
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene-enhanced platinum-catalysed hydrosilylation of amides and chalcones: a sustainable strategy allocated with in situ heterogenization and multitask application of H2PtCl6†
Ning Li‡
ab,
Xiao-Yun Dong‡ab,
Jing-Lei Zhanga,
Ke-Fang Yangb,
Zhan-Jiang Zhengb,
Wei-Qiang Zhang*a,
Zi-Wei Gao a and
Li-Wen Xu*ab
a and
Li-Wen Xu*ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education (MOE), School of Chemistry and Chemical Engineering, Shaanxi Normal University, No. 620, West Chang'an Avenue, Xi'an 710062, P. R. China. E-mail: liwenxu@snnu.edu.cn; liwenxu@hznu.edu.cn
bKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, No.1378 Wenyi West Road, Hangzhou 311121, P. R. China
First published on 31st October 2017
Abstract
We describe a new sustainable strategy for the comprehensive utilization of a platinum catalyst in different organic transformations, in which an organosilicon/graphene-supported platinum catalyst prepared from a simple hydrosilylation-type reduction could be further used in the 1,4-hydrosilylation of chalcones. The rationally designed and in situ formed Pt@G@Si nanocatalyst is demonstrated to be highly effective in the 1,4-hydrosilylation of α,β-unsaturated enones, allowing for the facile synthesis of a variety of otherwise inaccessible substituted silyl enolates. In addition, with the aid of platinum catalyst residue and TBAF, the one-pot downstream Michael addition of substituted silyl enolates to alkyl acrylates is also reported in this work.
Introduction
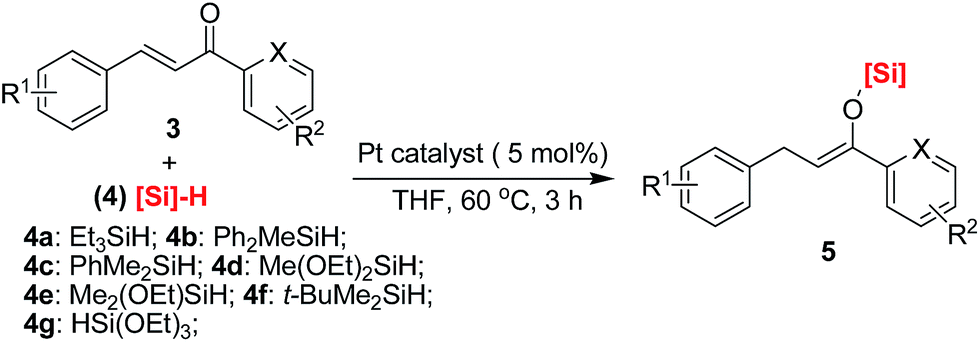
It is of great necessity to explore new strategies for the construction of sustainable catalyst materials from general transition-metal-based heterogeneous catalysts in the preparation of fine chemicals and numerous industrial intermediates for a variety of valuable compounds and materials.1 One of the key driving forces of heterogeneous catalysis is the increased demand for an environmentally friendly method, with a high level of efficiency and chemoselectivity as well as an atom-economic synthetic procedure.2 It is well recognized that an ideal catalyst, including a heterogeneous supported metal catalyst, should remain at least somewhat active until the last molecule of the reaction substrate has been consumed. However, a decrease in catalytic activity of recovered catalysts is unavoidable in both heterogeneous catalysis and homogeneous catalysis.3 Interestingly, there are few examples focused on the reuse of a recovered and deactivated metal catalyst from metal wastes in another new organic reaction.4 Therefore, we hypothesized that further exploration of the catalytic functions of a recovered metal catalyst in new and downstream organic transformations would be killing two birds with one stone, which would be beneficial to the development of new reactions and sustainable catalyst materials. In this regard, we have previously reported a multitask and maximum reuse approach in which the recovered palladium catalyst/residue could be successfully applied in different types of one-by-one and downstream reactions, from catalytic hydrogenation to cross-coupling reactions, including the Suzuki and Sonogashira reactions, and to Knoevenagel condensations.5 Similar to the concept of tandem catalysis,6 a multitask strategy with the same catalyst precursor could be applied in multiple reactions, which is quite valuable for green and sustainable chemistry.7 Nonetheless, such simple new catalyst preparations by a multitask strategy are infrequently reported, and methods making use of certain organic reaction-accessible deactivated metal catalysts in organic transformations are also rare. Inspired by this preliminary result, we believed that a multitask strategy based on one-by-one downstream organic reactions allocated to the in situ formation of transition-metal catalysts with different activities would find wide application in organic synthesis, in which the synthetic reactions could be used as a springboard for the facile and in situ construction of sustainable catalyst materials.Platinum catalysts capable of Si–H activation and hydrosilylation of carbon–carbon/heteroatom multiple bonds have inspired intense research efforts within the polymer chemistry, organosilicon material and synthetic chemistry communities.8 Although many advances have been made in the field of hydrosilylation reactions of alkenes or α,β-unsaturated aldehydes or ketones, there has been no report on the platinum-catalyzed hydrosilylation of chalcones.9 Notably, this type of hydrosilylation reaction continues to present a challenge because it usually generates mixtures of reductive carbonyl compounds, ketene silyl acetals, α- and β-adducts, silyl ethers, and other side-products depending on the catalyst systems (Fig. 1).10,11
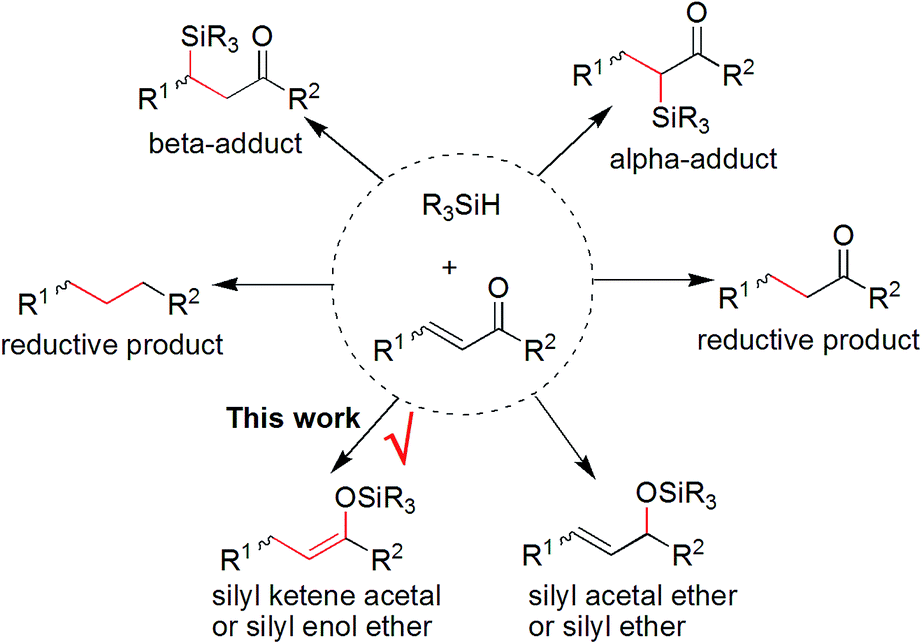 | ||
| Fig. 1 Possible products from the hydrosilylation of α,β-unsaturated carbonyl compounds. | ||
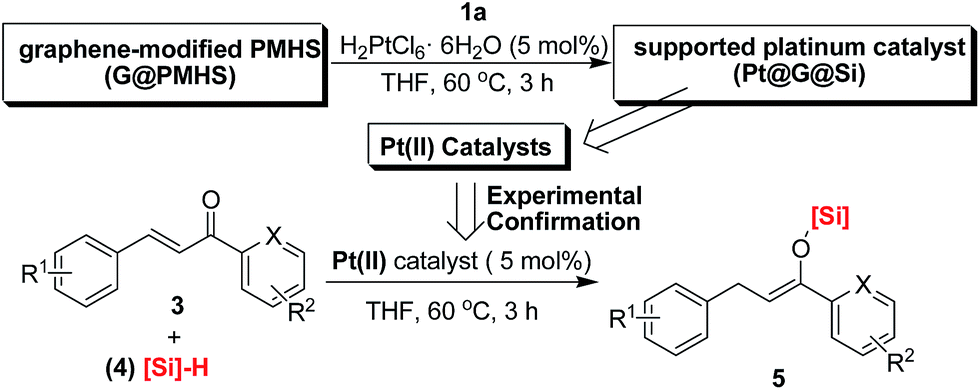
Herein, we confirm the powerful potential of the multitask strategy in platinum-catalyzed organic reactions, in which the new organosilicon/graphene-supported platinum(II) catalyst was in situ formed and immobilized on graphene-dispersed organosilicon material during the platinum (H2PtCl6)-catalyzed hydrogenation/hydrosilylation of amide, and then the in situ formed graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) is used in the catalytic hydrosilylation of chalcones (Fig. 2). On the basis of the multitask strategy, the rationally designed and in situ formed Pt@G@Si nanocatalyst material that is achieved from the H2PtCl6-catalyzed hydrosilylation-type reduction of amides in the presence of graphene-dispersed PMHS (polymethylhydrosiloxane) is demonstrated to be effective in the 1,4-hydrosilylation of α,β-unsaturated enones, allowing for the facile synthesis of a variety of otherwise inaccessible substituted silyl enolates in a straightforward manner.
