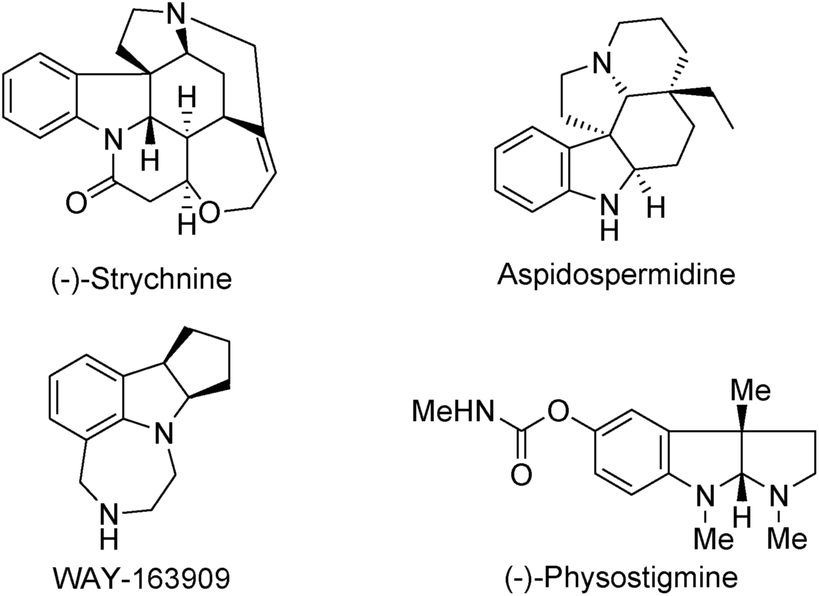 | ||
| Fig. 1 Representative chiral 2,3-disubstituted indoline derivatives. | ||
Consequently, diverse strategies have been developed for the synthesis of enantioenriched indolines.3 Among these, the most powerful tool for the asymmetric synthesis of 2,3-disubstituted indolines is the catalytic hydrogenation of substituted indole derivatives.4 Although asymmetric hydrogenation is one of the most straightforward methods in terms of simplicity and atom efficiency, this transformation affords only cis-2,3-disubstituted indolines. Catalytic kinetic resolution is also used to obtain such chiral indolines.5 Recently, some significant syntheses of optically active indolines have been achieved using asymmetric intramolecular cyclization reactions such as electrophilic cyclization.6 The benefit of these reactions is the easy introduction of various functional groups at the 2- and 3-positions of indoline. Kündig and co-workers reported that the reaction of N-aryl, N-branched alkyl carbamates with a Pd-catalyst bearing chiral N-heterocyclic carbene ligand, giving enantioenriched 2,3-disubstituted indolines.6c Xiao and co-workers developed an innovative cycloaddition strategy for the enantioselective synthesis of trans-2,3-disubstituted indolines, involving the Pd-catalyzed decarboxylation–cycloaddition reaction of vinyl benzoxazinanones with sulfur ylides.6b More recently, Buchwald and co-workers reported an efficient Cu–H-catalyzed strategy using 2-alkenylimine precursors obtained from alkenylanilines and aromatic aldehydes for the highly diastereo- and enantioselective synthesis of cis-2,3-disubstituted indolines.6a However, to the best of our knowledge, the synthesis of chiral 2,3-disubstituted indolines by asymmetric cycloaddition using organocatalyst has not been reported, even though asymmetric organocatalysis has significantly progressed over the past decade.7,8
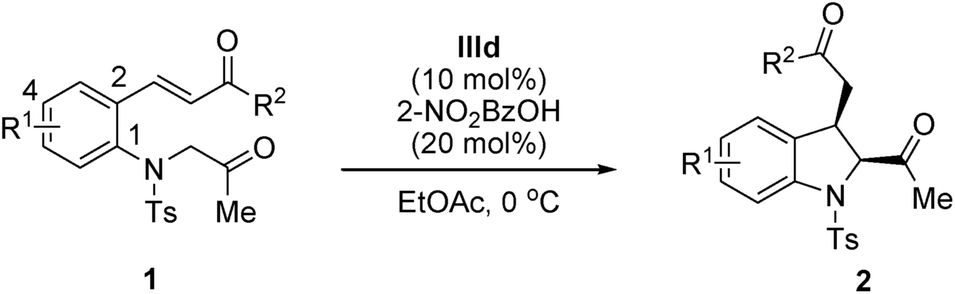
Within the framework of our program devoted to the development of stereoselective cascade reactions of o-aminophenyl α,β-unsaturated compounds,9 we recently reported an aza-alkylation/Michael cascade reaction of 2-(tosylamino)phenyl α,β-unsaturated ketones with α-bromoacetophenones to afford cis-2,3-disubstituted indolines in high yields and with excellent diastereoselectivities (Scheme 1, eqn (1)).10 In this reaction, the nitrogen atom of 2-(tosylamino)phenyl α,β-unsaturated ketone attacks α-bromoacetophenone through SN2 reaction to first afford 2-(2-oxoalkyltosylamino)phenyl α,β-unsaturated ketone, followed by an intramolecular Michael addition. These findings prompted us to investigate the asymmetric synthesis of 2,3-disubstituted indolines. We envisioned that optically active 2,3-disubstituted indoline derivatives can be accessed by the intramolecular Michael addition of 2-(2-oxoalkyltosylamino)phenyl α,β-unsaturated ketone using a chiral organocatalyst (Scheme 1, eqn (2)).
