
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu3Pt1–Cu2O nanocomposites: synergistic effect-dependent high activity and stability for the gas-phase selective oxidation of alcohols†
Kun Liu *a,
Houkun Longb,
Guangyi Wangb,
Yongbin Suna,
Chao Houa,
Jian Dong*a and
Xiaoqun Caoa
*a,
Houkun Longb,
Guangyi Wangb,
Yongbin Suna,
Chao Houa,
Jian Dong*a and
Xiaoqun Caoa
aSchool of Chemistry and Pharmaceutical Engineering, Taishan Medical College, Taian 271016, China. E-mail: liukun2436@126.com; dongjian@tsmc.edu.cn
bYan Kuang Lu Nan Chemicals Co. Ltd., China
First published on 1st December 2017
Abstract
The catalyst Cu3Pt1–Cu2O/SiC was facilely prepared via the in situ reaction of the corresponding compounds supported on SiC in the reaction stream. Cu3Pt1–Cu2O/SiC exhibits excellent catalytic activity for the oxidation of alcohols (conversion of benzyl alcohol and selectivity of benzyl aldehyde are 93% and 98% respectively). The reduction of active Cu2O to inactive Cu0 is the cause behind the deactivation of Cu/SiC. For Cu3Pt1–Cu2O-7/SiC, a Cu2O–Cu3Pt1 alloy formed under the reaction conditions plays an important role in the reaction. Active 5 nm Cu2O nanoparticles are stabilized by the inactive Cu3Pt1 alloy, which was confirmed by control experiments, characterization results and a three-step experiment.
Introduction
The oxidation of alcohols to aldehydes or ketones is very important in industrial processes and organic synthesis, and provides valuable intermediates and important fine chemicals.1–3 Traditionally, stoichiometric oxidants such as dichromate and permanganate are employed in this process, but they are toxic and expensive. The catalytic efficiency of the oxidation reaction of alcohols in liquid phase that adopt H2O2 or TBHP (tert-butyl hydroperoxide) as the oxidant is very low. For example, the preparation process of AuCu/Al2O3 or Au/Al2O3 is tedious and the low efficiency limits its applications.4,5 There needs to be an urgent fundamental shift from methods based on toxic and expensive inorganic oxidants to greener and more atom-efficient methods that adopt recyclable catalysts and molecular oxygen as the oxidant.4–7Recently, the selective oxidation of alcohols in gas phase using molecular oxygen as the oxidant has been widely investigated due to its high efficiency and easy separation process. Cu is widely used in alcohol oxidation reactions compared with other metal-based catalysts (such as Au, Pd and Rh), due to its high activity, selectivity and simple preparation method, but still faces many problems.4 AuCu/SiO2 is active in the liquid phase oxidation of alcohols, but a calcination process is necessary in order to re-form its active site, Au–Cu2O, after use.8 Conversion of benzyl alcohol is higher than 98% when using K–Cu/TiO2 as a catalyst, but the weight hourly space velocity (WHSV) used is very low and deactivation is inevitable within 50 h due to the temperature increase effect in the catalyst bed.9 The microstructured Au/Cu-fiber catalyst with excellent heat conductivity developed by Zhao et al. is highly active in alcohol oxidation reactions, but the high content of Au and the tedious preparation process for the Cu fiber impede its industrial applications.10,11 AuCuMgCrO4 exhibits excellent catalytic activity in the ethanol oxidation reaction due to the synergistic effect between Au and Cu2O, but the tedious preparation process of spinel hinders its applications.12 CoO@Cu2O developed by Zhao et al. exhibits excellent catalytic activity in the ethanol oxidation reaction; however, the low WHSV used (10 h−1) is not beneficial for industrial applications.13
The exothermic effect is a prominent problem in alcohol oxidation, which is adverse in industrial applications. Therefore, we should choose other thermally-conductive supports to prepare catalysts. In our previous report, the CuPd–Cu2O/Ti-powder catalyst exhibits high activity and selectivity in alcohol oxidation reactions and a synergic effect between the CuPd alloy and Cu2O was found.7 However, the conversion of alcohol is lower than 90% and the Pd content is higher than 3%, which is unfavorable for industrial applications. The Ag–Cu2O/SiC catalyst exhibits excellent catalytic activity in alcohol oxidation; however, deactivation is inevitable and the WHSV employed is much lower.15 As a continuation of our previous research, the strategy of stabilizing Cu2O using Pt delivers the catalyst CuPt–Cu2O/SiC, which shows high activity and stability. SiC powder was chosen as the support because of its excellent heat conductivity, low price and high oxidation/acid corrosion resistance behavior.14–16 Control-experiments and characterization results indicate that a synergistic effect between the CuPt alloy and Cu2O contributes to the activity and stability.