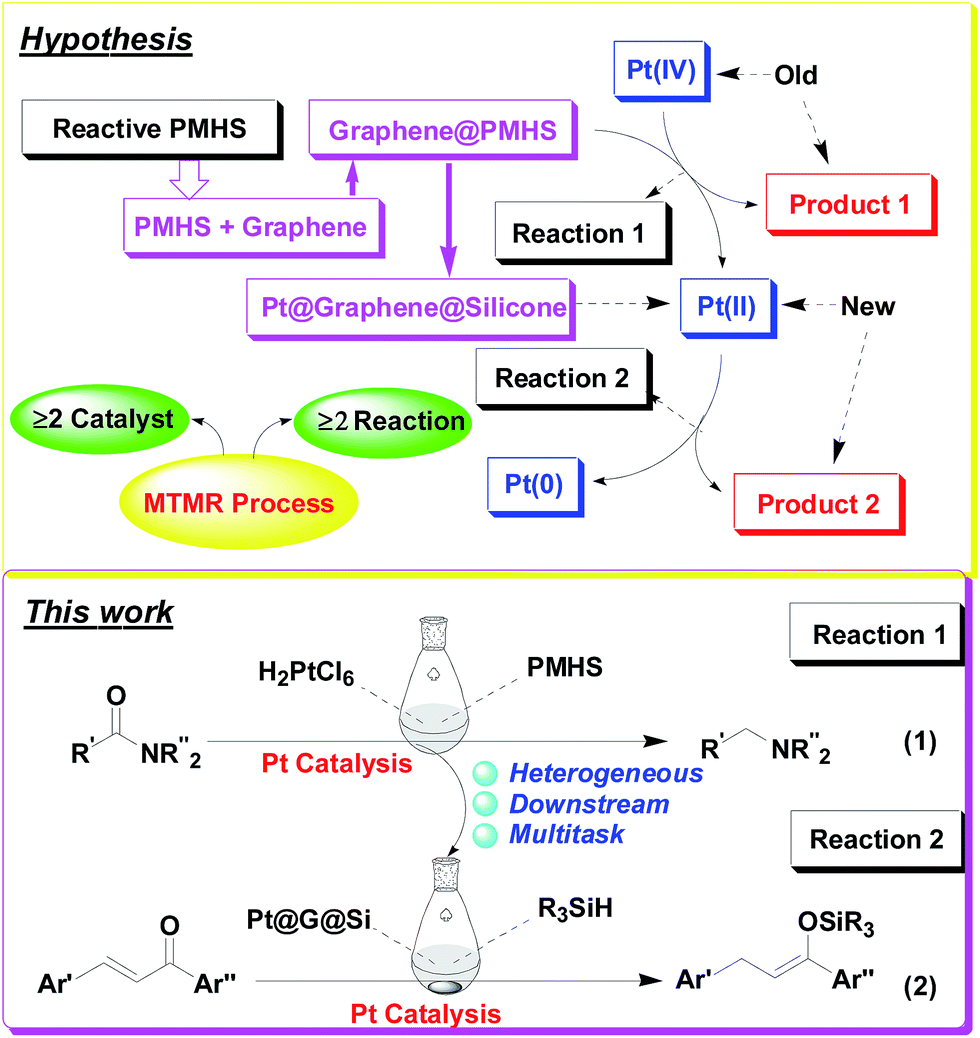 | ||
| Fig. 2 Our hypothesis for a multitask strategy with an in situ formed organosilicon-supported platinum catalyst: from hydrosilylation/hydrogenation of amides (eqn (1)) to hydrosilylation of chalcones (eqn (2)). | ||
Results and discussion
Initially, we wanted to develop a new method for the catalytic α-silylation of α,β-unsaturated ketones by platinum catalysis because there are few reports on the catalytic α-silylation of α,β-unsaturated ketones. However, when we tried to apply commercially available platinum catalysts in this reaction we found it really challenging because the desired α-silylated product was not obtained in these preliminary explorations (Fig. 1). In this case, the 1,4-conjugate reduction was a major reaction process when chalcone was used as a model substrate. Fortunately, in this work, we presented an unprecedented new platinum catalyst-controlled 1,4-silylation of chalcones based on the multitask process, in which the supported platinum catalyst is quite effective for the 1,4-hydrosilylation of chalcones to prepare a variety of otherwise inaccessible silyl enolates. It is well known that silyl enolates are extremely valuable and useful organosilicon compounds in organic synthesis.12 In the past few decades, transition-metal-catalyzed 1,4-hydrosilylation of α,β-unsaturated carbonyl compounds has become a powerful method for the preparation of silyl enolates.13 However, the stereoselectivities are not satisfactory in most of these reports. Subsequently, several improved methods have also been reported in past years. For example, Yorimitsu and Oshima reported a palladium-catalyzed 1,4-hydrosilylation of α,β-unsaturated ketones with hydrosilanes for the preparation of silyl enolates with high Z selectivity.14 Unfortunately, the substrate scope was not enough good because of the limitation of the hydrosilanes. In 2014, Takeuchi et al. investigated a cationic rhodium complex-catalyzed 1,4-hydrosilylation of α,β-unsaturated ketones with triethylsilane (Et3SiH) to prepare various silyl enolates in good yields.15 All these homogeneous catalyst systems seem actually to be complementary. For the 1,4-hydrosilylation of chalcones, there is only one example reported very recently, where Shishido et al.16 developed an Nb2O5-supported Pd–Au alloy catalyst which could be used as a selective catalyst for the hydrosilylation of α,β-unsaturated ketones. These works suggested that considerable attention has been focused on the transition-metal catalysed hydrosilylation of α,β-unsaturated ketones.In our previous studies, we have found that polymethylhydrosiloxane (PMHS) and related organosilicon materials were useful multifunctional supports for the preparation of supported transition-metal catalysts.5,10f,g And in this regard, we have also noticed that the platinum-catalyzed hydrosilylation/reduction of carboxamides with polymethylhydrosiloxane (PMHS) was reported by Nagashima et al.17 in 2009, in which the synergetic effect of two Si–H groups was found to be crucial to the reduction of amides to amines under mild reaction conditions. In addition, such a hydrosilylation reaction was accompanied by the removal of both organosilicon wastes and platinum as insoluble silicone resin. Therefore the hydrosilylation of amides could act as a springboard for the construction of an organosilicon material (silicone)-supported platinum catalyst. However, we found that the recovery and reuse of a platinum catalyst immobilized on PMHS is really not easy due to the formation of polysiloxane gel in this reaction.18
Therefore, inspired by our previous findings that PMHS could be used to initiate the cobalt-catalyzed polymerization of acrylates and subsequent multitask processes,5,10g we wanted to construct a graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) accompanied by the platinum-catalyzed reduction of amides to amines (Fig. 3). As shown in Fig. 3, we hypothesized that graphene that is accepted as specific alkenes that are built with an infinite number of benzene rings and with double carbon–carbon (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) bonds,19 could be linked with PMHS under cobalt-catalyzed silicon-carbon bond-forming reaction conditions (Fig. 3, step 1). In fact, the hydrosilanes, including pentamethyldisiloxane (TMSOSiMe2H) or tris(trimethylsilyl)silane (TTMSS), have been reported by Bandermann20 and Lalevée21 for use as an effective initiator for the radical polymerization of activated olefins. And we also found that the catalytic polymerization of activated olefins with PMHS could be completed by two different cobalt catalyst systems ([Co(OAc)2/TsOH] and [Co(acac)2]), in which the PMHS-derived semi-IPNs was formed by a coupling reaction involving hydrosilane.22