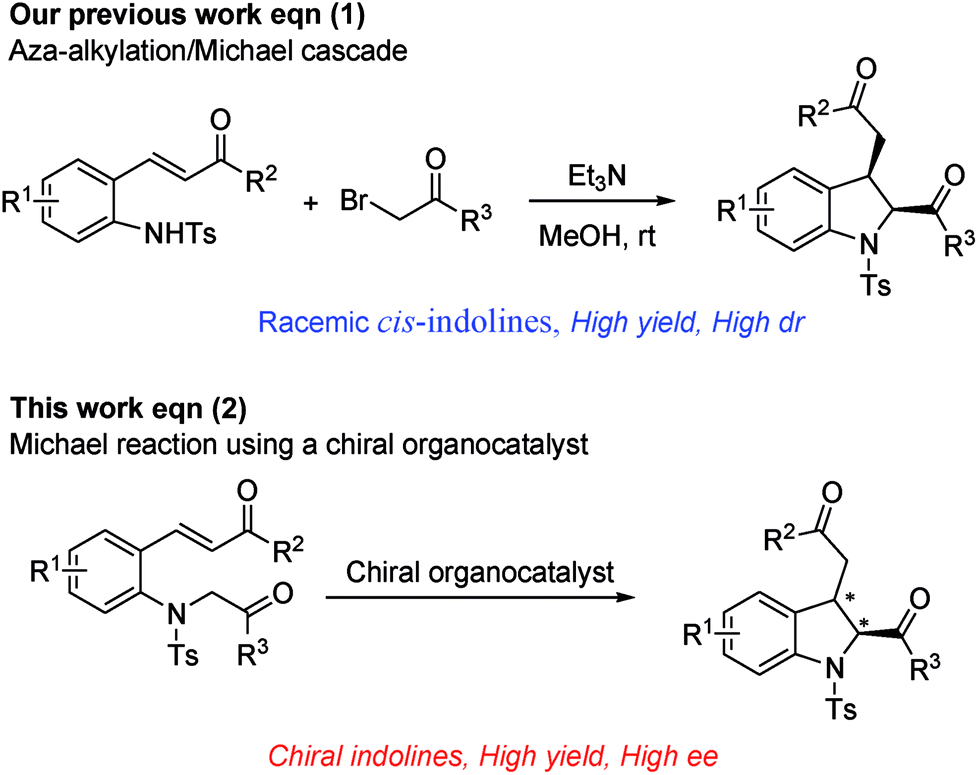 | ||
| Scheme 1 Stereoselective synthesis optically active 2,3-disubstituted indolines. | ||
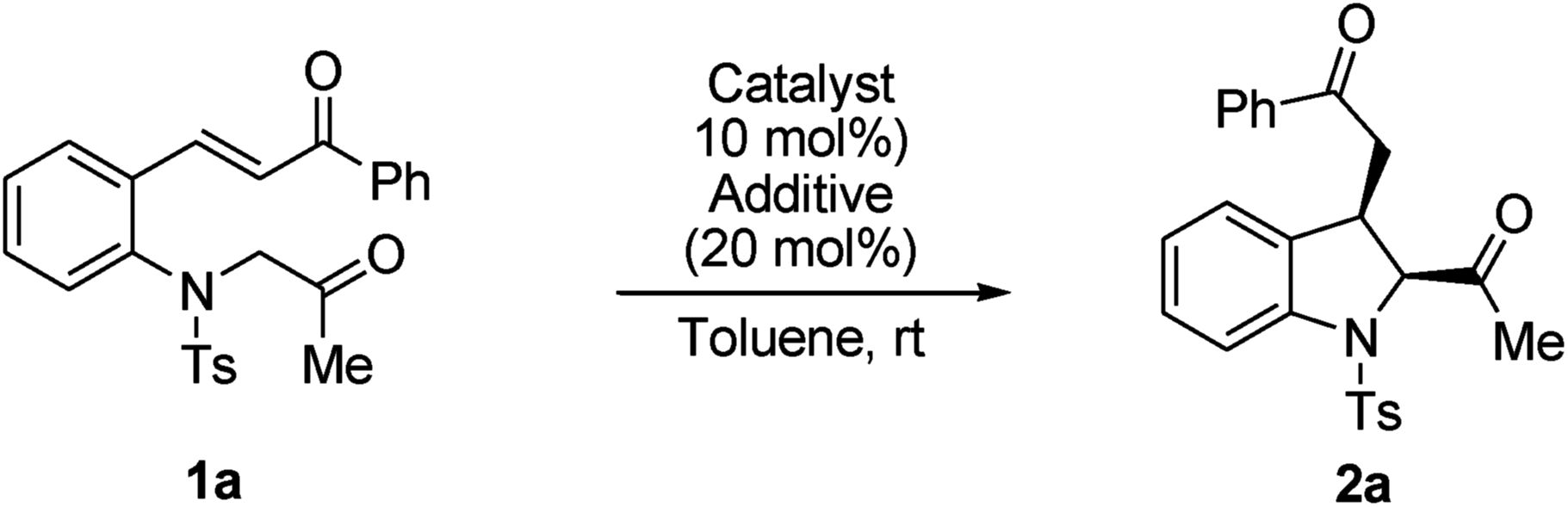
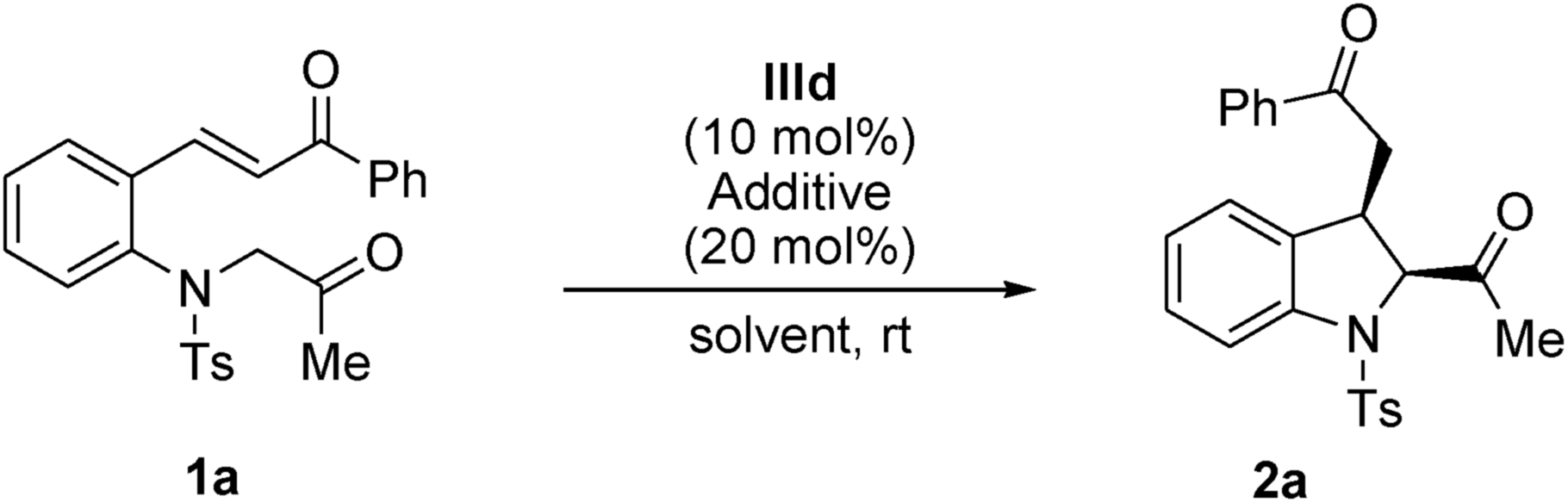
First, (E)-3-(2-(2-oxopropyltosylamino)phenyl)-1-phenylprop-2-en-1-one 1a was selected as the model substrate, and its intramolecular Michael addition was investigated using a bifunctional catalytic system (Fig. 2). Initially, the model reaction was performed using 10 mol% of Takemoto bifunctional thiourea catalyst I11 in toluene at room temperature (Table 1, entry 1). The Michael reaction was completed within 48 h and afforded the desired indoline 2a in 92% yield and with moderated diastereo- and enantioselectivities. Encouraged by these results, several bifunctional catalysts were screened to obtain a higher enantioselectivity. Cinchona-derived thiourea catalyst II12 gave almost the same results as catalyst I (Table 1, entry 1). Next, bifunctional primary amine catalysts III13 derived from cinchona alkaloids were screened; they were found to be promising catalysts for this intramolecular reaction (Table 1, entries 3–6). When primary amine catalysts IIIa–c were used, the reaction smoothly afforded the corresponding product 2a with enantioselectivities of 64, 62, and 77% ee for the major diastereomer, respectively (Table 1, entries 3–5). The use of a primary amine catalyst IIId significantly increased the ee value of product 2a (Table 1, entry 6, 88% ee for the major isomer). Although a low diastereoselectivity was obtained, catalyst IIId was the best choice for the model reaction in terms of the chemical yield and enantioselectivity of the major diastereomer. In addition, an acid additive was an important factor in this reaction; the reaction without using an additive showed lower reactivity and enantioselectivity than those using an additive (Table 1, entry 6 vs. entry 7).
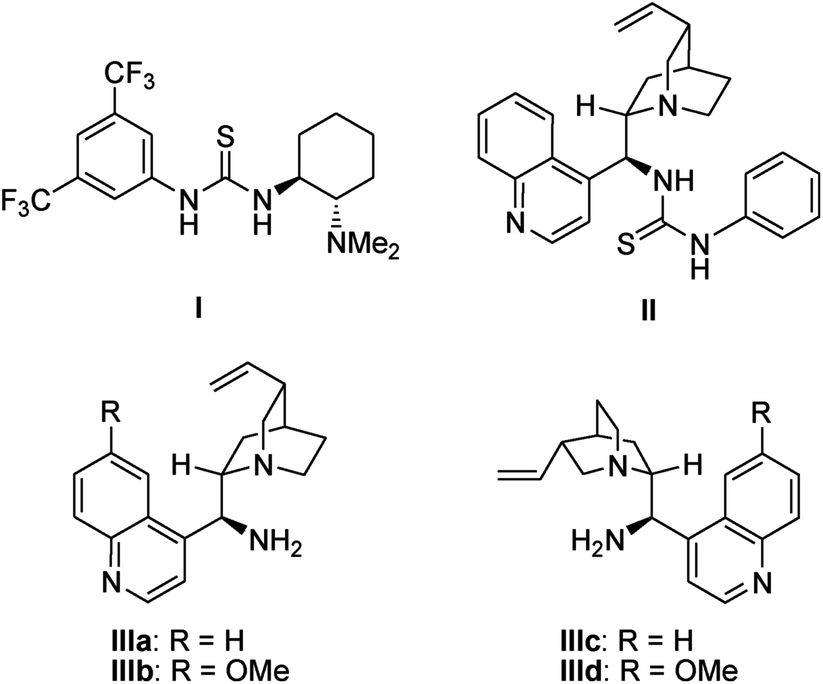 | ||
| Fig. 2 Evaluated bifunctional chiral organocatalysts. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Time (h) | Yieldb (%) | cis/transc | eed,e (%) |
| a All reactions were performed with 0.1 mmol of 1a, catalyst (10 mol%), and additive (20 mol%) in toluene (0.2 M).b Isolated yield after chromatographic purification.c Determined by 1H NMR analysis.d The ee values were determined by chiral HPLC analysis.e ee values cis/trans diastereomers. | ||||||