Experimental
Materials
SiC powder (200–300 mesh with a surface area of 0.15 m2 g−1), H2PtCl6·6H2O and Cu(NO3)2·3H2O were purchased from Sinopharm Chemical Reagent Co. Ltd without further treatment. Other substrates, such as 1-phenylethanol and 2-phenylethanol, were supplied by Aladdin Reagent Co. Ltd.Preparation of the catalysts
All catalysts were prepared using a simple impregnation and calcination method. If not specifically pointed out, the metal weight is 10%.The CuPt–Cu2O-7/SiC catalyst (metal loading of 10% and Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt molar ratio of 7:1) was prepared by impregnation of 1 g SiC powder, 266 mg Cu(NO3)2·3H2O, 80 mg H2PtCl6·6H2O and 10 mL H2O, followed by drying at 100 °C for 10 h and calcination at 300 °C in air for 1 h, and is denoted as CuPt–Cu2O-7/SiC fresh. Additionally, other catalysts such as CuPt–Cu2O-x (x indicates the molecular ratio of Cu:Pt), Cu/SiC and Pt/SiC were prepared following the same procedure, by simply tuning the amount of H2PtCl6·6H2O and copper salts in the corresponding solutions. CuO/SiC was prepared by impregnation of 1 g SiC powder and 400 mg Cu(NO3)2·3H2O, followed by drying at 100 °C for 10 h and calcination at 500 °C in air for 5 h.
:Pt molar ratio of 7:1) was prepared by impregnation of 1 g SiC powder, 266 mg Cu(NO3)2·3H2O, 80 mg H2PtCl6·6H2O and 10 mL H2O, followed by drying at 100 °C for 10 h and calcination at 300 °C in air for 1 h, and is denoted as CuPt–Cu2O-7/SiC fresh. Additionally, other catalysts such as CuPt–Cu2O-x (x indicates the molecular ratio of Cu:Pt), Cu/SiC and Pt/SiC were prepared following the same procedure, by simply tuning the amount of H2PtCl6·6H2O and copper salts in the corresponding solutions. CuO/SiC was prepared by impregnation of 1 g SiC powder and 400 mg Cu(NO3)2·3H2O, followed by drying at 100 °C for 10 h and calcination at 500 °C in air for 5 h.
Catalytic tests
The gas-phase selective oxidation process was carried out on a fixed-bed reactor (inner diameter 10 mm) under atmospheric pressure.7 The amount of catalyst used in each experiment was 0.3 g (200–300 mesh). Alcohols were fed continuously by a peristaltic pump, in parallel with the addition of O2 (oxidant) and N2 (diluted gas) using calibrated mass flow controllers, into the reactor, which was heated to the desired reaction temperature. The WHSV of alcohol was set to 20 h−1 and the molecular ratio of alcohol:O2:N2 was 1:0.6:2.4. The products were analyzed by Gas Chromatography (SP-7820 with TCD detector) and GC-MS (Agilent 6890 equipped with a HP-5 column). The content of negligible benzoic acid, toluene and COx was 0.8%, 0.2% and 0.3% respectively, so the selectivity of benzylaldehyde in our reaction conditions was higher than 98% and the carbon balance in the liquid phase was 99%.
In order to prevent the oxidation of our catalyst after the reaction, the used catalysts were collected after they had totally cooled down to room temperature under the protection of N2. After running for 60 h and 120 h, the deposited coke was removed by calcination of the catalysts in O2 at 400 °C for 2 h (regeneration process).