C–) bonds,19 could be linked with PMHS under cobalt-catalyzed silicon-carbon bond-forming reaction conditions (Fig. 3, step 1). In fact, the hydrosilanes, including pentamethyldisiloxane (TMSOSiMe2H) or tris(trimethylsilyl)silane (TTMSS), have been reported by Bandermann20 and Lalevée21 for use as an effective initiator for the radical polymerization of activated olefins. And we also found that the catalytic polymerization of activated olefins with PMHS could be completed by two different cobalt catalyst systems ([Co(OAc)2/TsOH] and [Co(acac)2]), in which the PMHS-derived semi-IPNs was formed by a coupling reaction involving hydrosilane.22
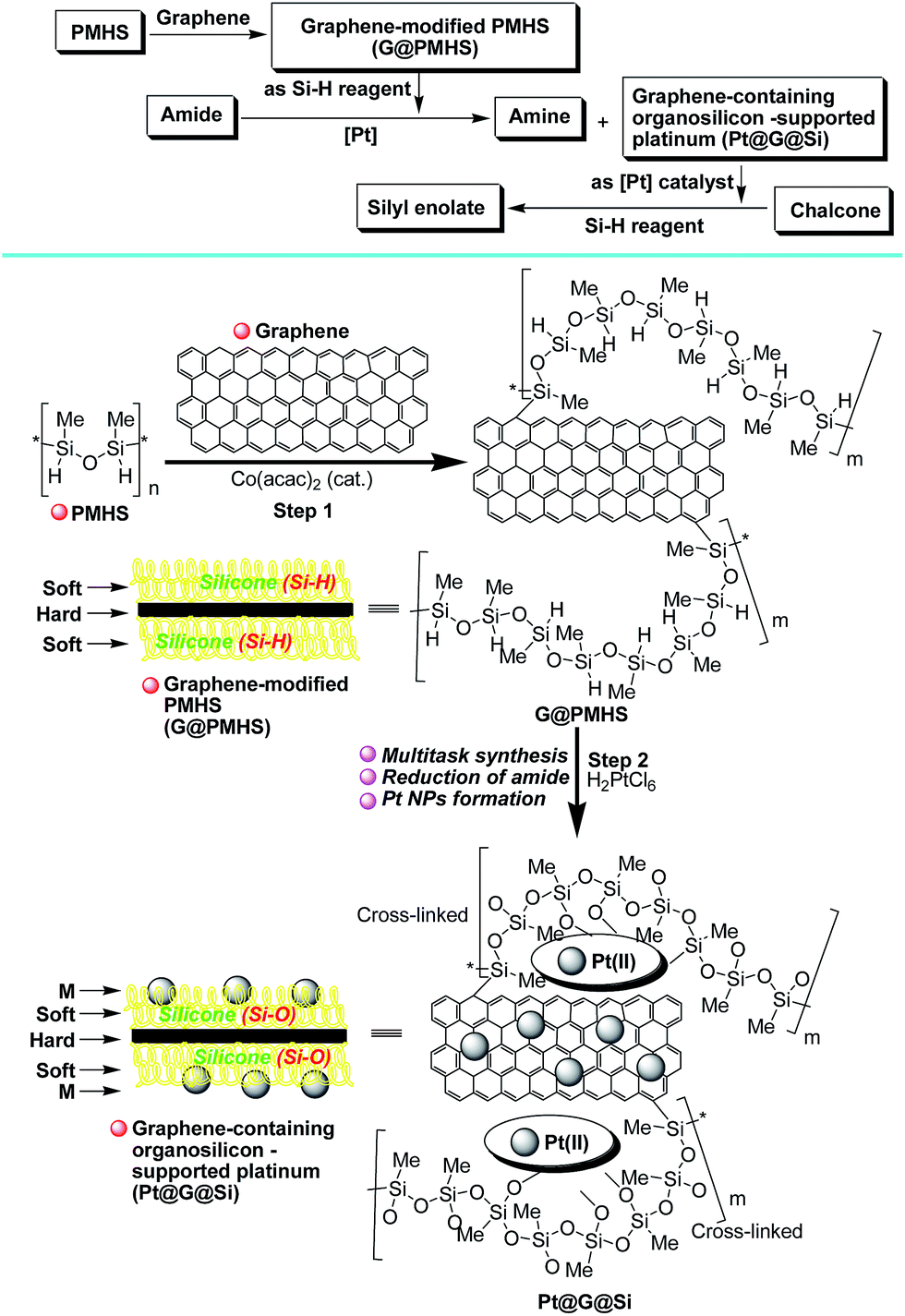 | ||
| Fig. 3 Construction of a graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) accompanied by the platinum-catalyzed reduction of amides to amines. | ||
Thus on the basis of previous reports and our experimental results on the cobalt-catalyzed radical polymerization of activated alkenes (Fig. 4, eqn (1)), especially with the decomposition analysis of these PMHS-based SIPNs by TBAF, we believed that the PMHS-involved cobalt-catalyzed polymerization of activated alkenes was a controlled radical reaction which could be used in the preparation of graphene-modified PMHS (Fig. 4, eqn (2)).23 In fact, we found that graphene-modified PMHS was easily obtained with good dispersion under the reported reaction conditions. However, the simple mixture of graphene and PMHS resulted in phase separation because of their inherent liquid and solid properties even at high temperature. This phenomenon indirectly supported the hypothesis that graphene and PMHS would be linked under cobalt-catalyzed radical reaction conditions. In addition, similar to our previous report,22 the cobalt catalyst used in this step could be easily washed by water and organic solvent and did not have an impact on the next application in the platinum-catalysed hydrosilylation of amides. Then, satisfied with the successful preparation of dispersed graphene-modified PMHS material, we believed the graphene could be an ideal supporter and additive for the immobilization of a platinum catalyst according to the microencapsulation method, that was first introduced by Kobayashi in 1998.24 Therefore, we prepared the cross-linked graphene-containing PMHS by microencapsulation of graphene with PMHS (G@PMHS, see ESI†) for further application in the construction of a soft-hard-soft sandwich-type graphene-containing organosilicon material (G@Si)-supported platinum catalyst, accompanied by the platinum-catalyzed reduction of amides to amines. As shown in Fig. 4, the construction of a graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) was proved to be successfully completed in the platinum-catalyzed reduction of amides to amines as in the following exploration.
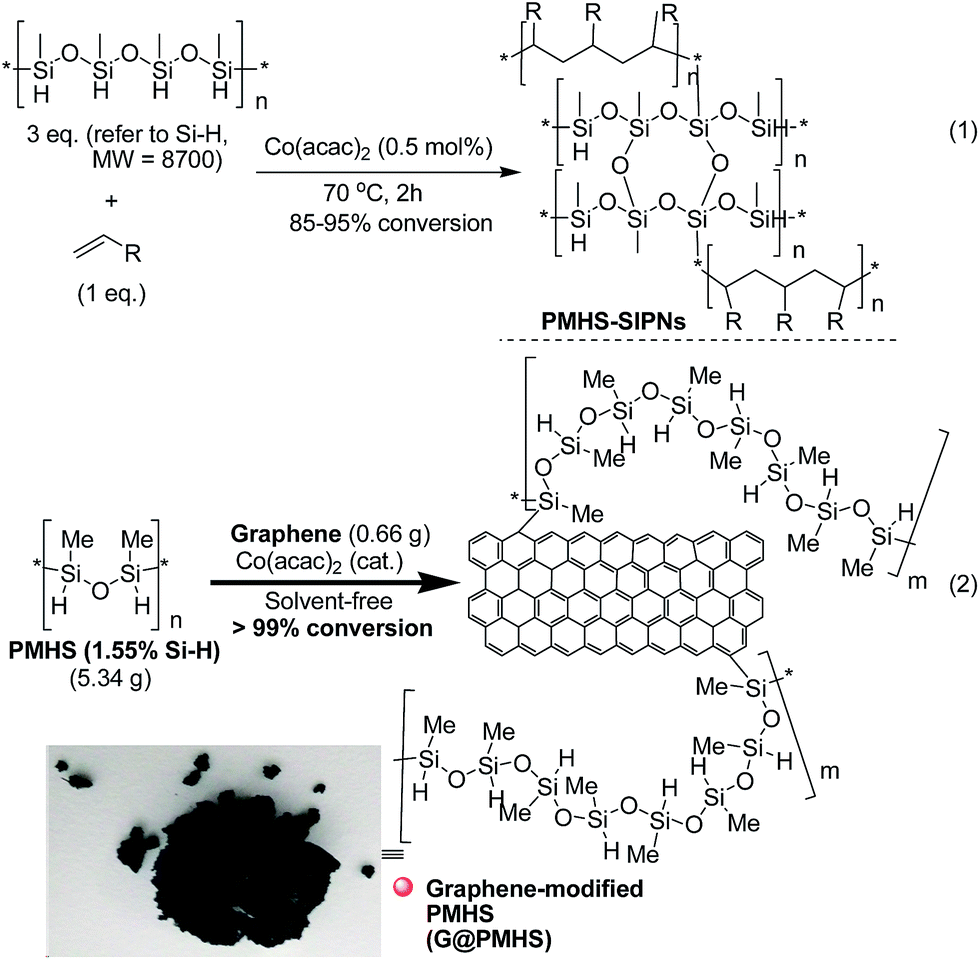 | ||
| Fig. 4 Synthesis of structurally diverse insoluble PMHS-based semi-interpenetrating networks (PMHS-SIPNs) via cobalt-catalyzed radical polymerization of terminal olefins. | ||
With the novel graphene-modified PMHS (G@PMHS) in hand, we then focused on the utilization of G@PMHS with the dual function of a novel hydrosilane (Si–H) reagent and catalyst support for the further evaluation of a multitask process by an in situ formed organosilicon-supported platinum catalyst in different hydrosilylation reactions. Initially, the platinum catalyst precursor (H2PtCl6, 1 mol% of Pt)-catalyzed hydrosilylation of amides with the aid of graphene-containing PMHS (G@PMHS) was carried out according to Nagashima's reduction reaction conditions (Fig. 5). Fortunately, it was found that yields of the desired product (2) were quite high in the presence of G@PMHS, which were better than those with graphite-containing PMHS. And we found that the use of 5 mol% of H2PtCl6 gave complete conversion without any side reactions in the presence of G@PMHS. More interestingly, the novel platinum catalyst attached to the carbon surface here was assisted by the cross-linked silicone material that has a high binding affinity for Pt ions in order to create a possibly recyclable hybrid platinum-graphene catalyst that exhibited minimal metal leaching. Notably, we have investigated the recovered platinum catalyst after the reduction of amide with PMHS, and, unfortunately, the resulting organosilicon material-supported platinum catalyst (Pt@G@Si) did not exhibit catalytic activity in the hydrosilylation-type reduction of amide under the optimized reaction conditions. These results suggested that the recovered platinum catalyst is different from the catalyst precursor or H2PtCl6 after the reaction. Therefore, further investigation of the recovered organosilicon material-supported platinum catalyst in new organic transformations is a valuable topic (Fig. 6).