| 1 | I | None | 48 | 92 | 67:33 |
41/23 |
| 2 | II | None | 168 | 94 | 66:34 |
33/31 |
| 3 | IIIa | AcOH | 168 | 62 | 15:85 |
38/64 |
| 4 | IIIb | AcOH | 240 | 76 | 31:69 |
52/62 |
| 5 | IIIc | AcOH | 168 | 61 | 20:80 |
36/77 |
| 6 | IIId | AcOH | 120 | 86 | 53:47 |
88/85 |
| 7 | IIId | None | 240 | 85 | 41:59 |
69/21 |
After confirming primary amine IIId as the optimum catalyst for this Michael reaction, other factors influencing the reaction such as the acid additive, solvent, and reaction temperature were thoroughly investigated (Table 2). First, an acid additive was determined as essential to both the reactivity and enantioselectivity of the reaction. 2-Nitrobenzoic acid gave superior results with respect to reaction rate and enantioselectivity (Table 2, entry 6). Moreover, this Michael reaction was found to be sensitive to the solvent. In toluene, CH2Cl2, ClCH2CH2Cl, CHCl3, and EtOAc, the reaction smoothly proceeded and completed within 10 h, affording the corresponding product 2a in high yields and with excellent enantioselectivities for both diastereomers (Table 2, entries 6–10). In CH3CN, THF and MeOH, the reaction was completed in a longer time than in other solvents with moderate yields and enantioselectivities, even though a high enantioselectivity was observed for the major diastereomer in THF (Table 2, entries 11–13). Although a low diastereoselectivity was observed in EtOAc, both the diastereomers were obtained in the highest yields and enantioselectivities (Table 2, entry 10). Therefore, EtOAc was selected as the most suitable solvent for this asymmetric intramolecular Michael addition. A lower reaction temperature provided a better stereocontrol. The desired product was achieved in an excellent yield and with enantioselectivities (98% ee cis isomer; 94% ee trans isomer) and a moderate diastereoselectivity (cis/trans, 63:37) (Table 2, entry 14). In addition, when the amount of 2-nitrobenzoic acid was reduced from 20 mol% to 10 mol%, the enantioselectivity decreased (Table 2, entry 14).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Solvent | Time (h) | Yieldb (%) | cis/transc | eed,e (%) |
| a Unless otherwise specified, the reactions were performed with 0.1 mmol of 1a, catalyst IIId (10 mol%), and additive (20 mol%) in solvent (0.2 M).b Isolated yield after chromatographic purification.c Determined by 1H NMR analysis.d The ee values were determined by chiral HPLC analysis.e ee values cis/trans diastereomers.f Reaction performed at 0 °C.g Reaction performed with 10 mol% 2-NO2BzOH at 0 °C. | ||||||