Characterization of the catalyst
X-ray diffraction patterns (XRD) of the catalysts were obtained on a Bruker D8 diffractometer with Cu-Kα radiation. Transmission electron microscopy (TEM) was performed on a JEOL 2100 F instrument operating at 30 kV. Inductively-coupled plasma atomic emission spectroscopy (ICP-AES) measurements were performed on a Thermo Scientific iCAP 6300 instrument. X-ray photoelectron spectroscopy (XPS) experiments were performed on a PHI-5500 spectrometer with Al Kα X-ray radiation as the X-ray source for excitation. Thermal gravimetric analysis (TGA) experiments were performed on Shimadzu TGA-50 equipment. H2 temperature programmed reduction (TPR) of the catalysts was performed on a Xianquan TP-5800 instrument.Results and discussion
Characterization of fresh catalysts
The characterization results of the fresh catalysts are illustrated in Fig. 1. For Cu/SiC fresh and CuPt–Cu2O-7/SiC fresh, the characteristic peaks of CuO (2θ = 38.7°) are found in the XRD patterns (Fig. 1A). The Cu 2p spectra show that peaks at 934.4 eV and 934.5 eV are assigned to CuO species (Fig. 1B).7,10 The Pt 4f spectrum indicates that the transformation of H2PtCl6 to metal Pt0 has occurred in the calcination process for CuPt–Cu2O-7/SiC fresh (Fig. 1C) and the corresponding diffraction peaks of Pt are not found in the XRD pattern due to its low content (Fig. 1A).17 The XRD results indicate that the characteristic peaks of Pt0 (2θ = 39.6° and 46.4°) are clearly observed in the Pt/SiC fresh catalyst (Fig. 1A). TEM indicates that the particle size of Cu/SiC fresh is 6 nm and the particles are distributed randomly on the support (Fig. S1A in ESI†).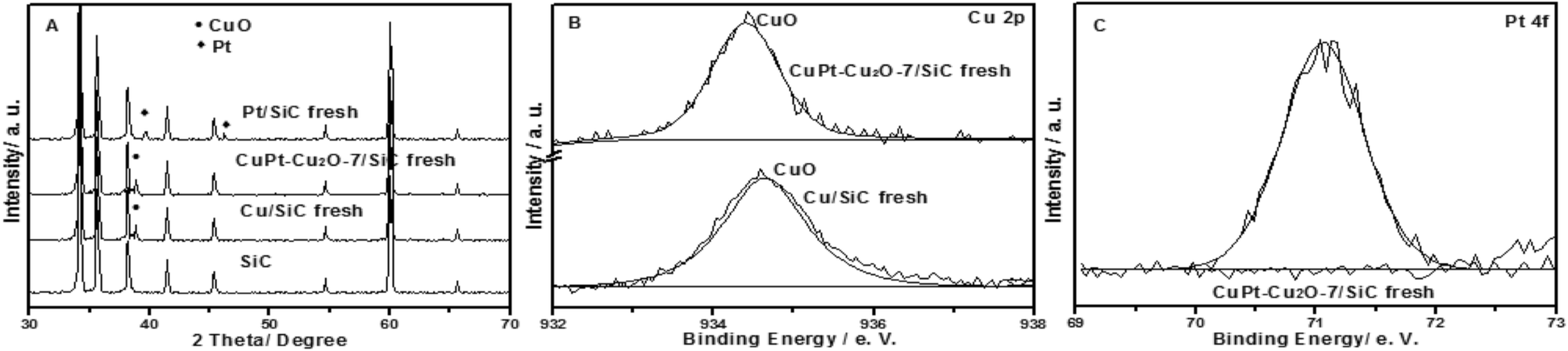 | ||
| Fig. 1 XRD and XPS spectra of the fresh catalyst: XRD patterns of SiC, Cu/SiC fresh, CuPt–Cu2O-7/SiC fresh and Pt/SiC fresh (A); Cu 2p of Cu/SiC fresh and CuPt–Cu2O-7/SiC fresh (B); Pt 4f of CuPt–Cu2O-7/SiC fresh (C). | ||
Catalytic activity
The catalytic activity of pure SiC powder (200–300 mesh) with excellent heat conductivity and corrosion resistance is rather low (Table 1, entry 1), which is similar to a Ti support.7 With an increase in the Cu loading from 5% to 10%, the conversion of benzyl alcohol increases from 65% to 85% (Table 1, entries 2 and 3). Given the excellent catalytic activity of Cu/SiC with 10% metal loading, we further investigated its stability. The conversion of benzyl alcohol decreases after just 6 hours (Fig. 2), which is in accordance with our previous report.7 When the metal loading is 10 wt% with a Cu:Pt molecular ratio of 1:1, the conversion of benzyl alcohol is 7% and the selectivity of benzyl aldehyde is 99%. The conversion of benzyl alcohol increases to 93% when the molar ratio increases to 7:1 (Table 1, entries 4–7).