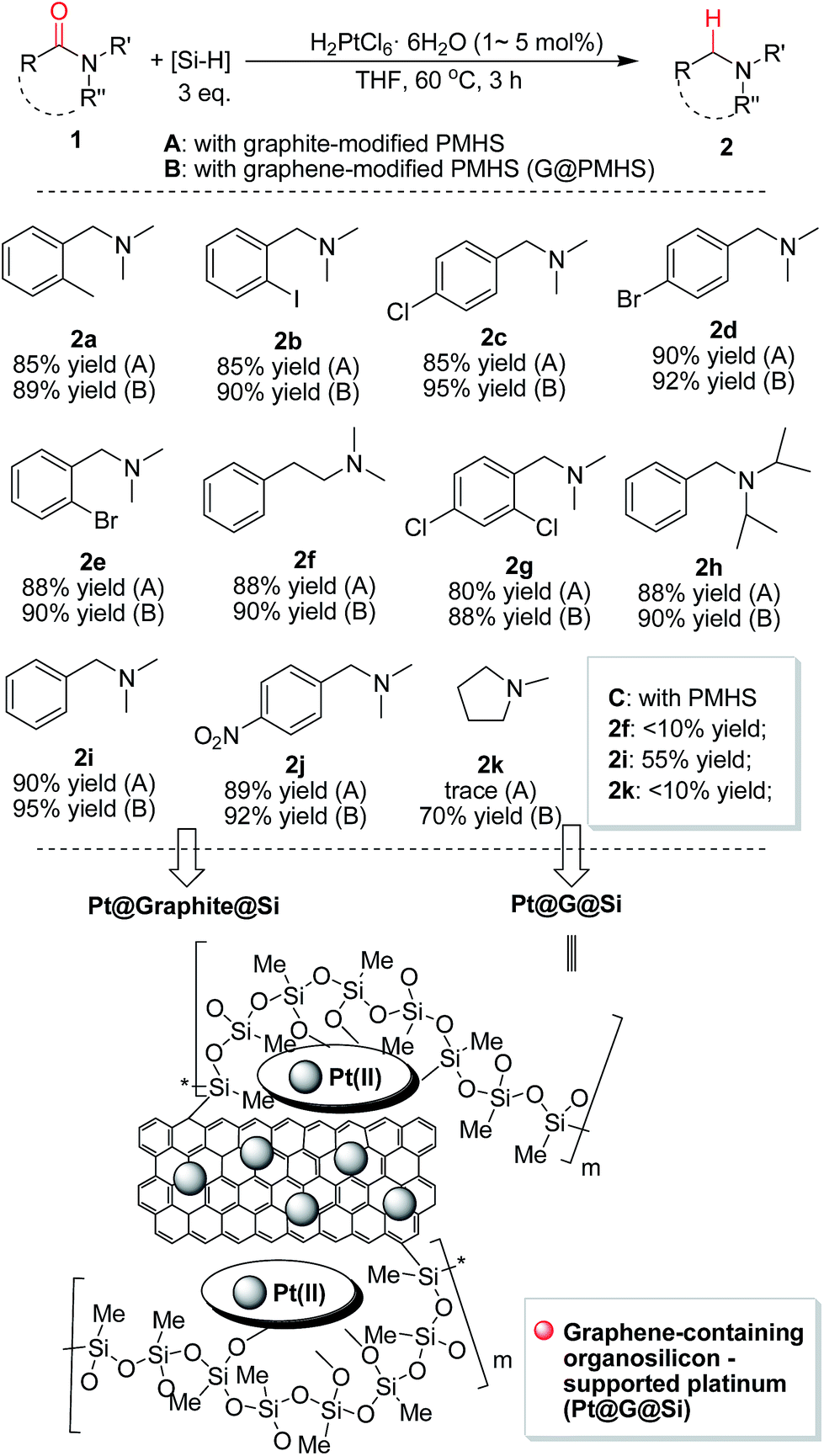 | ||
| Fig. 5 Platinum (H2PtCl6)-catalyzed hydrosilylation of amides with the aid of graphene-containing PMHS: (1) the determination of the superiority of graphene-modified PMHS (G@PMHS) over graphite-modified PMHS (graphite@PMHS) and commercially available PMHS in this reaction. (2) The recovery of in situ formed graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) and graphite-containing organosilicon material (graphite@Si)-supported platinum catalyst (Pt@graphite@Si). | ||
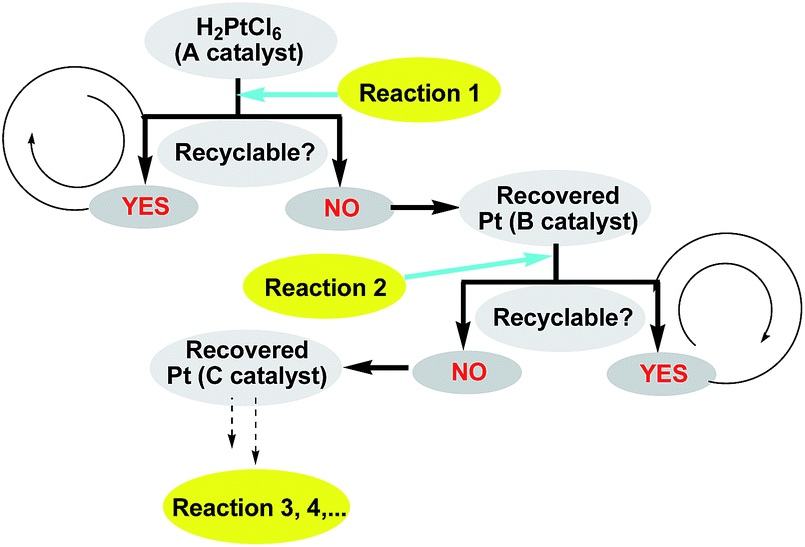 | ||
| Fig. 6 Multitask processes of recovered platinum catalyst in different types of downstream reactions. | ||
The resulting graphene-containing organosilicon material (G@Si)-supported platinum catalyst (Pt@G@Si) and other supported platinum catalysts (for example, Pt@graphite@Si generated from graphite-modified PMHS) evaluated in this work were characterized using X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Raman spectroscopy, and/or X-ray diffraction (XRD). The detailed spectra for these characterizations are provided in the ESI (Fig. S1–S13†). In addition, graphite- and graphene-modified PMHS material and the recovered Pt catalyst for reuse have also been adequately characterized by ICP-MS analysis (Table S3 of ESI†). All these characteristic data supported the finding that the regular structure and morphology of Pt@G@Si are different from those of Pt@graphite@Si. Thus it could indirectly provide a reason why graphene-modified PMHS (G@PMHS) is better than graphite-modified PMHS (graphite@PMHS) in the platinum-catalyzed hydrosilylation of amides (Fig. 5).
Accompanying the change in the structure of the surface, the morphology of Pt@G@Si and Pt@graphite@Si is shown in the SEM and TEM images (Fig. S1–S6†). From the TEM images of the catalyst, one can conclude that the distribution of platinum nanoparticles on the graphene-containing silicone material surface, with the hard–soft interface of carbon-based 2D material and silicon-based cross-linked polymer, creates a confined space for platinum nanoparticles (Fig. 7).
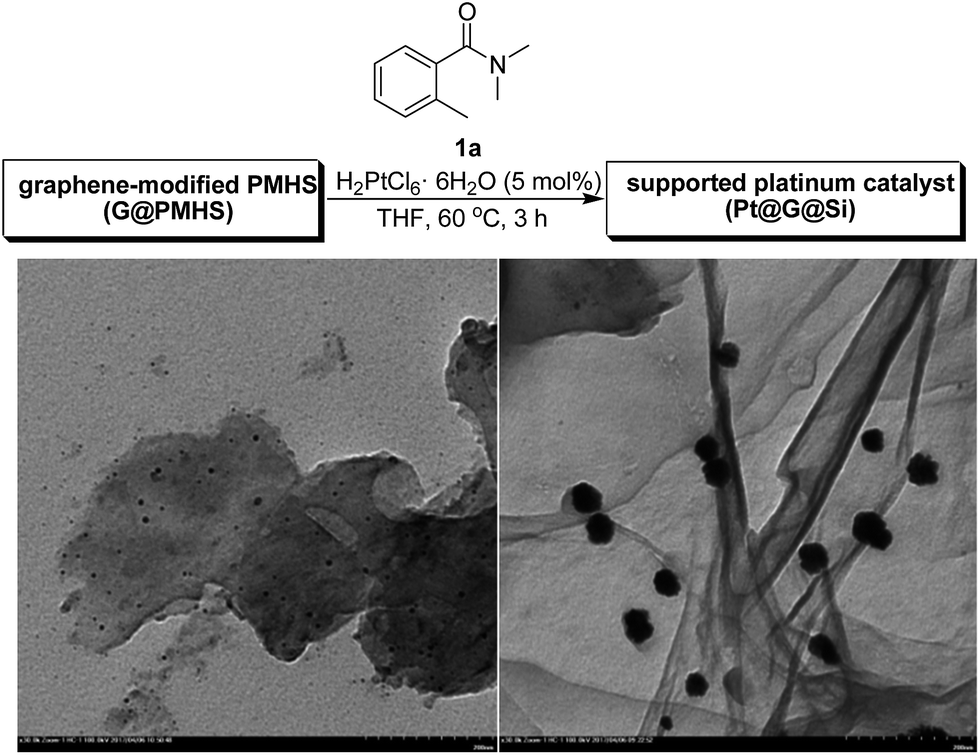 | ||
| Fig. 7 TEM images for a Pt@graphite@Si catalyst (left) and Pt@G@Si catalyst (right, for Pt nanoparticles, 30–40 nm). | ||
XRD patterns of Pt@G@Si and Pt@graphite@Si are shown in Fig. S9 and S10.† The main peak at 26.3° is ascribed to the (002) plane of the graphitic-type lattice. No obvious diffraction peak assigned to Pt particles and graphene is found in the XRD patterns, indicating that platinum nanoparticles (NPs) and graphene exist with good dispersion or with very small size. In addition, Raman, IR, and solid-state NMR analysis of the Pt@G@Si and graphene material also gave useful structural information of the corresponding organosilicon-supported graphene-based platinum catalyst (Fig. S7, S8 and S13†). This is also attributed to the residual platinum NPs being encapsulated into the graphene-containing organosilicon material (G@Si) after catalytic hydrosilylation of amides.