| 1 | AcOH | Toluene | 120 | 85 | 41:59 |
88/85 |
| 2 | CF3CO2H | Toluene | 72 | 91 | 59:41 |
91/88 |
| 3 | CCl3CO2H | Toluene | 48 | 88 | 63:37 |
93/88 |
| 4 | PhCO2H | Toluene | 24 | 95 | 59:41 |
94/88 |
| 5 | 4-NO2BzOH | Toluene | 6 | 85 | 40:60 |
93/89 |
| 6 | 2-NO2BzOH | Toluene | 8 | 94 | 64:36 |
96/88 |
| 7 | 2-NO2BzOH | CH2Cl2 | 10 | 98 | 56:44 |
95/90 |
| 8 | 2-NO2BzOH | ClCH2CH2Cl | 8 | 97 | 51:49 |
95/90 |
| 9 | 2-NO2BzOH | CHCl3 | 6 | 96 | 60:40 |
96/87 |
| 10 | 2-NO2BzOH | EtOAc | 4 | 96 | 60:40 |
96/90 |
| 11 | 2-NO2BzOH | CH3CN | 96 | 41 | 23:77 |
51/36 |
| 12 | 2-NO2BzOH | THF | 96 | 73 | 52:48 |
89/68 |
| 13 | 2-NO2BzOH | MeOH | 96 | 50 | 20:80 |
48/45 |
| 14f | 2-NO2BzOH | EtOAc | 24 | 98 | 63:37 |
98/94 |
| 15g | 2-NO2BzOH | EtOAc | 28 | 97 | 62:38 |
96/89 |
With the optimized reaction conditions in hand (1 equiv. of 1, 10 mol% catalyst IIId, and 20 mol% 2-nitrobenzoic acid in EtOAc at 0 °C), the substrate scope of the reaction was investigated, and the results are summarized in Table 3. This transformation has a broad substrate scope. The reactions of all the substrates smoothly afforded the corresponding indolines in moderate-to-high yields (62–97%) and with excellent enantioselectivities (93–99% ee) for the major diastereomer in all the cases except p-Me-substituted substrate 1m. Gratifyingly, the electronic nature of the R1 group slightly affected the reaction efficiencies with regard to enantioselectivities and yields. Both electron-withdrawing (Table 3, entries 2–4) and electron-donating (Table 3, entries 5 and 6) groups were well tolerated; the obtained diastereoselectivities were still low in all the cases. For the R2 group, the reactions proceeded well with electron-withdrawing substituents (Table 3, entries 7–12) and electron-donating substituents (Table 3, entries 12–14). Notably, a substrate bearing an ortho- and para-substituted halogen group on the phenyl R2 group afforded the corresponding product with a higher diastereoselectivity than the other substrates. A substrate bearing both ortho-substituted chloro group provided trans-2,3-disubstituted indoline 2j in the major diastereomer (cis/trans, 33:67) (Table 3, entry 10). Moreover, a heteroaromatic group was introduced at the R2 position; the reaction proceeded rapidly, affording the corresponding products 2o and 2p in high yields and with good enantiocontrol (over 91% ee, Table 3, entries 15 and 16).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | 2 | Yieldb (%) | cis/transc | eed,e (%) |
| a All reactions were performed with 0.1 mmol of 1, catalyst IIId (10 mol%), and 2-NO2BzOH (20 mol%) in EtOAc (0.2 M) at 0 °C.b Isolated yield after chromatographic purification.c Determined by 1H NMR analysis.d The ee values were determined by chiral HPLC analysis.e ee values cis/trans diastereomers. | |||||||
| 1 | H | Ph | 28 | 2a | 97 | 63:37 |
98/94 |
| 2 | 4-Cl | Ph | 36 | 2b | 96 | 58:42 |
95/93 |
| 3 | 5-Cl | Ph | 48 | 2c | 88 | 58:42 |
99/95 |
| 4 | 4-Br | Ph | 48 | 2d | 97 | 56:44 |
99/95 |
| 5 | 4-Me | Ph | 56 | 2e | 91 | 60:40 |
99/93 |
| 6 | 4,5-MeO | Ph | 72 | 2f | 62 | 58:42 |
95/97 |
| 7 | H | p-ClC6H4 | 36 | 2g | 86 | 61:39 |
92/76 |
| 8 | H | m-ClC6H4 | 56 | 2h | 93 | 51:49 |
92/90 |
| 9 | H | o-ClC6H4 | 48 | 2i | 96 | 73:27 |
94/92 |
| 10 | H | 2,6-ClC6H3 | 96 | 2j | 83 | 33:67 |
91/90 |
| 11 | H | p-BrC6H4 | 48 | 2k | 82 | 70:30 |
98/94 |
| 12 | H | p-FC6H4 | 36 | 2l | 95 | 65:35 |
95/86 |
| 13 | H | p-MeC6H4 | 36 | 2m | 85 | 54:46 |
90/84 |
| 14 | H | p-MeOC6H4 | 48 | 2n | 91 | 55:45 |
96/89 |
| 15 | H | 2-Furanyl | 36 | 2o | 92 | 47:53 |
92/91 |
| 16 | H | 2-Thienyl | 24 | 2p | 86 | 43:57 |
94/94 |
To evaluate the practical applications of this methodology, a gram-scale reaction using the standard conditions was conducted; product 2a was obtained without any significant changes in the yield or stereoselectivity (Scheme 2). The absolute configuration of product cis-2d was determined by X-ray crystallographic analysis and found to be 2S,3R.14 The configurations of the other products were assigned by analogy.
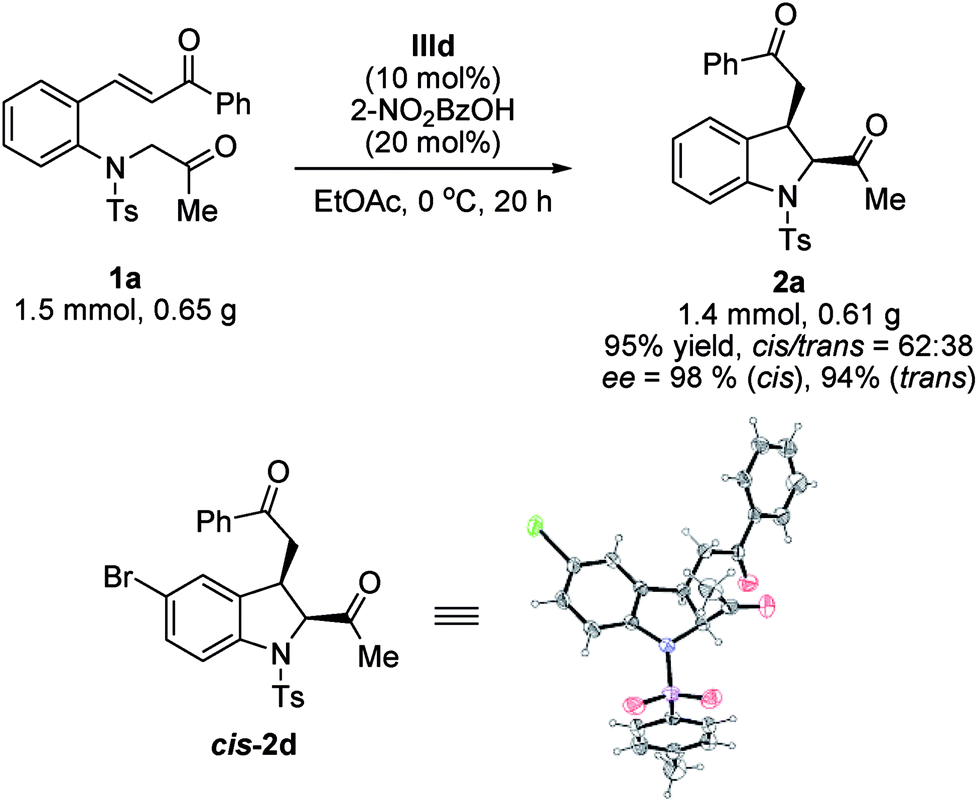 | ||
| Scheme 2 Gram-scale synthesis of indoline 2a and X-ray crystal structure of cis-2d. | ||
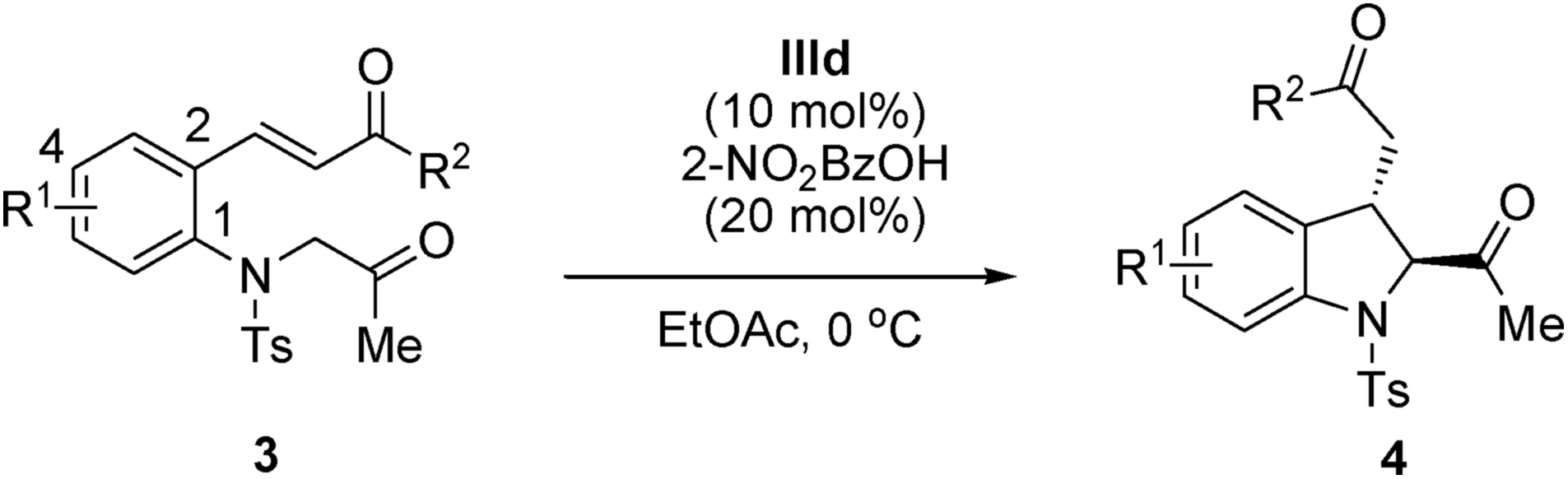
Furthermore, the reactions of (E)-3-(2-(2-oxopropylamino)aryl)-1-alkylprop-2-en-1-ones bearing an alkyl group at the R2 position, such as methyl, cyclopropyl and t-butyl, smoothly afforded the corresponding 2,3-disubstituted indolines in high yields and with excellent enantioselectivities (Table 4, entries 1–3). To our delight, t-butyl α,β-unsaturated ketone provided the desired trans-2,3-disubstituted indoline 4c with an excellent diastereomeric ratio (trans/cis, 13:1) and enantioselectivity (98% ee; Table 4, entry 3).15 Encouraged by the excellent results obtained using t-butyl α,β-unsaturated ketone, the scope of our strategy was further extended. No obvious impact was observed on either the reaction efficiency or stereoselectivity, regardless of the electronic nature, bulkiness, or position of the substituent on the benzene ring of (E)-3-(2-(2-oxopropylamino)aryl)-1-t-butylprop-2-en-1-ones. Both electron-withdrawing groups (Table 4, entries 4–6) and electron-donating groups (Table 4, entries 7 and 8) were well tolerated, and all the products were obtained in good yields and with excellent diastereoselectivities (trans/cis, 13:1 to >25:1) and enantioselectivities (93–99% ee).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | 4 | Yieldb (%) | trans/cisc | eed (%) |
| a Unless otherwise specified, the reactions were performed with 0.1 mmol of 3, catalyst IIId (10 mol%), and 2-NO2BzOH (20 mol%) in EtOAc (0.2 M) at 0 °C.b Isolated yield after chromatographic purification.c Determined by 1H NMR analysis.d The ee values were determined by chiral HPLC analysis. | |||||||
| 1 | H | Me | 72 | 4a | 65 | 1:1.5 |
84/87 |
| 2 | H | Cyclopropyl | 48 | 4b | 93 | 4:1 |
94 |
| 3 | H | t-Bu | 18 | 4c | 98 | 13:1 |
98 |
| 4 | 4-Cl | t-Bu | 12 | 4d | 97 | 13:1 |
95 |
| 5 | 5-Cl | t-Bu | 48 | 4e | 78 | >25:1 |
93 |
| 6 | 4-Br | t-Bu | 12 | 4f | 92 | 15:1 |
98 |
| 7 | 4-Me | t-Bu | 18 | 4g | 98 | 20:1 |
99 |
| 8 | 4,5-MeO | t-Bu | 18 | 4h | 85 | >25:1 |
99 |
On the basis of our experimental results, a plausible transition state for this asymmetric Michael reaction is shown in Scheme 3. The Si-face attack of anti-enamine to the Re-face of t-butyl α,β-unsaturated ketone led to the formation of the S,S-configurated trans-indoline 4c.
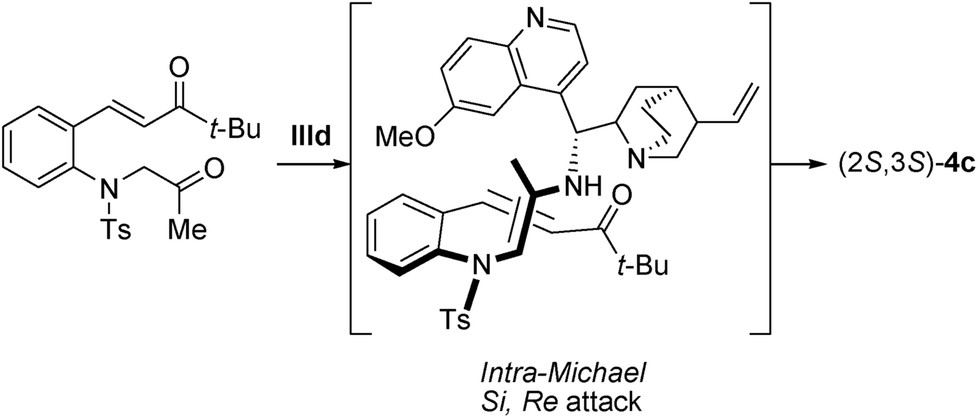 | ||
| Scheme 3 Plausible transition state. | ||
To further demonstrate the synthetic utility of this methodology, we have illustrated a representative procedure to convert the enantioenriched idoline product into the corresponding alkenyl ester 5 (Scheme 4). The reaction of trans-4c with methyl(triphenylphosphoranylidene)acetate provided Wittig product 5 in moderate yield without affecting the enantioselectivity.
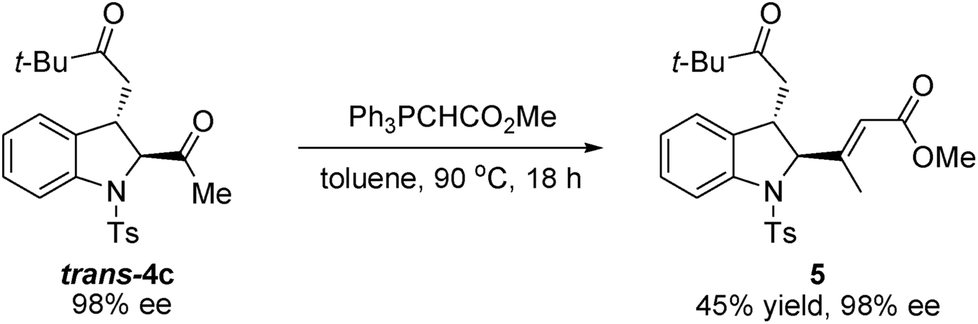 | ||
| Scheme 4 The synthetic transformation of indoline trans-4c. | ||
Conclusions
In conclusion, we have developed an efficient method for the asymmetric synthesis of 2,3-disubstituted indolines through an organocatalytic intramolecular Michael addition. The reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-arylprop-2-en-1-ones afforded the corresponding cis-2,3-disubstituted indoline derivatives in high yields and with moderate diastereoselectivities and excellent enantioselectivities (up to 2.7:1 dr and 99% ee) using a primary amine derived from a cinchona alkaloid as the catalyst. Moreover, the catalytic reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-alkylprop-2-en-1-ones helped to develop an unprecedented approach for the synthesis of chiral trans-2,3-disubstituted indolines in high yields and with good-to-excellent diastereo- and enantioselectivities (up to >25:1 dr, and 99% ee).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Nanomaterial Technology Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2012M3A7B4049645) and the Basic Science Research Program through NRF funded by the Ministry of Education (NRF-2016R1D1A1A09916621).Notes and references
- For recent reviews, see: (a) E. Fattorusso and O. Taglialatela-Scafati, Modern Alkaloids, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) G. M. Cragg, D. G. I. Kingston and D. J. Newman, Anticancer Agents from Natural Products, CRC, Boca Raton, FL, 2012 Search PubMed; (c) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151 CrossRef CAS PubMed; (d) D. L. Boger, C. W. Boyce, R. M. Garbaccio and J. A. Goldberg, Chem. Rev., 1997, 97, 787 CrossRef CAS PubMed.
- (a) T. Bui, S. Syed and C. F. Barbas III, J. Am. Chem. Soc., 2009, 131, 8758 CrossRef CAS PubMed; (b) H. Zhang, J. Boonsombat and A. Padwa, Org. Lett., 2007, 9, 279 CrossRef CAS PubMed; (c) K. L. Marquis, A. L. Sabb, S. F. Logue, J. A. Brennan, M. J. Piesla, T. A. Comery, S. M. Grauer, C. R. Ashby Jr, H. Q. Nguyen, L. A. Dawson, J. E. Barret, G. Stack, H. Y. Meltzer, B. L. Harrison and S. Rosenzweig-Lipson, J. Pharmacol. Exp. Ther., 2007, 320, 486 CrossRef CAS PubMed; (d) R. Iyengar, K. Schildknegt, M. Morton and J. Aube, J. Org. Chem., 2005, 70, 10645 CrossRef CAS PubMed.
- (a) D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447 CrossRef CAS PubMed; (b) D. Liu, G. Zhao and L. Xiang, Eur. J. Inorg. Chem., 2010, 3975 CrossRef CAS , and references therein.
- For recent selected examples, see: (a) Z. Yang, F. Chen, Y. He, N. Yang and Q.-H. Fan, Angew. Chem., Int. Ed., 2016, 55, 13863 CrossRef CAS PubMed; (b) Y.-C. Zhang, J.-J. Zhao, F. Jiang, S.-B. Sun and F. Shi, Angew. Chem., Int. Ed., 2014, 53, 13912 CrossRef CAS PubMed; (c) C. Romano, M. Jia, M. Monari, E. Manoni and M. Bandini, Angew. Chem., Int. Ed., 2014, 53, 13854 CrossRef CAS PubMed; (d) L. Chen, C. Wang, L. Zhou and J. Sun, Adv. Synth. Catal., 2014, 356, 2224 CrossRef CAS; (e) Y. Duan, M.-W. Chen, Q.-A. Chen, C.-B. Yu and Y.-G. Zhou, Org. Biomol. Chem., 2012, 10, 1235 RSC; (f) Y.-C. Xiao, C. Wang, Y. Yao, J. Sun and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 10661 CrossRef CAS PubMed; (g) Y. Duan, M.-W. Chen, Z.-S. Ye, D.-S. Wang, Q.-A. Chen and Y.-G. Zhou, Chem.–Eur. J., 2011, 17, 7193 CrossRef CAS PubMed; (h) M.-W. Chen, C.-B. Yu, Y. Duan and G.-F. Jiang, Chem. Sci., 2011, 2, 803 RSC.
- (a) K. Saito, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 11740 CrossRef CAS PubMed; (b) M. Lopez-Iglesias, E. Busto, V. Gotor and V. Gotor-Fernández, J. Org. Chem., 2012, 77, 8049 CrossRef CAS PubMed; (c) X. L. Hou and B. H. Zheng, Org. Lett., 2009, 11, 789 CrossRef PubMed; (d) F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 14264 CrossRef CAS PubMed; (e) V. Gotor-Fernández, P. Fernández-Torres and V. Gotor, Tetrahedron: Asymmetry, 2006, 17, 2558 CrossRef.
- (a) E. Ascic and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 4666 CrossRef CAS PubMed; (b) T.-R. Li, F. Tan, L.-Q. Lu, Y. Wei, Y.-N. Wang, Y.-Y. Liu, Q.-Q. Yang, J.-R. Chen, D.-Q. Shi and W.-J. Xiao, Nat. Commun., 2014, 5, 5500 CrossRef CAS PubMed; (c) D. Katayev, M. Nakanishi, T. Bürgi and E. T. Kündig, Chem. Sci., 2012, 3, 1422 RSC; (d) K. H. Kang, J. Do and Y. S. Park, J. Org. Chem., 2012, 77, 808 CrossRef CAS PubMed; (e) J. L. García Ruano, A. Parra, V. Marcos, C. del Pozo, S. Catalán, S. Monteagudo, S. Fustero and A. Poveda, J. Am. Chem. Soc., 2009, 131, 9432 CrossRef PubMed; (f) J. L. García Ruano, J. Alemán, S. Catalán, V. Marcos, S. Monteagudo, A. Parra, C. del Pozo and S. Fustero, Angew. Chem., Int. Ed., 2008, 47, 7941 CrossRef PubMed.
- For examples of organocatalytic asymmetric syntheses of 2-substituted indolines via cycloaddition, see: (a) R. Miyaji, K. Asano and S. Matsubara, Org. Lett., 2013, 15, 3658 CrossRef CAS PubMed; (b) S. Fustero, C. del Pozo, C. Mulet, R. Lazaro and M. Sánchez-Roselló, Chem.–Eur. J., 2011, 17, 14267 CrossRef CAS PubMed; (c) S. Fustero, J. Moscardó, D. Jiménez, M. D. Pérez-Carrión, M. Sánchez-Roselló and C. del Pozo, Chem.–Eur. J., 2008, 14, 9868 CrossRef CAS PubMed; (d) E. C. Carlson, L. K. Rathbone, H. Yang, N. D. Collett and R. G. Carter, J. Org. Chem., 2008, 73, 5155 CrossRef CAS PubMed.
- For selected reviews on organocatalysis, see: (a) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed; (b) H. Pellissier, Recent Developments in Asymmetric Organocatalysis, RSC, Cambridge, 2010 Search PubMed; (c) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (d) A. Dondoni and A. Massi, Angew. Chem., Int. Ed. Engl., 2008, 47, 4638 CrossRef CAS PubMed; (e) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) Y. Lee, S. Heo and S.-G. Kim, Adv. Synth. Catal., 2015, 357, 1545 CrossRef CAS; (b) H. Kim and S.-G. Kim, Tetrahedron Lett., 2015, 56, 4819 CrossRef CAS; (c) M. Yu and S.-G. Kim, Tetrahedron Lett., 2015, 56, 4159 CrossRef CAS; (d) A. Kim, C. Kim and S.-G. Kim, Bull. Korean Chem. Soc., 2015, 36, 417 CrossRef CAS; (e) Y. Lee and S.-G. Kim, J. Org. Chem., 2014, 79, 8234 CrossRef CAS PubMed; (f) K.-T. Kang and S.-G. Kim, Synthesis, 2014, 46, 3365 CrossRef CAS; (g) S. Kim, K.-T. Kang and S.-G. Kim, Tetrahedron, 2014, 70, 5114 CrossRef CAS; (h) C. Kim and S.-G. Kim, Tetrahedron: Asymmetry, 2014, 25, 1376 CrossRef CAS; (i) S. Heo, S. Kim and S.-G. Kim, Tetrahedron Lett., 2013, 54, 4978 CrossRef CAS.
- M. Yu and S.-G. Kim, Tetrahedron Lett., 2015, 56, 7034 CrossRef CAS.
- (a) S. Sakamoto, T. Inokuma and Y. Takemoto, Org. Lett., 2011, 13, 6374 CrossRef CAS PubMed; (b) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed.
- For selected reviews on bifunctional cinchona thiourea catalysis, see: (a) B. Han, J.-L. Li, Y.-C. Xiao, S.-L. Zhou and Y.-C. Chen, Curr. Org. Chem., 2011, 15, 4128 CrossRef CAS; (b) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593 CrossRef CAS PubMed; (c) C. E. Song, Cinchona Alkaloids in Synthesis & Catalysis, Wiley-VCH, Weinheim, 2009 Search PubMed; (d) S. J. Connon, Chem. Commun., 2008, 2499 RSC.
- For selected reviews on primary amine catalysis, see: (a) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS PubMed; (b) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC; (c) F. Peng and Z. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1 CrossRef CAS.
- CCDC 1527668 (2d) contains the supplementary crystallographic data for this paper.†.
- The relative stereochemistry of compound 4 (trans-isomer) was established by the distinguished chemical shift in 1H NMR of the trans-isomer and by NOESY.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1527668. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10775g |
| This journal is © The Royal Society of Chemistry 2017 |