:O2:benzyl alcohol = 2.4:0.6:1)
| Entry | Catalysts | Weight of Cuc (%) | Weight of Ptc (%) | Conv.d (%) | Sel.e (%) |
|---|---|---|---|---|---|
| a Metal content is 5%.b Catalyst is reduced with H2 at 300 °C for 3 h.c Weight ratio is detected by ICP.d Conversion of benzyl alcohol.e Selectivity of benzyl aldehyde, by-products are benzoic acid with trace amount of toluene and COx. | |||||
| 1 | SiC | 0 | 0 | 5 | 98 |
| 2a | Cu/SiC | 4.8 | 0 | 65 | 97 |
| 3 | Cu/SiC | 9.3 | 0 | 85 | 98 |
| 4 | CuPt–Cu2O-1/SiC | 2.4 | 7.5 | 7 | 99 |
| 5 | CuPt–Cu2O-3/SiC | 5.3 | 4.6 | 13 | 98 |
| 6 | CuPt–Cu2O-5/SiC | 6.2 | 3.8 | 78 | 99 |
| 7 | CuPt–Cu2O-7/SiC | 6.6 | 3.3 | 93 | 98 |
| 8 | CuPt–Cu2O-9/SiC | 7.5 | 2.4 | 92 | 99 |
| 9 | Pt/SiC | 0 | 9.7 | 13 | 98 |
| 10 (ref. 5) | Au/Cu-fiber | — | 0 | 85 | 98 |
| 11 (ref. 4) | K–Cu–TiO2 | 2 | 0 | 99 | 98 |
| 12 (ref. 7) | CuPd–Cu2O-4/Ti powder | 8.9 | 0 | 89 | 97 |
| 13 | CuO/SiC | 9.1 | 0 | 8 | 96 |
| 14b | Cu/SiC | 9.3 | 0 | 5 | 96 |
| 15b | CuPt–Cu2O-7/SiC | 6.6 | 3.3 | 7 | 98 |
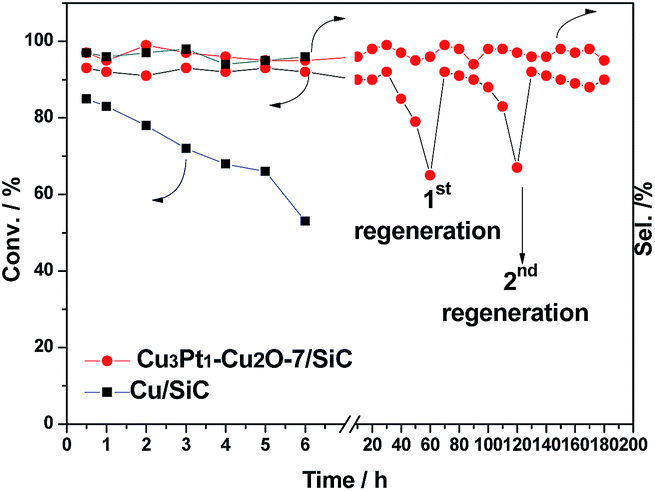 | ||
| Fig. 2 Catalytic activity of Cu/SiC and CuPt–Cu2O-7/SiC. | ||
The catalytic activity of CuPt–Cu2O-9/SiC decreases after just a 6 h run, though the initial activity is excellent compared with the aforementioned catalysts (Table 1, entry 8), while the catalytic activity of Pt/SiC is rather low (Table 1, entry 9), so our catalyst CuPt–Cu2O-7/SiC was selected as the optimal catalyst, given its fine activity/selectivity, excellent heat conductivity and low noble metal loading. Table 1 shows that our catalyst exhibits higher activity and selectivity than Au/Cu-fiber,10 K–Cu/TiO2,9 and CuPd–Cu2O-4/Ti-powder.7 It is notable that the noble metal loading is much lower, which is beneficial for industrial applications (Table 1, entries 10–12). For benzyl alcohol oxidation at 280 °C, CuPt–Cu2O-7/SiC delivers a single-run lifetime of 60 hours with excellent activity and selectivity, and is able to maintain its activity after calcination in air at 400 °C to burn away the deposited coke (4 wt% of carbon after first run, identified by TGA) (Fig. 2).