XPS was employed to investigate the electronic interaction between platinum and supports based on the binding energy shift of Pt 4f. In the XPS spectra of Pt@G@Si material (see Fig. S11 and S12 of ESI†), the Pt 4f XPS spectra of the hybrid catalyst present two main peaks at about 72.5 and 75.5 eV, corresponding to the spin–orbit split doublet of Pt 4f7/2 and 4f5/2, respectively. It is well known that Pt 4f XPS spectra are deconvoluted into three different components, including the predominant metallic Pt(0), oxide states Pt(II), and Pt(IV). Peaks at 72.5 eV (Pt 4f7/2) and 75.5 (Pt 4f5/2) are assigned to Pt(II), which probably anchored with the Si–O groups on the surface of the G@Si material. Of course, the functional species on the graphene surface have a great influence on the distribution of Pt(II) species. Therefore, after this reaction, the graphene-containing organosilicon material (G@Si) reduced a certain amount of the Pt(IV) (H2PtCl6) to Pt(II), which was strongly chemisorbed at the surfaces of the G@Si material. Meanwhile, returning to the H2Pt(IV)Cl6-catalyzed hydrosilylation of amides, it was clearly determined that the Pt(IV) intermediate plays a crucial role in this reaction, and the recovered organosilicon material-supported platinum catalyst (Pt@G@Si) did not exhibit any catalytic activity in the hydrosilylation-type reduction of amide, which gives indirect support to the previous report on the theoretical study of the reaction mechanism of platinum-catalyzed reduction of amides with hydrosilanes bearing dual Si–H groups. In 2015, Nakatani et al. reported that a key to accomplishing the reduction of amides via the present mechanism is the formation of a five-coordinate Pt(IV) species (Pt(IV)-disilyl-dihydride intermediate).25
On the other hand, the XPS analysis for Pt@G@Si supports the presence of Pt(II) species (Fig. S11†). Thus it is obvious that the platinum nanoparticles are possibly not metallic Pt but exist as an aggregation of Pt(II) on the surface of the graphene-containing organosilicon material, which differs from previous reports on the preparation of Pt nanoparticles.26 Of course, it is difficult to exclude the possibility that Pt(IV) or Pt(II) species aggregate to metallic platinum nanoparticles in the reductive hydrosilylation of amides.26 However, the XPS spectrum of Pt@G@Si (Pt 4f region) shows a typical contribution from Pt(II). This fact indicates that the Pt nanoparticles are possibly oxidized.27 According to the large decrease in Si–H groups on the graphene-modified PMHS (G@PMHS) during the reductive hydrosilylation of amides, it is a rational to suppose that the Si–H groups transformed into Si–O groups, thus forming Pt(II) groups in this case. Based on the distribution of Si–O groups on the graphene-containing organosilicon material (G@Si), the Pt(II) groups may occur on the edge plane or basal plane of the G@Si material.27a Another possible explanation for this apparent contradiction is that the graphene-containing organosilicon material (G@PMHS) partially reduces Pt(II) to Pt(0), in which case mixed valence nanoparticles form. Alternatively, the presence of “oxidized” Pt(II) species may reflect the strong chemisorption between the surface atoms of the Pt nanoparticles and the graphene-containing organosilicon material. Such chemical interactions can strongly change the binding energy of Pt. In both scenarios, the Pt(II) species are undoubtedly formed in situ during the induction period.26a Therefore, the novel graphene-modified PMHS (G@PMHS) appears to serve two crucial roles: one as a support for the Pt catalyst during the reduction of amides and the other as a nanostructured matrix responsible for controlling platinum nanoparticle growth.
Following characterization of the in situ generation of platinum(II) catalysts, the efficiency of heterogeneous Pt@G@Si or Pt@graphite@Si were evaluated as catalysts for the hydrosilylation of chalcone and triethylsilane under mild reaction conditions (Fig. 8 and Table S1, see ESI†). It should be noted that chalcone is a common, simple and privileged scaffold in numerous naturally occurring compounds,28 and, unexpectedly, there has been no report on the catalytic hydrosilylation of enones up to the present. We initially screened the reaction parameters (temperature and solvent effect) with Pt@graphite@Si as a model catalyst in this reaction. While Pt@graphite@Si was slightly less efficient compared to Pt@G@Si, the hydrosilylation reaction still reached 56% yield of silyl enolate 5a with moderate chemoselectivity (the ratio of Z/E selectivity is 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, see Fig. 8). As expected, the graphene-derived Pt@G@Si catalyst was a highly efficient catalyst, with the reaction reaching complete conversion and an excellent isolated yield (94%) in 3 h at 60 °C, and there are almost no other side products shown in Fig. 5. Notably, in a series of control experiments at 60 °C (Fig. 8), it was found that the Karstedt catalyst or H4PtCl6 also exhibited no activity in this silicon-oxygen bond-forming hydrosilylation reaction, which further confirmed the privileged role of the in situ formed Pt@G@Si nanocatalyst in the synthesis of a silyl enolate product. An important feature commonly seen in the well-dispersed and confined Pt@G@Si in silicone material is the moderate size of the platinum nanoparticles (30–40 nm, see the TEM image of Fig. 7). Interestingly, the recovered Pt@Si that was directly obtained from the H2PtCl6-catalyzed hydrosilylation of amide without graphene or graphite exhibited almost no catalytic activity in the hydrosilylation reaction of chalcone (Fig. 8). Although the active catalytic species of the Pt@G@Si nanocatalyst is not observed under the rapid reaction conditions, experimental results indicate that the Pt@G@Si nanocatalyst is distinct from that of organosilicon-encapsulated platinum in terms of the identity of the synergistic effect of Pt NPs on 2D graphene surface combined with cross-linked silicone material and the active Pt(II) nanoparticle species. In addition, the stability of the Pt@G@Si catalyst formed during the hydrosilylation using a mixture of H2PtCl6 and G@PMHS was also investigated, in which the fresh Pt@G@Si catalyst obtained by hot filtration exhibited the same level of high yield (94%) in the 1,4-hydrosilylation of chalcone with triethylsilane. Unfortunately, although the used catalysts could be easily recovered by simple filtration for recycling experiments, an obvious drop in yield was observed in the second cycle (20% yield). As shown in Table S3 (see ESI†), the results of ICP-MS analyses indicated that a large majority of the Pt species (about 81%) in Pt@G@Si catalyst were detached from the supported platinum catalyst.
:30, see Fig. 8). As expected, the graphene-derived Pt@G@Si catalyst was a highly efficient catalyst, with the reaction reaching complete conversion and an excellent isolated yield (94%) in 3 h at 60 °C, and there are almost no other side products shown in Fig. 5. Notably, in a series of control experiments at 60 °C (Fig. 8), it was found that the Karstedt catalyst or H4PtCl6 also exhibited no activity in this silicon-oxygen bond-forming hydrosilylation reaction, which further confirmed the privileged role of the in situ formed Pt@G@Si nanocatalyst in the synthesis of a silyl enolate product. An important feature commonly seen in the well-dispersed and confined Pt@G@Si in silicone material is the moderate size of the platinum nanoparticles (30–40 nm, see the TEM image of Fig. 7). Interestingly, the recovered Pt@Si that was directly obtained from the H2PtCl6-catalyzed hydrosilylation of amide without graphene or graphite exhibited almost no catalytic activity in the hydrosilylation reaction of chalcone (Fig. 8). Although the active catalytic species of the Pt@G@Si nanocatalyst is not observed under the rapid reaction conditions, experimental results indicate that the Pt@G@Si nanocatalyst is distinct from that of organosilicon-encapsulated platinum in terms of the identity of the synergistic effect of Pt NPs on 2D graphene surface combined with cross-linked silicone material and the active Pt(II) nanoparticle species. In addition, the stability of the Pt@G@Si catalyst formed during the hydrosilylation using a mixture of H2PtCl6 and G@PMHS was also investigated, in which the fresh Pt@G@Si catalyst obtained by hot filtration exhibited the same level of high yield (94%) in the 1,4-hydrosilylation of chalcone with triethylsilane. Unfortunately, although the used catalysts could be easily recovered by simple filtration for recycling experiments, an obvious drop in yield was observed in the second cycle (20% yield). As shown in Table S3 (see ESI†), the results of ICP-MS analyses indicated that a large majority of the Pt species (about 81%) in Pt@G@Si catalyst were detached from the supported platinum catalyst.
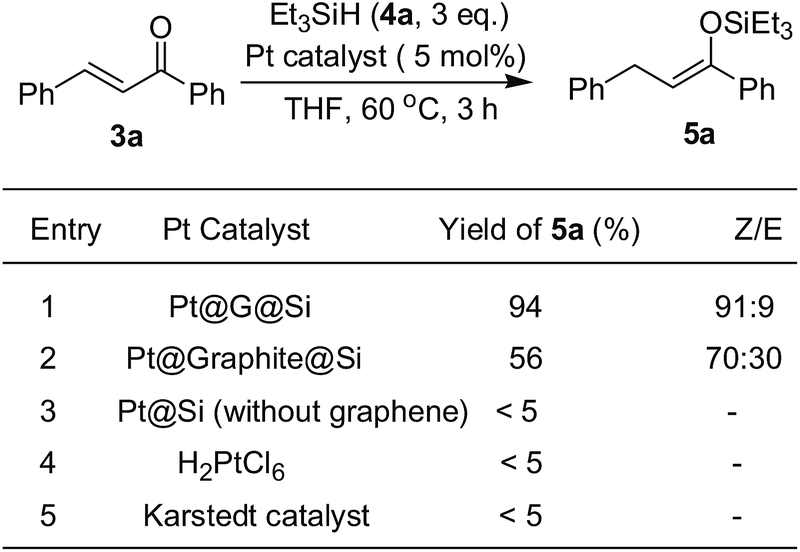 | ||
| Fig. 8 The catalytic activity of different platinum catalysts in the 1,4-hydrosilylation of chalcone with triethylsilane. | ||
Next, having the optimized conditions for the hydrosilylation of chalcone in hand, we determined the privileged role of graphene-derived Pt@G@Si in this reaction, and then examined the scope of the 1,4-hydrosilylation reaction of various chalcones bearing a substituent on the aromatic ring.
As can be seen from Table 1, the scope of Pt-catalyzed 1,4-hydrosilylation of various chalcones with triethylsilane appears to be very broad, and the silicon–oxygen bond-forming 1,4-hydrosilylation reaction is satisfactory in isolated yield and Z selectivity even with versatile hydrosilanes. Since the ability of hydrosilane to hydrogenate enones, including chalcones, to saturated ketones or alcohols has been recognized in the past,28,29 this is a rare example where a heterogeneous platinum catalyst (Pt@G@Si) could be used to promote the silicon–oxygen bond-forming 1,4-hydrosilylation of aromatic enones.
|
||||
|---|---|---|---|---|
| Entrya | R1/R2 | [Si]–H | Ratio of Z/Eb | Yieldc (%) |
| a Reactions were performed with chalcone (0.5 mmol), hydrosilane ([Si]–H, 1.5 mmol), Pt@G@Si catalyst (5 mol%), THF (1 mL).b The ratio of Z/E is determined by 1H-NMR.c Isolated yield. | ||||
| 1 | H/H | 4a | 91:9 |
5a: 94 |
| 2 | 4-OMe/H | 4a | 79:21 |
5b: 83 |
| 3 | 4-Br/H | 4a | 96:4 |
5c: 86 |
| 4 | 2-OMe/H | 4a | 88:12 |
5d: 85 |
| 5 | 4-Cl/4-Cl | 4a | 80:20 |
5e: 89 |
| 6 | 3-OMe/4-F | 4a | 75:25 |
5f: 90 |
| 7 | 4-Ph/H | 4a | 80:20 |
5g: 88 |
| 8 | H/4-Me | 4a | 78:22 |
5h: 90 |
| 9 | H/4-Br | 4a | 93:7 |
5i: 88 |
| 10 | H/4-Cl | 4a | 82:18 |
5j: 91 |
| 11 | 4-Cl/H | 4a | 89:11 |
5k: 90 |
| 12 | 3-OMe/H | 4a | 77:23 |
5l: 89 |
| 13 | H/4-OMe | 4a | 85:15 |
5m: 84 |
| 14 | 4-Me/H (X = N) | 4a | 97:3 |
5n: 89 |
| 15 | 3-Br/H (X = N) | 4a | 99:1 |
5o: 84 |
| 16 | H/H | 4b | 90:10 |
5p: 76 |
| 17 | H/H | 4c | 90:10 |
5q: 71 |
| 18 | H/H | 4d | 91:9 |
5r: 81 |
| 19 | H/H | 4e | 90:10 |
5s: 73 |
| 20 | H/H | 4f | 90:10 |
5t: 78 |
| 21 | H/H | 4g | 99:1 |
5u: 83 |
As shown in Table 1, a number of substituted chalcones were initially exposed to the 1,4-hydrosilylation under the optimized reaction conditions. Substrates bearing a variety of functional groups on the aryl ring (for example, Cl, Br, F, MeO, Me) on the meta-, para-, or ortho-position of the chalcones were tolerated in this reaction. And there is almost no difference in reactivity for these chalcones because good to excellent yields (71–95% yields) and high ratio of Z selectivities (up to 99:1) could be achieved (entries 1–13). Interestingly, the pyridine-containing chalcones are also reactive substrates: for example, 1-pyridin-2-yl-3-p-tolyl-propenone and 3-(3-bromo-phenyl)-1-pyridin-2-yl-propenone were smoothly converted to the corresponding silyl enolate products 5n and 5o with 89% and 84% yields, respectively (entries 14 and 15). When the hydrosilanes were substituted with other groups, such as methyl, ethoxyl, phenyl, tert-butyl, the hydrosilylation of chalcone 3a went smoothly and furnished the desired silyl enolate products in good yields (71–83%), thus showing the high tolerance for structurally diverse and functional hydrosilanes as well as the high 1,4-silylation capacity with a superior active Pt-based nanocatalyst (Pt@G@Si).
Notably, these results showed that the really active platinum is Pt(II) because the XPS spectrum of Pt@G@Si (Pt 4f region) shows a typical contribution from Pt(II). In fact, inspired by this finding, we then investigated the catalytic effect of (PPh3)2PtCl2 on the 1,4-hydrosilylation of chalcones under similar reaction conditions. Gratifyingly, (PPh3)2PtCl2 is proved to be a highly efficient catalyst in this reaction (Table 2), in which it exhibited better Z-selectivity and achieved the same level of yields in comparison to that of a heterogeneous Pt@G@Si catalyst. Therefore, the (PPh3)2PtCl2-catalyzed 1,4-hydrosilylation of chalcones provided direct evidence in support of the key catalyst of Pt@G@Si being Pt(II). Meanwhile, this work also provides a strategy in which the structural analysis of heterogeneous catalyst materials would give a new opportunity to design new catalysts, even for homogeneous catalysis.
|
||||
|---|---|---|---|---|
| Entrya | R/R2 | [Si]–H | Ratio of Z/Eb | Yieldc (%) |
| a Reactions were performed with chalcone (0.5 mmol), hydrosilane ([Si]–H, 1.5 mmol), (PPh3)2PtCl2 catalyst (5 mol%), THF (1 mL).b The ratio of Z/E is determined by 1H-NMR.c Isolated yield. | ||||
| 1 | H/H | 4a | 98:2 |
94 |
| 2 | 4-OMe/H | 4a | 95:5 |
87 |
| 3 | 4-Br/H | 4a | 96:4 |
85 |
| 4 | 2-OMe/H | 4a | 95:5 |
88 |
| 5 | 4-Cl/4-Cl | 4a | 99:1 |
86 |
| 6 | 3-OMe/4-F | 4a | 99:1 |
91 |
| 7 | 4-Ph/H | 4a | 97:3 |
86 |
| 8 | H/4-Me | 4a | >99:1 |
83 |
| 9 | H/4-Br | 4a | 97:3 |
82 |
| 10 | H/4-Cl | 4a | 99:1 |
89 |
| 11 | 4-Cl/H | 4a | 92:8 |
84 |
| 12 | 3-OMe/H | 4a | 95:5 |
84 |
| 13 | H/4-OMe | 4a | 97:3 |
86 |
| 14 | 4-Me/H (X = N) | 4a | 98:2 |
85 |
| 15 | 3-Br/H (X = N) | 4a | 99:1 |
81 |
| 16 | H/H | 4b | 97:3 |
81 |
| 17 | H/H | 4c | 99:1 |
87 |
| 18 | H/H | 4f | 99:1 |
83 |
In addition, we have also investigated the possibility of the recovery and re-use of Pt@G@Si after the catalytic 1,4-hydrosilylation of chalcone. However, the recovered Pt@G@Si is not the same platinum catalyst as that used freshly in this reaction, which was determined by TEM and XPS analysis (see Fig. S14†). The platinum catalyst is possibly dispersed into the inner organosilicon material. Although the change in chemical structure of the catalyst support (graphene-containing organosilicon material) cannot be ruled out at this time, we favor the deactivation of the platinum catalyst on the basis of the above results. We believe that a further evaluation of the multitask process could be a solution to the catalytic application of recovered Pt@G@Si catalyst. Therefore, in performing these reactions with the same catalyst precursor, we anticipated that the multitask strategy would be a major advantage from the point of view of the catalyst-economic process. We have shown that there is no border between homogeneous catalysis and heterogeneous catalysis, and the multitask strategy could be considered as an environmentally benign process to reuse a transition-metal catalyst from a homogeneous reaction system, and an investigation into the corresponding in situ formed supported metal catalyst in a heterogeneous reaction system would give useful information for developing a new transition-metal catalyst system.
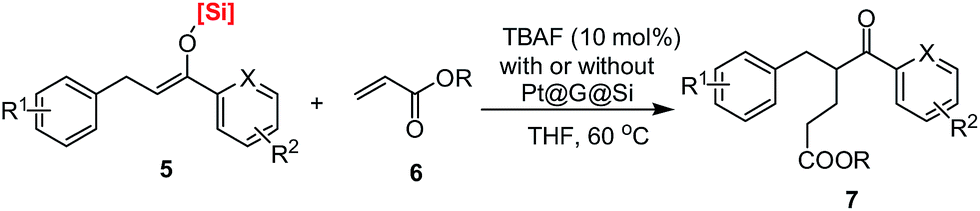
Finally, in order to obtain more information on the synthetic utilization of the corresponding products, silyl enolates, that were obtained in this work, TBAF-promoted conjugate addition of silyl enolates 5 to ethyl acrylate 6 was investigated in THF at room temperature. It should be noted that there has been no report on the conjugate addition of such silyl enolates to alkyl acrylate in the past.30 As shown in Table 3, the conjugate addition occurred smoothly to give the desired product in good to excellent yields (76–90% isolated yields). Various silyl enolates with different silicon-based bulky groups could be used in this reaction and gave the α-branched products in good yields. And notably, the same level of conversion could be achieved by the one-pot Pt@G@Si-catalyzed 1,4-hydrosilylation of chalcones and then the TBAF-promoted conjugate addition of silyl enolates 5 to alkyl acrylates 6 (Table 3, entry 21). Thus, it is an interesting and useful example that aromatic 5-oxohexanoate products (1,5-keto esters)31 could be prepared by a new way with the conjugate addition of silyl enolates to ethyl acrylate.
|
||||
|---|---|---|---|---|
| Entrya | SM | [Si] | R | Yieldb (%) |
| a Reactions were performed with silyl enolate (0.5 mmol), alkyl acrylate (0.5 mmol), TBAF (0.1 eq.), THF (1 mL).b Isolated yield.c With the addition of TBAF in presence of Pt@G@Si catalyst, total yield for one-pot two-step. | ||||
| 1 | 5a | SiEt3 | Et | 7a: 89 |
| 2 | 5p | SiMePh2 | Et | 7a: 83 |
| 3 | 5q | SiMe2Ph | Et | 7a: 81 |
| 4 | 5r | SiMe(OEt)2 | Et | 7a: 80 |
| 5 | 5s | SiMe2(OEt) | Et | 7a: 76 |
| 6 | 5t | SiMe2t-Bu | Et | 7a: 78 |
| 7 | 5u | Si(OEt)3 | Et | 7a: 81 |
| 8 | 5b | SiEt3 | Et | 7b: 85 |
| 9 | 5c | SiEt3 | Et | 7c: 82 |
| 10 | 5d | SiEt3 | Et | 7d: 80 |
| 11 | 5e | SiEt3 | Et | 7e: 81 |
| 12 | 5f | SiEt3 | Et | 7f: 82 |
| 13 | 5g | SiEt3 | Et | 7g: 83 |
| 14 | 5h | SiEt3 | Et | 7h: 81 |
| 15 | 5i | SiEt3 | Et | 7i: 83 |
| 16 | 5j | SiEt3 | Et | 7j: 89 |
| 17 | 5k | SiEt3 | Et | 7k: 90 |
| 18 | 5l | SiEt3 | Et | 7l: 88 |
| 19 | 5m | SiEt3 | Et | 7m: 89 |
| 20 | 5n | SiEt3 | Et | 7n: 89 |
| 21 | 5a | SiEt3 | Et | 7a: 91c |
Experimental
General procedure for the platinum-catalyzed 1,4-hydrosilylation reaction of chalcones
The triethylsilane (0.17 mL, 1.5 mmol, 3.0 equiv.), Pt@G@Si (10.2 mg, 25 μmol, 5 mol%), and chalcone (104 mg, 0.5 mmol, 1.0 equiv.) were dissolved in anhydrous THF (1.0 mL). And then the reaction mixture was heated to 60 °C and monitored by TLC until total conversion of the starting materials (about 3 hours). The reaction mixture was cooled to room temperature. After filtration under diminished pressure, the organic layer was diluted with 10.0 mL EtOAc, and the aqueous layer was extracted with EtOAc (5.0 mL × 2). The combined organic layer was dried over Na2SO4 and concentrated. Purification by silica gel flash chromatography (using petroleum:EtOAc = 10:1) gave the desired products with good yields. All the products were confirmed by MS, and the usual spectral methods (1H-NMR, 13C-NMR) (see ESI†). For example, (Z)-(1,3-diphenylprop-1-enyloxy)triethylsilane (5a): 94% yield; 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 6.9 Hz, 2H), 7.19 (t, J = 5.2 Hz, 3H), 7.17–7.15 (m, 3H), 7.15–7.05 (m, 2H), 5.22 (t, J = 7.2 Hz, 1H), 3.50 (d, J = 7.2 Hz, 2H), 0.85 (t, J = 7.9 Hz, 9H), 0.54 (q, J = 7.9 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 150.3, 141.7, 139.55, 128.5, 128.1, 127.7, 125.9, 109.8, 77.4, 77.1, 76.8, 32.4, 6.8, 5.5, 5.2. HRMS (ESI): m/z: [M + H]+ calculated for C21H29OSi: 325.1982, found: 325.1971.