Given the high catalytic activity of CuPt–Cu2O-7/SiC, we extended the experiments by using various substrates, and the results show that the catalytic activity is influenced by the structure of the substrate (Table S1 in ESI†). A faster oxidation of aromatic alcohols to aldehydes or ketones is observed, compared to the oxidation of aliphatic alcohols. CuPt–Cu2O-7/SiC could oxidize 1-phenylethanol to the corresponding aldehyde at a high conversion of 85% at 300 °C, but only 45% for 2-phenylethanol at 340 °C. The conversion of octanol is higher than 2-octanol, which is in accordance with our previous report.7 Interestingly, cyclohexanol and cyclopropyl carbinol are very reactive among the aliphatic alcohols. Cyclohexanol is selectively oxidized to cyclohexanone at a conversion of 83% with a selectivity of 93% at 340 °C. For cyclopropyl carbinol, a conversion of 87% is obtained at 280 °C with 94% selectivity to cyclopropyl aldehyde.
Identification of active Cu3Pt1–Cu2O species in CuPt–Cu2O-7/SiC
:Cu0 ratio decreases sharply from 87% to 30% (Table S2 in ESI†). Just like the CuPd–Cu2O/Ti-powder catalyst,7 we conclude that the reduction of Cu2O to Cu0 contributes to the deactivation of Cu/SiC and the support effect (Ti powder or SiC powder) is ruled out.
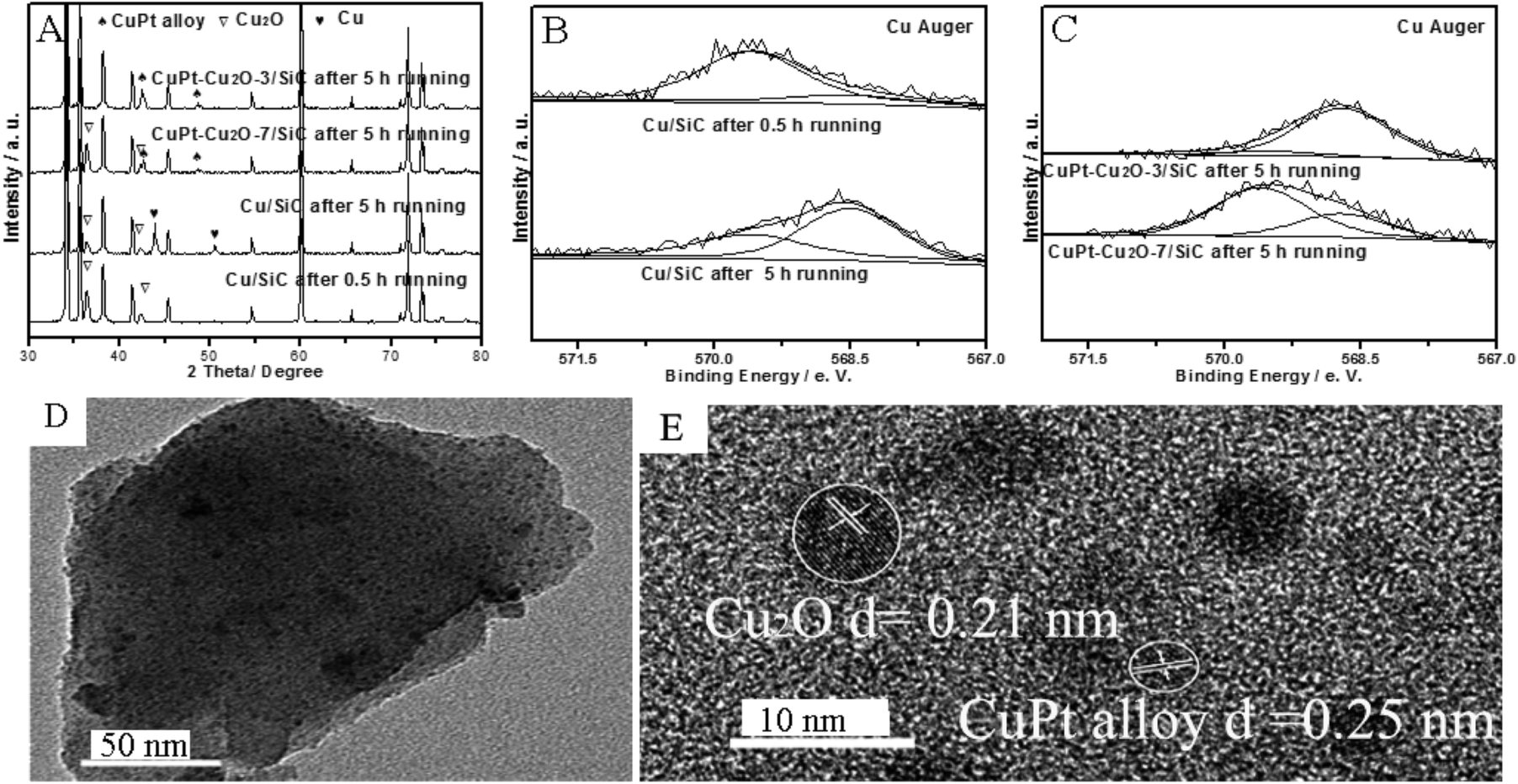 | ||
| Fig. 3 XRD, XPS and TEM images of used catalysts: XRD patterns of Cu/SiC after a 0.5 h run, Cu/SiC after a 5 h run, CuPt–Cu2O-7/SiC after a 5 h run and CuPt–Cu2O-3/SiC after a 5 h run (A). Cu Auger spectra of Cu/SiC after a 0.5 h run and Cu/SiC after a 5 h run (B). Cu Auger spectra of CuPt–Cu2O-7/SiC after a 5 h run and CuPt–Cu2O-3/SiC after a 5 h run (C). TEM and HRTEM images of CuPt–Cu2O-7/SiC after a 5 h run (D and E). | ||
The Cu 2p spectrum indicates that the corresponding peak of CuO (934.5 eV) is ambiguous, while the Cu Auger spectrum indicates that the new phases of Cu2O (569.5 eV) or Cu0 (568.6 eV) are formed in the reaction conditions for CuPt–Cu2O-7/SiC after a 5 h run (Fig. 3C and S1D in ESI†).10,12 The Pt 4f spectrum indicates that metallic Pt0 is formed in the reactions (Fig. S1E in ESI†).17 Apart from active Cu2O, Cu3Pt1 alloy is formed from metallic Cu0 and Pt0 due to the Cu0:Pt0 molecular ratio and the initial Cu:Pt molar ratio in the preparation process on the surface, which agrees with the XRD patterns (Fig. 3A and C and Table S2 in ESI†). TEM and HRTEM images indicate that the particles are not sintered and the lattice fringes of the Cu3Pt1 alloy and Cu2O are 0.25 nm and 0.21 nm respectively, which is in accordance with the XRD results (Fig. 3D and E).7,18 For the catalyst CuO/SiC, it seems that the catalytic activity is very low and the XRD pattern shows that Cu2O is not formed after a run of just 2 min, so CuO is not active in alcohol oxidation (Table 1, entry 13 and Fig. S1C in ESI†). For CuPt–Cu2O-3/SiC after a 5 h run, the XRD and XPS results indicate that Cu3Pt1 alloy is formed under the reaction conditions and the catalytic activity is extremely low (Fig. 3C and Table 1, entry 5). The catalytic activity of hydrogen reduced CuPt–Cu2O-7/SiC and Cu/SiC is very low (Table 1, entries 14 and 15). In order to further investigate the synergistic effect between Cu2O and CuPt alloy, we performed H2-TPR tests. The temperature of the Cu2O reduction peak for the Cu/SiC catalyst (360 °C) is lower than that of the CuPt–Cu2O/SiC catalyst (485 °C), so we conclude that Cu2O is active for alcohol oxidation (Fig. S2 in ESI†).
The Cu2O/SiC catalyst is stabilized by the inactive Cu3Pt1 alloy given to the excellent activity of CuPt–Cu2O-7/SiC after a 5 h run and the poor activity of CuPt–Cu2O-3/SiC after a 5 h run. We conclude that the higher electronegative properties of AuCu alloy,10,11 PdCu alloy,7 PtCu alloy, and Ag15 and Au12 nanoparticles prevent the reduction of Cu2O.
Identification of Cu3Pt1–Cu2O by three-step experiment
To further elucidate the catalytic essence of CuPt–Cu2O-7/SiC, surface sensitive XPS was used to illustrate the evolution behavior of our catalyst after undergoing a specifically designed three-step experiment.19 The reaction was carried out with an alcohol/O2/N2 mixture feedstock, and subsequently with alcohol alone by cutting-off O2 and then re-feeding O2. On CuPt–Cu2O-7/SiC after a 5 h run, apart from the Cu3Pt1 alloy, a large amount of Cu2O specimens are formed during the reaction (Fig. 3A and C). On sample (a) (CuPt–Cu2O-7/SiC after a 5 h run, undergoing the reaction by cutting-off O2 for 1 h), we can see that the reduction of Cu2O to Cu0 is inevitable and the Cu3Pt1 alloy is stable under the reaction conditions, and the conversion of alcohol is just 5% (Fig. 4A and B). On sample (b) (sample (a) undergoing reaction by re-feeding O2 for 1 h), the conversion of alcohol is about 90% (Fig. 4B). CuO is ruled out in the three-step experiment given to the disappearance of the corresponding peak of CuO at 934.4 eV in the Cu 2p spectrum (Fig. S1F in ESI†).7 The XRD results indicate that the Cu3Pt1 alloy and Cu0 are formed by cutting-off O2 for sample (a), and the active Cu3Pt1 alloy–Cu2O is formed again by re-feeding O2 (Fig. 4A), so the three-step experiment confirms that active Cu2O is crucial to the catalytic activity and the Cu3Pt1 alloy prevents the reduction of Cu2O to Cu0.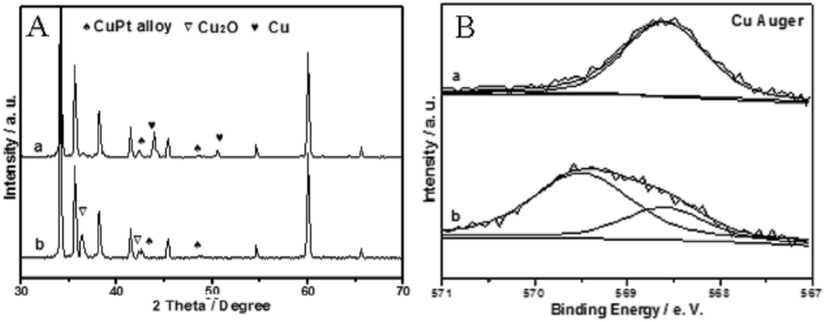 | ||
| Fig. 4 XRD and XPS results of the three-step experiment. XRD patterns of samples (a and b) (A). Cu Auger spectra of samples (a and b) (B) (a) CuPt–Cu2O-7/SiC undergoing reaction by cutting off O2 for 1 h, (b) sample (a) undergoing reaction by re-feeding O2 for 1 h. | ||
Conclusions
Cu3Pt1–Cu2O-7/SiC was successfully prepared using a simple impregnation method, and shows excellent activity and stability in alcohol oxidation reaction (conversion of benzyl alcohol is 93% and selectivity of aldehyde is 98%). Active Cu2O is formed under the reaction conditions from CuO and the reduction of Cu2O to Cu0 contributes to the deactivation of Cu/SiC. For the CuPt based catalyst, active Cu2O is stabilized by the inactive Cu3Pt1 alloy, which is crucial to the oxidation of substrates. The Cu3Pt1 alloy and Cu0 are formed by cutting-off O2 and the active Cu3Pt1 alloy–Cu2O is formed again by re-feeding O2, which was confirmed by XRD, XPS and a three-step experiment.Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. W. Li, J. L. Xu and Y. Ding, ChemCatChem, 2013, 5, 1705–1708 CrossRef CAS.
- A. Corma, S. Lborra and A. Velty, Chem. Rev., 2017, 107, 2411–2502 CrossRef PubMed.
- R. P. Unnikrishnan and S. D. Endalkachew, J. Catal., 2012, 211, 434–444 Search PubMed.
- M. J. Ndolomingo and R. Meijboom, Appl. Surf. Sci., 2017, 38, 19–32 CrossRef.
- G. F. Zhao, L. Min and Y. Lu, Green Chem., 2011, 13, 3088–3092 RSC.
- K. Liu, X. J. Yan and L. Y. Dai, Catal. Commun., 2015, 58, 132–136 CrossRef CAS.
- K. Liu, Z. X. Chen and L. Y. Dai, Catal. Commun., 2015, 67, 54–58 CrossRef CAS.
- W. J. Li, A. Q. Wang and T. Zhang, Appl. Catal., A, 2012, 433–434, 146–151 CrossRef CAS.
- J. Fan, Y. H. Dai and G. D. Stucky, J. Am. Chem. Soc., 2009, 131, 15568–15569 CrossRef CAS PubMed.
- G. F. Zhao, H. Y. Hu and Y. Lu, Green Chem., 2011, 13, 55–58 RSC.
- G. F. Zhao, H. Y. Hu and Y. Lu, ChemCatChem, 2011, 3, 1629–1636 CrossRef CAS.
- P. Liu and J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS PubMed.
- G. F. Zhao, S. Y. Fan and Y. Lu, ChemSusChem, 2017, 10, 1380–1384 CrossRef CAS PubMed.
- J. K. Wang, Y. Z. Zhang and S. W. Zhang, Powder Technol., 2017, 15, 209–215 CrossRef.
- L. Zhao, L. P. Kong and L. Y. Dai, Catal. Commun., 2017, 98, 1–4 CrossRef CAS.
- X. L. Fan, X. X. Qu and N. Batail, Catal. Today, 2016, 278, 350–360 CrossRef CAS.
- P. P. Zou, M. S. Wang and Y. Y. Wang, Appl. Organomet. Chem., 2016, 30, 722–725 CrossRef CAS.
- T. Komatsu and A. Tamura, J. Catal., 2008, 258, 306–314 CrossRef CAS.
- G. F. Zhao, H. Y. Hu and Y. Lu, Appl. Catal., A, 2013, 467, 171–177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 contains gas phase oxidation of other substrates by CuPt–Cu2O-7/SiC, Table S2 contains the surface atom contents calculated by XPS, Fig. S1 contains XRD, XPS and TEM results of the catalysts, Fig. S2 contains H2-TPR results of Cu/SiC and Cu3Pt1–Cu2O/SiC. See DOI: 10.1039/c7ra10511h |
| This journal is © The Royal Society of Chemistry 2017 |