Conclusions
In summary, we have confirmed the powerful potential of the multitask process for a recovered platinum catalyst in situ formed in the reduction of amides and its sustainable catalytic application in downstream 1,4-hydrosilylation reactions. In detail, we described an interesting finding that an organosilicon/graphene-supported platinum nanocatalyst could be in situ prepared from the simple H2PtCl6-catalyzed reduction reaction of amides, and the corresponding graphene- and organosilicon-supported Pt nanoparticles (NPs) were found to be a highly efficient catalyst in the silicon-carbon bond-forming 1,4-hydrosilylation of chalcones, which is clearly beneficial to the development of a new multitask strategy with multitask and comprehensive utilization of a recovered and deactivated transition metal catalyst. Apart from the advantages generated from the novel multitask strategy, the corresponding platinum catalyst systems that were in situ formed in the H2PtCl6-catalyzed reduction of amides provide the following features: (i) a high-yield and enhanced deoxygenated reduction of amides with the aid of carbon-based materials, such as graphene or graphite, was observed under mild reaction conditions; (ii) an example of highly efficient platinum-catalyzed 1,4-hydrosilylation of α,β-unsaturated enones is characterized by exquisite chemoselectivity, allowing for the facile synthesis of a variety of otherwise inaccessible substituted silyl enolates in a straightforward manner. And in view of the new catalytic function of a rationally designed platinum catalyst supported on a graphene/organosilicon-based composite material in the downstream reaction, we anticipate that the multitask process and the in situ formed or newly generated platinum catalyst as well as the hydrosilylation reaction of chalcones will offer the possibility for new application of waste or deactivated transition metal catalysts for new downstream organic transformations. Finally, in order to obtain more information of the synthetic utilization of the corresponding products, silyl enolates, we found that the silyl enolates could be used as nucleophiles in the conjugate addition of alkyl acrylates, which proceeded smoothly to give a new type of 1,5-keto esters. Further investigations of related mechanism on platinum-promoted hydrosilylation transformations and an asymmetric version are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This Project was supported by the National Natural Science Foundation of China (No. 21472031, 21503060, and 21773051), Shaanxi Provincial Natural Science Foundation of China (2017JM2001), Zhejiang Provincial Natural Science Foundation of China (LZ18B020001, LY16E030009, and LY17E030003), and Science and Technology Department of Zhejiang Province (2015C31138). This work is also supported partially by the Program of “One Hundred Talented People” of Shaanxi Province. The authors also thank Dr Z. R. Qu, Dr C. Q. Sheng, Dr K. Z. Jiang, and Dr Q. H. Pan (all at HZNU) for their technical and analytical support. The authors also sincerely thank the reviewers for their help in the improvement of this manuscript.Notes and references
- (a) J. A. Gladysz, Chem. Rev., 2002, 102, 3215–3216 CrossRef CAS PubMed; (b) D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed; (c) M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed; (d) Z. Wang, G. Chen and K. L. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS PubMed; (e) Y. Yang, C. J. Sun, D. E. Brown, L. Zhang, F. Yang, H. Zhao, Y. Wang, X. Ma, X. Zhang and Y. Ren, Green Chem., 2016, 18, 3558–3566 RSC; (f) P. Zhang, Z. Zhao, B. Dyatkin, C. Liu and J. Qiu, Green Chem., 2016, 18, 3594–3599 RSC.
- (a) V. K. Dioumaev and R. M. Bullock, Nature, 2003, 424, 530–532 CrossRef CAS PubMed; (b) D. E. Bergbreiter and S. D. Sung, Adv. Synth. Catal., 2006, 348, 1352–1266 CrossRef CAS; (c) C. A. M. Afonso, L. C. Branco, N. R. Candeias, P. M. P. Gois, N. M. T. Lourenço, N. M. M. Mateus and J. N. Rosa, Chem. Commun., 2007, 2669–2679 RSC; (d) N. Janssens, L. H. Wee, S. Bajpe, E. Breynaert, C. E. A. Kirschhock and J. A. Martens, Chem. Sci., 2012, 3, 1847–1850 RSC; (e) D. B. Lao, B. R. Galan, J. C. Linehan and D. J. Heldebrant, Green Chem., 2016, 18, 4871–4874 RSC.
- For recent reviews, see: (a) A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS; (b) F. X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718–724 CrossRef CAS PubMed; (c) N. Shindoh, Y. Takemoto and K. Takasu, Chem.–Eur. J., 2009, 15, 12168–12179 CrossRef CAS PubMed; (d) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 RSC; (e) I. V. Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2017, 17, 2012–2026 RSC.
- (a) F. X. Felpin, O. Ibarguren, L. Nassar-Hardy and E. Fouquet, J. Org. Chem., 2009, 74, 1349–1352 CrossRef CAS PubMed; (b) F. X. Felpin, J. Coste, C. Zakri and E. Fouquet, Chem.–Eur. J., 2009, 15, 7238–7245 CrossRef CAS PubMed; (c) O. Ibarguren, C. Zakri, E. Fouquet and F. X. Felpin, Tetrahedron Lett., 2009, 50, 5071–5074 CrossRef CAS; (d) J. Laudien, E. Fouquet, C. Zakri and F. X. Felpin, Synlett, 2009, 1539–1543 Search PubMed; (e) P. J. Aliamo, R. O'Brien III, A. W. Johnson, S. R. Slauson, J. M. O'Brien, E. L. Tyson, A. L. Marshall, C. E. Ottinger, J. G. Chacon, L. Wallace, C. Y. Paulino and S. Connell, Org. Lett., 2008, 10, 5111–5114 CrossRef PubMed; (f) B. H. Lipshutz, D. M. Nihan, E. Vinogradova, B. R. Taft and Ž. V. Bošković, Org. Lett., 2008, 10, 4279–4282 CrossRef CAS PubMed; (g) A. Zulauf, M. Mellah and E. Schulz, Chem. Commun., 2009, 6574–6576 RSC; (h) V. Escande, A. Velati, C. Garel, B. L. Renard, E. Petit and C. Grison, Green Chem., 2015, 17, 2188–2199 RSC.
- H. Wang, L. Li, X. F. Bai, W. H. Deng, Z. J. Zheng, K. F. Yang and L. W. Xu, Green Chem., 2013, 15, 2349–2355 RSC.
- For representative reviews, see: (a) C. J. Chapman and C. G. Frost, Synthesis, 2007, 1–21 CAS; (b) F.-X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718–724 CrossRef CAS PubMed; (c) J. A. Mata, F. Ekkehardt and E. Peris, Chem. Sci., 2013, 5, 1723–1732 RSC; (d) T. L. Lohr, Z. Li and T. J. Marks, Acc. Chem. Res., 2016, 49, 824–834 CrossRef CAS PubMed; (e) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed; (f) J.-L. Renaud and S. Gaillard, Synthesis, 2016, 48, 3659–3683 CrossRef CAS; (g) Y. B. Huang, J. Liang, X. S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–147 RSC.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practise, Oxford University Press, Oxford, 1998 Search PubMed; (b) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- For representative reviews, see: (a) M. Pagliaro, R. Ciriminna, V. Pandarus and F. Béland, Eur. J. Org. Chem., 2013, 6227–6235 CrossRef CAS; (b) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (c) S. Putzien, O. Nuyken and F. E. Kühn, Prog. Polym. Chem., 2010, 35, 687–713 CrossRef CAS. For recent examples in the field of green chemistry, see: (d) F. P. Bouxin, A. McVeigh, F. Tran, N. J. Westwood, M. C. Jarvis and S. D. Jackson, Green Chem., 2015, 17, 1235–1242 RSC; (e) P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016, 18, 842–847 RSC; (f) Z. Bai, M. Shi, Y. Zhang, Q. Zhang, L. Yang, Z. Yang and J. Zhang, Green Chem., 2016, 18, 386–391 RSC.
- (a) A. P. Barlow, N. M. Boag and F. G. A. Stone, J. Organomet. Chem., 1980, 191, 39–47 CrossRef CAS; (b) C. R. Johnson and R. K. Raheja, J. Org. Chem., 1994, 59, 2287–2288 CrossRef CAS.
- (a) N. L. Lampland, A. Pindwal, S. R. Neal, S. Schlauderaff, A. Ellern and A. D. Sadow, Chem. Sci., 2015, 6, 6901–6907 RSC; (b) K. Takeshita, Y. Seki, K. Kawamoto, S. Murai and N. Sonoda, J. Org. Chem., 1987, 52, 4864–4868 CrossRef CAS; (c) E. Yoshii, Y. Kobayashi, T. Koizumi and T. Oribe, Chem. Pharm. Bull., 1974, 22, 2767–2769 CrossRef CAS; (d) S. A. Bruno, US pat., 4785126A, 1988S. A. Bruno, US pat., 5332852A, 1994 (e) Recently, Chang et al. reported that the B(C6F5)3 could be used to promote the addition of tertiary silanes to α,β-unsaturated esters giving silyl ketene acetals, in which it isomerize to α-silyl esters through B(C6F5)3-catalyzed rearrangement process. See: Y. Kim and S. Chang, Angew. Chem., Int. Ed., 2016, 55, 218–222 (Angew. Chem., 2016, 128, 226–230) CrossRef CAS PubMed; (f) H. Wang, L. Li, X. F. Bai, J. Y. Shang, K. F. Yang and L. W. Xu, Adv. Synth. Catal., 2013, 355, 341–347 CAS; and polymerization is also a possible pathway when acrylates were used a substrate, see: ref. 5 and (g) H. Wang, K. F. Yang, L. Li, Y. Bai, Z. J. Zheng, W. Q. Zhang, Z. W. Gao and L. W. Xu, ChemCatChem, 2014, 6, 580–591 CrossRef CAS.
- A. Pindwal, S. Patnaik, W. C. Everett, A. Ellern, T. L. Windus and A. D. Sadow, Angew. Chem., Int. Ed., 2017, 56, 628–631 (Angew. Chem, 2017, 129, 643–646) CrossRef CAS PubMed.
- (a) S. Kobayashi, K. Manabe, H. Ishitani and J. Matsuno, Science of Synthesis, ed. I. Fleming, Thieme, Stuttgart, Germany, 1999, vol 4, ch. 4.16 Search PubMed; (b) K. Miura and A. Hosomi, Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley-VCH, Weinheim, Germany, 2004, vol. 2, ch. 10.2 Search PubMed; (c) J.-I. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109–9118 CrossRef CAS PubMed; (d) S. B. J. Kan, K. K.-H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097–9108 CrossRef CAS PubMed; (e) T. Kitanosomo and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 3095–3118 CrossRef PubMed; (f) F. Ye, Z. J. Zheng, W. H. Deng, L. S. Zheng, Y. Deng, C. G. Xia and L. W. Xu, Chem.–Asian J., 2013, 8, 2242–2253 CrossRef CAS PubMed.
- (a) I. Ojima and T. Kogure, Organometallics, 1982, 1, 1390–1399 CrossRef CAS; (b) G. Z. Zheng and T. H. Chan, Organometallics, 1995, 14, 70–79 CrossRef CAS; (c) A. Mori and T. Kato, Synlett, 2002, 1167–1169 CrossRef CAS; (d) B. H. Lipshutz, W. Chrisman, K. Noson, P. Papa, J. A. Sclafani, R. W. Vivian and J. M. Keith, Tetrahedron, 2002, 56, 2779–2788 CrossRef; (e) J. M. Blackwell, D. J. Morrison and W. E. Piers, Tetrahedron, 2002, 58, 8247–8254 CrossRef CAS; (f) M. Anada, M. Tanaka, K. Suzuki, H. Nambu and S. Hashimoto, Chem. Pharm. Bull., 2006, 54, 1622–1623 CrossRef CAS PubMed; (g) A. Tsubouchi, K. Onishi and T. Takeda, J. Am. Chem. Soc., 2006, 128, 14268–14269 CrossRef CAS PubMed; (h) M. Anada, M. Tanaka, T. Washio, M. Yamawaki, T. Abe and S. Hashimoto, Org. Lett., 2007, 9, 4559–4562 CrossRef CAS PubMed; (i) M. Rubio, J. Campos and E. Carmona, Org. Lett., 2011, 13, 5236–5239 CrossRef CAS PubMed; (j) J. Koller and R. G. Bergman, Organometallics, 2012, 31, 2530–2533 CrossRef CAS.
- Y. Sumida, H. Yorimitsu and K. Oshima, J. Org. Chem., 2009, 74, 7986–7989 CrossRef CAS PubMed.
- G. Onodera, R. Hachisuka, T. Noguchi, H. Miura, T. Hashimoto and R. Takeuchi, Tetrahedron Lett., 2014, 55, 310–313 CrossRef CAS.
- H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543–1553 CrossRef CAS.
- S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032–15040 CrossRef CAS PubMed.
- B. P. S. Chauhan and J. S. Rathore, J. Am. Chem. Soc., 2005, 127, 5790–5791 CrossRef CAS PubMed.
- L. Liao, H. Peng and Z. Liu, J. Am. Chem. Soc., 2014, 136, 12194–12200 CrossRef CAS PubMed , and references cited therein.
- R. Imann, F. Bandermann and H. Korth, Macromol. Chem. Phys., 1996, 197, 921–935 CrossRef CAS.
- J. Lalevée, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2003–2010 CrossRef.
- X. Y. Dong, Y. Lin, Y. M. Cui, K. F. Yang, Z. J. Zheng and L. W. Xu, ChemistrySelect, 2016, 1, 2400–2404 CrossRef CAS , and references cited therein.
- The graphene-modified PMHS (G@PMHS) was also confirmed by solid NMR analysis, see Fig. S13 (see ESI†). The chemical shift of graphene (1H NMR) is obviously changed after the introduction of PMHS. (a) M. Leskes, G. Kim, T. Liu, A. L. Michan, F. Aussenac, P. Dorffer, S. Paul and C. P. Grey, J. Phys. Chem. Lett., 2017, 8, 1078–1085 CrossRef CAS PubMed; (b) A. R. MacIntosh, K. J. Harris and G. R. Goward, Chem. Mater., 2016, 28, 360–367 CrossRef CAS.
- (a) S. Kobayashi and S. Nagayama, J. Am. Chem. Soc., 1998, 120, 2985–2986 CrossRef CAS; (b) R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS PubMed; (c) Y. Chen, W. Q. Zhang, B. X. Yu, Y. M. Zhao, Z. W. Gao, Y. J. Jian and L. W. Xu, Green Chem., 2016, 18, 6357–6366 RSC , and references cited therein.
- N. Nakatani, J.-Y. Hasegawa, Y. Sunada and H. Nagashima, Dalton Trans., 2015, 44, 19344–19356 RSC.
- (a) H. E. V. Dam and H. V. Bekkum, J. Catal., 1991, 131, 335–349 CrossRef; (b) J. N. Kuhn, W. Huang, C. K. Tsung, Y. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026–14027 CrossRef CAS PubMed; (c) C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed; (d) R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194–7195 CrossRef CAS PubMed; (e) H. Lang, R. A. May, B. L. Iversen and B. D. Chandler, J. Am. Chem. Soc., 2003, 125, 14832–14836 CrossRef CAS PubMed; (f) Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS; (g) W. Chen, S. G. Sun, Z. Y. Zhou and S. P. Chen, J. Phys. Chem. B, 2003, 107, 9808–9812 CrossRef CAS; (h) Y. B. He, G. R. Li, Z. L. Wang, Y. N. Ou and Y. X. Tong, J. Phys. Chem. C, 2010, 114, 19175–19181 CrossRef CAS; (i) J. Chen, Y. Xiong, Y. Yin and Y. Xia, Small, 2006, 2, 1340–1343 CrossRef CAS PubMed.
- (a) M. Y. Yen, C. C. Teng, M. C. Hsiao, P. I. Liu, W. P. Chuang, C. C. M. Ma, C. K. Hsieh, M. C. Tsai and C. H. Tsai, J. Mater. Chem., 2011, 21, 12880–12888 RSC; (b) S. Ikeda, S. Ishino, T. Harada, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., 2006, 118, 7221–7224 CrossRef; (c) S. N. Sidorov, I. V. Volkov, V. A. Davankov, M. P. Tsyurupa, P. M. Valesky, L. M. Bronstein, R. Karlinsey, J. W. Zwanziger, V. G. Matveeva, E. M. Sulman, N. V. Lakina, E. A. Wilder and R. J. Spontak, J. Am. Chem. Soc., 2001, 123, 10502–10510 CrossRef CAS PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117(12), 7762–7810 CrossRef CAS PubMed.
- J. Y. Shang, F. Li, X. F. Bai, J. X. Jiang, K. F. Yang, G. Q. Lai and L. W. Xu, Eur. J. Org. Chem., 2012, 2809–2815 CrossRef CAS.
- (a) E. Nakamura, M. Shimizu, I. Kuwajima, J. Sakata, K. Yokoyama and R. Noyori, J. Org. Chem., 1983, 48, 932–945 CrossRef CAS; (b) J. Enda and I. Kuwajima, J. Am. Chem. Soc., 1985, 107, 5495–5501 CrossRef CAS; (c) D. A. Evans, T. Rovis, M. C. Kozlowski, C. W. Downey and J. S. Tedrow, J. Am. Chem. Soc., 2000, 122, 9134–9142 CrossRef CAS; (d) M. Yasuda, K. Chiba, N. Ohigashi, Y. Katoh and A. Baba, J. Am. Chem. Soc., 2003, 125, 7291–7300 CrossRef CAS PubMed; (e) T. Wedel and J. Podlech, Org. Lett., 2005, 7, 4013–4015 CrossRef CAS PubMed; (f) N. Takenaka, J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 742–743 CrossRef CAS PubMed; (g) T. E. Reynolds, M. S. Binkley and K. A. Scheidt, Org. Lett., 2008, 10, 2449–2452 CrossRef CAS PubMed.
- (a) K. W. Lin, C. Y. Chen, W. F. Chen and T. H. Yan, J. Org. Chem., 2008, 73, 4759–4761 CrossRef CAS PubMed; (b) G. X. Cai, J. Wen, T. T. Lai, D. Xie and C. H. Zhou, Org. Biomol. Chem., 2016, 14, 2390–2394 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10541j |
| ‡ N. Li and X. Y. Dong contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |