
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mimicking horseradish peroxidase and oxidase using ruthenium nanomaterials†
Gao-Juan Caoab,
Xiumei Jiangb,
Hui Zhangb,
Timothy R. Croleyb and
Jun-Jie Yin *b
*b
aDepartment of Applied Chemistry, College of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China
bDivision of Analytical Chemistry, Office of Regulatory Science, Center for Food Safety and Applied Nutrition, U.S. Food and Drug Administration, College Park, Maryland 20740, USA. E-mail: junjie.yin@fda.hhs.gov
First published on 10th November 2017
Abstract
Although important progress has been achieved for the study of noble metal-based enzyme-like catalysts, there are rare reports on the enzyme mimicking applications of ruthenium nanoparticles (Ru NPs). In this work, we investigated the horseradish peroxidase (HRP) and oxidase mimetic activity of Ru NPs. Mimicking HRP, Ru NPs could catalyze the oxidation of substrates 3,3,5,5-tetramethylbenzidine (TMB), o-phenylenediamine (OPD) and dopamine hydrochloride (DA) in the presence of exogenously added H2O2 to generate the products with blue, yellow and orange colors, respectively. We also report the first evidence that Ru NPs possess intrinsic oxidase-like activity, which could catalyze the oxidization of TMB and sodium L-ascorbate (NaA) by dissolved oxygen. The HRP-like and oxidase-like activities of Ru NPs were found to be related to the concentrations of Ru NPs. The catalytic mechanism was analyzed by electron spin resonance spectroscopy (ESR), which suggested that the enzyme mimicking activities of the Ru NPs might originate from their characteristic of accelerating electron transfer between substrates and H2O2 or O2. Our findings offer a better understanding of enzyme-mimicking Ru NPs and should provide important insights for future applications.
Introduction
Natural enzymes, which have efficient catalytic activity and substrate specificity under mild conditions, are the true biocatalysts mediating each biological process in living organisms. They are able to accelerate chemical reactions up to 1019 times for specific substrates and reactions and have been commercially used in various areas such as sewage treatment, textile finishing, household product preparation, and energy conversion.1–5 However, the widespread practical applications of natural enzymes have been severely hampered by their intrinsic drawbacks, i.e., low stability, high costs in preparation, difficulty in purification, and high sensitivity of catalytic activity to environments. Therefore, a lot of efforts have been made to extend the natural enzymes to enzyme mimetics.6–8As a promising candidate for artificial enzymes, nanomaterials with intrinsic enzyme-like activity known as nanozymes ignite intensive research interest due to their ability to replace specific enzymes in enzyme-based applications.9–13 The design and development of novel nanostructured materials mimicking the catalytic role of natural enzymes has emerged as a promising field for both fundamental and applied research. Since the discovery by Yan and coworkers that Fe3O4 nanoparticles possess intrinsic peroxidase-like activity and show potential to replace natural peroxidase in bioassays,14 substantial research is directed toward the development of nanomaterial catalysts that mimic functions of peroxidase, oxidase or other natural enzymes with the advantages of low cost, controlled synthesis, tunable catalytic activities, and high stability even under severe reaction conditions. Till now, a variety of nanoscale materials have been developed as the efficient enzyme alternatives15–29 and their diverse applications cover from sensing, imaging, and therapeutics, to pollutant removal, water treatment and beyond.30–32 Among these, much work is centered on the noble metal-based nanozymes, particularly gold, platinum, palladium, and iridium, because of their well-developed synthesis techniques, easy modification of surface, excellent catalytic activity, good bio-compatibility and so on.33–48 For instance, gold nanoparticles with different surface modifications or their combined nanomaterials have been reported to demonstrate different enzyme-like activities. Platinum nanostructures are outstanding catalysts and have been found to possess diverse functional activities similar to peroxidase, catalase, superoxide dismutase, polyphenol oxidase, ascorbate oxidase and ferroxidase.
Compared with other noble metal-based enzyme mimics, there is a large space for us to explore the potential of Ru NPs as nanozymes, which is limited by the relatively less research activities. Recently, Ru nanoframes are found to show peroxidase-like properties for the first time.49 Very recently, our group reported that Ru NPs have catalase-like and superoxide dismutase-like activities by scavenging hydrogen peroxide and superoxide, which may play important roles in maintaining redox balance in living organisms by scavenging excess reactive oxygen species.50 In this work, we discovered that Ru NPs possess intrinsic HRP-like and oxidase-like mimetic activities. The Ru NPs could catalyze the oxidation of the substrates 3,3,5,5-tetramethylbenzidine (TMB), o-phenylenediamine (OPD) and dopamine hydrochloride (DA) by H2O2 to produce the color products in aqueous solution. More interestingly, we found that Ru NPs could catalyze the oxidation of the colorless substrate TMB to the blue-colored product in the absence of H2O2. This study also demonstrated that the Ru NPs mimicking ascorbic acid oxidase (AAO) can oxidize NaA under ambient condition, as measured by using both UV-vis and ESR spectroscopy. It was speculated that the nature of enzyme mimicking activities of Ru NPs were attributed to their ability of facilitating electron transfer between substrates and H2O2 or O2.
Experimental
Chemicals and materials
All chemicals were from commercial suppliers without further purification unless otherwise mentioned. Ruthenium nanopowder (Ru NPs, 20–30 nm) was purchased from US Research Nanomaterials Inc. (Houston, TX). Hydrogen peroxide (H2O2, 30%), horseradish peroxidase (HRP), ascorbate oxidase (AAO), 3,3,5,5-tetramethylbenzidinedihydrochloride (TMB·2HCl), o-phenylenediamine (OPD), dopamine hydrochloride (DA), sodium L-ascorbate (NaA), 3-carbamoyl-2,5-dihydro-2,2,5,5-tetramethyl-1H-pyrrol-1-yloxyl (CTPO) were all purchased from Sigma-Aldrich (St. Louis, MO). 5-tert-Butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) was purchased from Bioanalytical Labs (Sarasota, FL). 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was obtained from Dojindo Molecular Technologies, Inc. (Rockville, MD). PBS 7.4 buffer, 1-hydroxy-3-carboxy-2,2,5,5-tetramethylpyrrolidine hydrochloride (CPH) were obtained from Enzo Life Sciences (Farmingdale, NY). The concentration of buffer stock solution (pH 1.12 HCl–KCl, pH 3.2 HAc–NaAc, pH 10.96 KOH–KCl) was 0.1 M.Characterization
The hydrodynamic size (dynamic light scattering, DLS) and surface charge (zeta potential, mV) were detected using a ZetaSizer Nano series Nano-ZS (Malvern, UK). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were captured on a Hitachi SU-70 SEM (Hitachi, USA) and a Jeol JEM 2100 TEM (Jeol, USA), respectively. The dispersity of Ru NPs is not good but Ru NPs are commercially used (see Table S1 and images in Fig. S1 and S2†). UV-vis absorption spectra were obtained using a Varian Cary 300 spectrophotometer (Santa Clara, CA) (Fig. 1–5 and S3†). Electron spin resonance (ESR) measurements were carried out using a Bruker EMX ESR spectrometer (Billerica, MA) (Fig. 6, 7 and S7†).TMB oxidation studies
In order to investigate the enzyme-like activities of Ru NPs, the absorbance variation of the reaction solution was monitored in a time-scan mode at 652 nm. Similar to HRP and oxidase, Ru NPs can catalyze the oxidation of substrate TMB in the presence or absence of H2O2 to produce a blue-colored product with maximum absorbance at 652 nm (ε = 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). The HRP-like activity of Ru NPs was studied by catalyzing the oxidation of TMB in the presence of H2O2. For a typical oxidation reaction, the time-dependent absorbance of the solution of 0.1 mM TMB and 0.1 mM H2O2 with 1 μg mL−1 HRP or variable concentrations of Ru NPs (2.5, 5, 10, 15, 20 μg mL−1) was recorded for 15 min. The control experiments were carried out in the absence of Ru NPs. The steady-state kinetic assays were conducted at room temperature in a reaction solution with 10 μg mL−1 Ru NPs as catalyst in the presence of H2O2 and TMB. The kinetic assays of Ru NPs with TMB as the substrate were performed with 0.1 mM H2O2 and different amounts of TMB (0.05, 0.075, 0.1, 0.15 mM) aqueous solution. The kinetic assays of Ru NPs with H2O2 as the substrate were performed with 0.1 mM TMB and different amounts H2O2 solution (0.05, 0.1, 0.15, 0.2 mM). Reaction systems containing 0.1 mM TMB and Ru NPs at different concentrations (10, 25, 50, 75, 100 μg mL−1) were used to show the chromogenic reactions implying oxidase-like activity. The steady-state kinetic assays of 50 μg mL−1 Ru NPs with TMB (0.05, 0.1, 0.2, 0.5 mM) as the substrate were performed at room temperature. The kinetic parameters were calculated using Lineweaver–Burk plots of the double reciprocal of Michaelis–Menten equation: 1/v = (Km/Vmax) × {1/[S] + 1/Km}, where v is the initial velocity, Vmax is the maximal reaction velocity, [S] is the concentration of substrate and Km is the Michaelis constant.
000 M−1 cm−1). The HRP-like activity of Ru NPs was studied by catalyzing the oxidation of TMB in the presence of H2O2. For a typical oxidation reaction, the time-dependent absorbance of the solution of 0.1 mM TMB and 0.1 mM H2O2 with 1 μg mL−1 HRP or variable concentrations of Ru NPs (2.5, 5, 10, 15, 20 μg mL−1) was recorded for 15 min. The control experiments were carried out in the absence of Ru NPs. The steady-state kinetic assays were conducted at room temperature in a reaction solution with 10 μg mL−1 Ru NPs as catalyst in the presence of H2O2 and TMB. The kinetic assays of Ru NPs with TMB as the substrate were performed with 0.1 mM H2O2 and different amounts of TMB (0.05, 0.075, 0.1, 0.15 mM) aqueous solution. The kinetic assays of Ru NPs with H2O2 as the substrate were performed with 0.1 mM TMB and different amounts H2O2 solution (0.05, 0.1, 0.15, 0.2 mM). Reaction systems containing 0.1 mM TMB and Ru NPs at different concentrations (10, 25, 50, 75, 100 μg mL−1) were used to show the chromogenic reactions implying oxidase-like activity. The steady-state kinetic assays of 50 μg mL−1 Ru NPs with TMB (0.05, 0.1, 0.2, 0.5 mM) as the substrate were performed at room temperature. The kinetic parameters were calculated using Lineweaver–Burk plots of the double reciprocal of Michaelis–Menten equation: 1/v = (Km/Vmax) × {1/[S] + 1/Km}, where v is the initial velocity, Vmax is the maximal reaction velocity, [S] is the concentration of substrate and Km is the Michaelis constant.
OPD and DA oxidation studies
The HRP-like activity of Ru NPs was also investigated by the catalytic oxidation of the HRP substrate OPD in the presence of H2O2. The absorbance of the color reaction at 417 nm for OPD was recorded to express the HRP-like activity. For a typical oxidation reaction, the time-dependent absorbance of the solution of 0.2 mM OPD and 1 mM H2O2 with 1 μg mL−1 HRP or variable concentrations of Ru NPs (10, 25, 50, 75, 100 μg mL−1) was recorded during 20 min. Steady-state kinetics assays of Ru NPs toward OPD oxidation were carried out with varied concentrations of the substrate OPD or H2O2 at room temperature. The control experiments were carried out in the absence of Ru NPs. The kinetic assays of 75 μg mL−1 RuNPs with OPD as the substrate were performed with 1 mM H2O2 and different amounts of OPD aqueous solution (0.05, 0.1, 0.15, 0.2, 0.25, 0.3 mM). The kinetic assays of 25 μg mL−1 Ru NPs with H2O2 as substrate were performed with 0.2 mM OPD and different amounts H2O2 solution (1, 2, 3, 4, 6 mM).The Ru NPs were then applied as catalyst for the oxidation of DA to aminochrome (AC) by H2O2. The time-scan mode measurements were performed by monitoring the absorbance change of DA at 480 nm. In a typical experiment, 1 μg mL−1 HRP or various concentrations of Ru NPs (10, 25, 50, 75, 100 μg mL−1) were respectively reacted with 0.1 mM DA and 1 mM H2O2. The control experiments were carried out in the absence of Ru NPs. The kinetics behavior of Ru NPs was also evaluated by applying the same procedure described. The kinetic assays of 25 μg mL−1 Ru NPs with DA as the substrate were performed with 1 mM H2O2 and different amounts of DA (0.25, 0.5, 0.75, 1, 2 mM) in 10 mM PBS 7.4 buffer solution. The kinetic assays of 25 μg mL−1 Ru NPs with H2O2 as the substrate were performed with 0.1 mM DA and different amounts H2O2 solution (0.5, 1, 2, 3 mM).
NaA oxidation studies
The oxidation of NaA catalyzed by AAO (1 U mL−1) or Ru NPs (25, 50, 75, 100, 150 μg mL−1) in 10 mM PBS 7.4 were performed at room temperature. The absorbance variation of the reaction solution was monitored in a time-scan mode at λmax (265 nm, ε = 54200 M−1 cm−1). The apparent steady-state kinetic parameters were determined by varying the concentration of NaA (25, 50, 100, 150, 200 μM) in presence of 50 μg mL−1 Ru NPs.
ESR spectroscopic measurements
All ESR measurements were carried out using a Bruker EMX ESR spectrometer (Billerica, MA) at ambient temperature (27 °C). 50 μL of aliquots of control or sample solutions were taken in glass capillary tubes with internal diameters of 1 mm and sealed. The capillary tubes were inserted in the ESR cavity, and the spectra were recorded at selected times.The oxidation of NaA can also be monitored by ESR spectroscopy. The ascorbyl radical is an intermediate formed during the oxidation of NaA by molecular oxygen. ESR can directly detect this radical which has a 10 min half-life. Ru NPs at different concentrations were added to 5 mM NaA to start the reaction. The peak-to-peak value of the first line of ascorbyl radical ESR spectrum was recorded to indicate the amount of ascorbyl formed. Spectra were recorded under the following conditions: 20 mW microwave power, 1 G field modulation, and 25 G scan width.
The catalytic mechanism of Ru NPs was first evaluated in the H2O2/DMPO or H2O2/BMPO system in the presence and absence of Ru NPs. Samples containing 50 mM DMPO, 10 mM H2O2, and different concentrations of the Ru NPs (25, 50, 75, 100 μg mL−1) were prepared in pH 1.12, 3.2, 7.4 and 10.96 buffer, then transferred to a quartz capillary tube and placed in the ESR cavity for spectra recording. The H2O2/BMPO system was applied the same procedure but varying the BMPO concentration (25 mM). The instrument settings (20 mW microwave power, 1 G modulation amplitude, and 100 G scan range) were used for the present ESR experiment.
ESR spectra were recorded from the sample mixture, containing spin probe 200 μM CPH and Ru NPs at different concentration (25, 50, 75, 100 μg mL−1). The ESR measurements were carried out using the following settings: 20 mW microwave power, 100 G scan range, and 1 G field modulation.
Results and discussion
Noble metal nanoparticles were reported to act as catalysts mimicking the native enzyme. Currently, research is highly focused on mimicking peroxidases and oxidase. HRP, a widely recognized peroxidase, has been largely explored as a sensor for H2O2 detection, a tracer in enzyme-linked immunosorbent assay (ELISA), and for detection of some peroxidase inhibitors. Oxidases that can catalyze the oxidation of substrates by simply employing molecular oxygen as the electron acceptor are more facile, economic and environment friendly. In this study, the catalytic activity of Ru NPs and their ability of mimicking HRP were evaluated using the chromogenic substrates TMB, OPD and DA (Scheme 1a–c). We also report the first evidence that Ru NPs possess intrinsic oxidase-like activity, which could catalyze the oxidization of TMB and NaA by dissolved oxygen (Scheme 1a and d).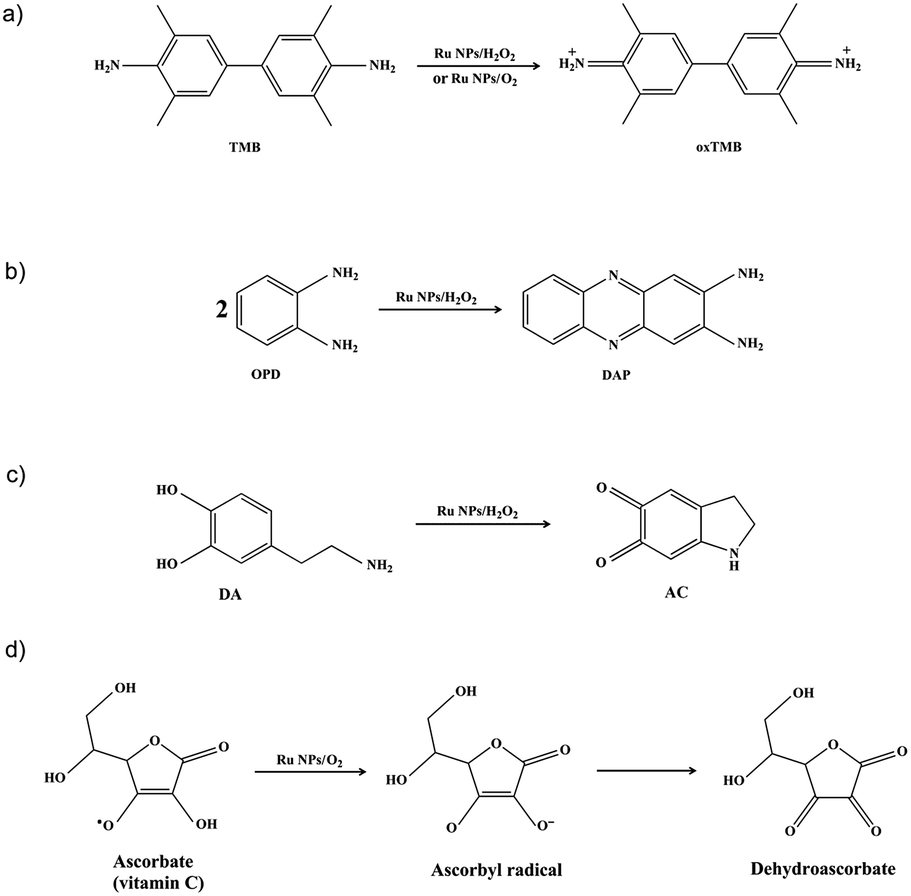 | ||
| Scheme 1 Ru NPs catalyzed oxidation of TMB (a), OPD (b), DA (c) and ascorbate (d). | ||
Enzyme-like activity of Ru NPs during oxidation of TMB
The Ru NPs were subjected to reaction conditions typical for HRP and oxidase. TMB is a chromophoric substrate commonly used in HRP mimetic studies and was firstly selected as a substrate to be oxidized in this study. The Ru NPs with varied concentrations can quickly catalyze the oxidation of TMB to produce a typical blue color either in the presence or absence of H2O2. Control reactions in the absence of Ru NPs were performed and showed negligible color changes over the same time period. The TMB cation free radical, a one-electron oxidation product, would be formed during the reaction procedure, which is responsible for the blue color (maximum absorbance at 652 nm), similar to the phenomena observed for the commonly used HRP enzyme. Time- and dose-dependent absorbance changes at 652 nm were measured as shown in Fig. 1a and b, 2a and b. The observed color changes also support UV-vis spectral analysis. The results showed that Ru NPs could catalyze the oxidation of TMB by H2O2 or dissolved O2 to produce a blue colored product, suggesting HRP-like and oxidase-like activity.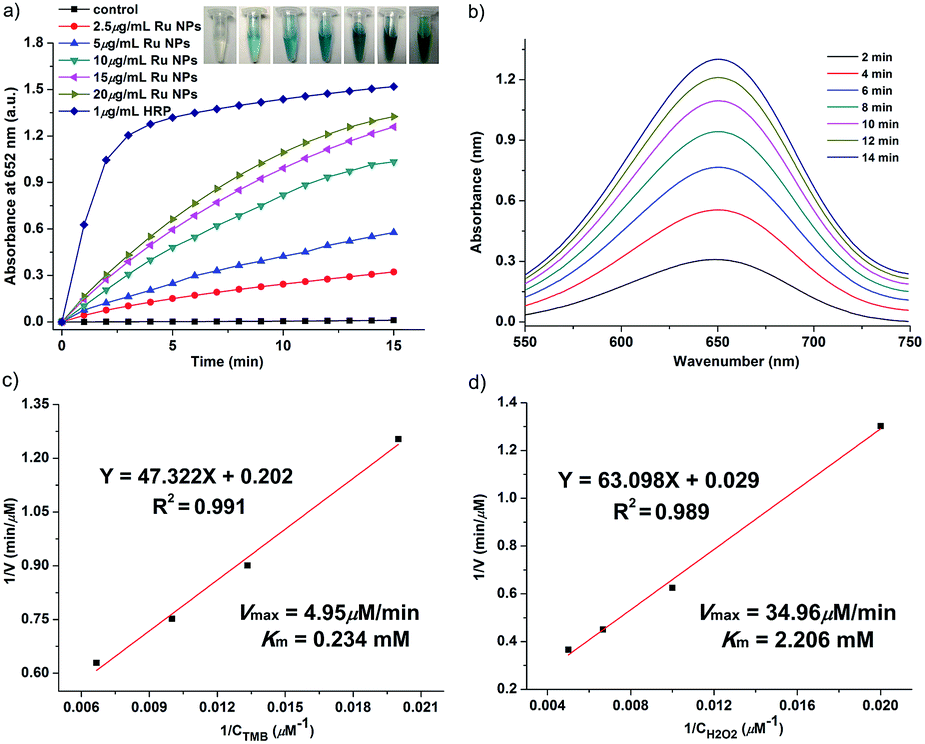 | ||
| Fig. 1 HRP-like activity of Ru NPs during oxidation of TMB. (a) Time-dependent absorbance changes upon oxidation of TMB to oxTMB by H2O2 using 1 μg mL−1 HRP or variable concentrations of Ru NPs in presence of 0.1 mM TMB and 0.1 mM H2O2. Insets are color evolution of TMB oxidation by 0.1 mM H2O2 and different concentrations of Ru NPs or 1 μg mL−1 HRP. (b) Time-dependent absorbance spectra of oxTMB generated upon the oxidation of 0.1 mM TMB in presence of 20 μg mL−1 Ru NPs and 0.1 mM H2O2. (c) Lineweaver–Burk plot of the activity in the presence of 10 μg mL−1 Ru NPs, 0.1 mM H2O2 and different concentrations of TMB. (d) Lineweaver–Burk plot of the activity in the presence of 10 μg mL−1 Ru NPs, 0.1 mM TMB and different concentrations of H2O2. | ||
 | ||
| Fig. 2 Oxidase-like activity of Ru NPs during oxidation of TMB. (a) Time-dependent absorbance changes upon oxidation of TMB using variable concentrations of Ru NPs in presence of 0.1 mM TMB. Insets are color evolution of TMB oxidation by different concentrations of Ru NPs. (b) Time-dependent absorbance spectra of oxTMB generated upon the oxidation of 0.1 mM TMB in presence of 100 μg mL−1 Ru NPs. (c) Time-dependent absorbance changes corresponding to the 50 μg mL−1 Ru NPs-catalyzed oxidation of TMB using variable concentrations of TMB. (d) Lineweaver–Burk plot of the activity in the presence of 50 μg mL−1 Ru NPs and different concentrations of TMB. | ||
In order to regulate enzyme activity and understand the relationship between nanomaterials and enzyme kinetic parameters, the Michaelis–Menten behaviors of Ru NPs were studied with H2O2 and TMB as substrates (Fig. 1c, d, 2c, d, S4 and S5†). A series of experiments were performed by changing the concentration of one substrate and keeping the other constant. The reaction rates of TMB oxidation both in the presence and absence of H2O2 were calculated. Typical Michaelis–Menten curves were received in a certain concentration range of TMB and H2O2 (Fig. S4 and S5†). Lineweaver–Burk plots of 1/v vs. Vmax were constructed and fitted to the Michaelis–Menten equation to calculate the Michaelis constant Km and the maximal reaction velocity Vmax. From the Lineweaver–Burk plots, the kinetic parameters Vmax and Km were obtained and shown in Table 1, Fig. 1d and 2d. In natural enzyme, Km is an indicator of affinity between the enzyme and substrate where a lower Km value indicates a higher affinity and catalytic activity. In the case of the nanostructure enzyme mimetics, the Km value is often used to compare the enzyme-like performance of the nanoparticles. The Km value of Ru NPs with TMB as the substrate is higher than that of Ru frames,49 Pd nanoplates42 and Fe3O4 NPs.14 The Km value of Ru NPs with H2O2 as the substrate is much lower than that of Ru frames and Fe3O4 NPs, and comparable with Pd nanoplates. These results indicated that Ru NPs had a low binding affinity towards TMB, and high binding affinity towards H2O2. It is also obvious that the oxidase-like activity of Ru NPs was much lower than its HRP-like activity.
| Catalyst | Substrate | Km (mM) | Vmax (μM min−1) | Reference |
|---|---|---|---|---|
| Ru NPs | TMB | 0.234 | 4.95 | This work |
| H2O2 | 2.206 | 34.96 | This work | |
| Ru frames | TMB | 0.0603 | 8.04 | 49 |
| H2O2 | 318 | 4.45 | ||
| Pd nanoplates | TMB | 0.1098 | 3.492 | 42 |
| H2O2 | 4.398 | 3.906 | ||
| Fe3O4 NPs | TMB | 0.098 | 2.064 | 14 |
| H2O2 | 154 | 5.868 | ||
| Ru NPs | TMB | 54.92 | 0.57 | This work |
| Pt NPs | TMB | 0.6 | 16.2 | 38 |
| Ru NPs | NaA | 0.155 | 2.331 | This work |
| Pt NPs (30 nm) | NaA | 0.022 | 12 | 40 |
Enzyme-like activity of Ru NPs during oxidation of OPD and DA
Typically, HRP as the catalyst via H2O2 for the oxidation of OPD to 2,3-diaminophenazine (DAP) has been successfully applied for more than a century.51,52 In the presence of HRP and H2O2, OPD is oxidized to DAP by H2O2, resulting in a yellow solution. The oxidation of chromogenic OPD has been employed for evaluating the HRP-like activity of Ru NPs, which is also investigated by UV-vis spectroscopy (Fig. 3). Similar to the enzymatic peroxidase activity observed from the HRP, OPD can be oxidized to a yellow reaction product in the presence of H2O2 upon addition of Ru NPs catalysts. Moreover, the color deepened with increasing reaction time and the concentration of Ru NPs. This process was also confirmed by absorption spectra. The catalytic reaction can be monitored by following the changing of absorbance at 417 nm, which originates from the oxidation products DAP. Fig. 3a and b presented the time course curves of the absorbance at 417 nm within 20 min. The reaction was also monitored while changing the substrate concentration of OPD or H2O2 and fixing the other two concentrations (Fig. 3c and d). | ||
| Fig. 3 HRP-like activity of Ru NPs during oxidation of OPD. (a) Time-dependent absorbance changes upon oxidation of OPD by H2O2 using 1 μg mL−1 HRP or variable concentrations of Ru NPs in presence of 0.2 mM OPD and 1 mM H2O2. Insets are color evolution of 0.2 mM OPD oxidation by 1 mM H2O2 and different concentrations of Ru NPs or 1 μg mL−1 HRP. (b) Time-dependent absorbance spectra generated upon the oxidation of 0.2 mM OPD in presence of 75 μg mL−1 Ru NPs and 1 mM H2O2. (c) Time-dependent absorbance changes corresponding to the 75 μg mL−1 Ru NPs-catalyzed oxidation of OPD using variable concentrations of OPD in presence of 1 mM H2O2. (d) Time-dependent absorbance changes corresponding to the 25 μg mL−1 Ru NPs-catalyzed oxidation of 0.2 mM OPD using variable concentrations of H2O2. | ||
Dopamine (DA) is a neurotransmitter in the catecholamine and phenethylamine families that plays a number of important roles in the brain and body of animals. HRP/H2O2 couple is commonly selected as a biomimetic oxidizing agent to examine the early stages of dopamine oxidation.53 The Ru NPs can catalyze the H2O2-driven oxidation of DA to aminochrome (AC), in analogy to HRP. The time-dependent absorbance change of AC, generated upon oxidation of dopamine, was shown in Fig. 4. Fig. 4a and b depicted the absorption spectra corresponding to AC generated within a fixed time interval using HRP or Ru NPs in the presence of DA and H2O2. There was an obvious increase of absorbance intensity at 480 nm with the increasing of the Ru NPs concentration, accompanying the progression of the orange color. With the increase of the concentration of Ru NPs, the oxidation reaction proceeded faster, resulted in the enhanced oxidation of dopamine. Fig. 4c illustrated the time-dependent absorbance changes of AC upon oxidation of dopamine using variable concentrations of dopamine and a fixed concentration of Ru NPs and H2O2. Evidently, the Ru NPs catalyzed oxidation of dopamine was controlled by the concentration of H2O2, and as its concentration increased, the rate of the oxidation of dopamine was enhanced (Fig. 4d).
 | ||
| Fig. 4 HRP-like activity of Ru NPs during oxidation of DA. (a) Time-dependent absorbance changes upon oxidation of DA by H2O2 using 1 μg mL−1 HRP or variable concentrations of Ru NPs in presence of 1 mM DA and 1 mM H2O2. Insets are color evolution of DA oxidation by 1 mM H2O2 and different concentrations of Ru NPs or 1 μg mL−1 HRP. (b) Time-dependent absorbance spectra generated upon the oxidation of 1 mM DA in presence of 100 μg mL−1 Ru NPs and 1 mM H2O2. (c) Time-dependent absorbance changes corresponding to the 50 μg mL−1 Ru NPs-catalyzed oxidation of DA using variable concentrations of OPD in presence of 1 mM H2O2. (d) Time-dependent absorbance changes corresponding to the 50 μg mL−1 Ru NPs-catalyzed oxidation of 0.2 mM DA using variable concentrations of H2O2. | ||
Enzyme-like activity of Ru NPs during oxidation of NaA
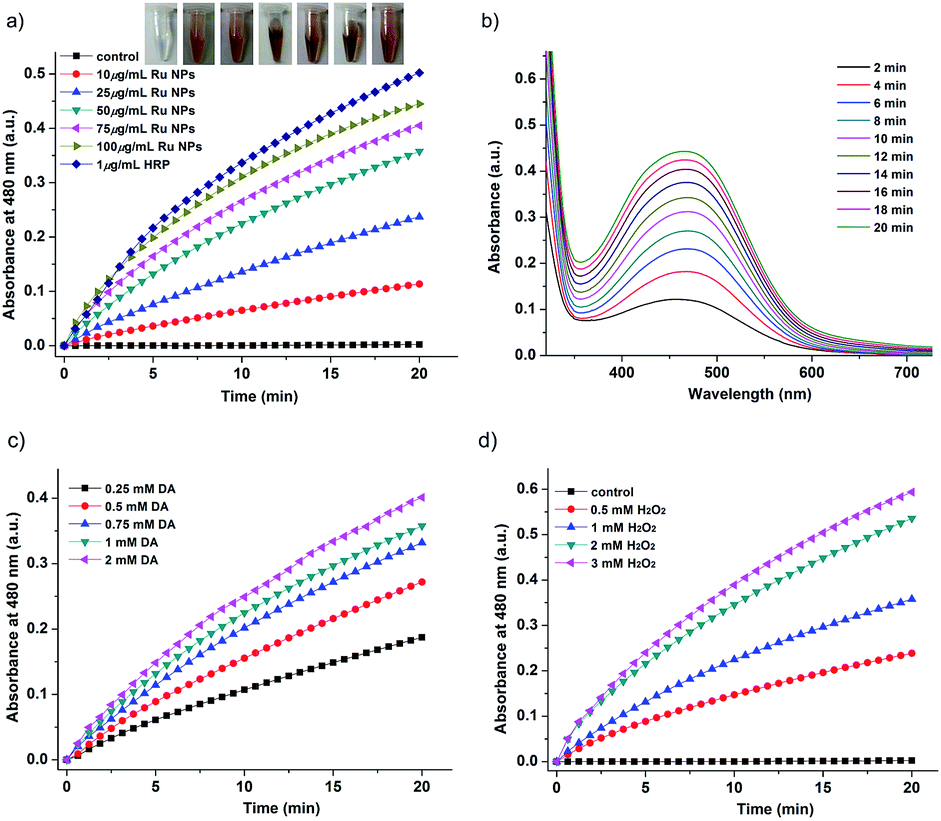
NaA is an essential nutrient and a well-known antioxidant that protects other important biological structures against oxidative damage by many oxidants. Numerous studies have shown the beneficial effects of NaA on human health.54–56 However, NaA can be oxidized very slowly by dioxygen to produce ascorbyl radical (AA˙). The presence of AAO greatly accelerated this process. Here, we investigated whether Ru NPs possess AAO-like enzyme-mimetic properties by oxidizing NaA (Fig. 5 and S6†). The oxidation of NaA to AA˙ is accompanied by a decrease in the characteristic absorption of NaA at 265 nm. Therefore, we used the absorbance change of NaA at 265 nm to monitor its oxidation. A typical set of experiment investigating Ru NPs with NaA was shown in Fig. 5a. No evident change at 265 nm was observed in Ru NPs-free reference solution during a 20 min monitoring. While mixing of NaA with Ru NPs (25, 50, 75, 100, 150 μg mL−1) resulted in decreased absorbance at 265 nm during time monitoring measurements (Fig. 5a and b). It is obvious that Ru NPs exhibited a significant ability to oxidize NaA in a time and dose- dependent manner. We determined the apparent steady-state kinetic parameters by varying the concentration of NaA in the presence of Ru NPs (Fig. 5c). The Km value of Ru NPs with NaA as a substrate was higher than that of AAO and Pt NPs (30 nm),40 suggesting that Ru NPs have a lower affinity than NaA (Fig. 5d). | ||
| Fig. 5 AAO-like activity of Ru NPs during oxidation of NaA. (a) Time-dependent absorbance changes upon oxidation of NaA using 1 U mL−1 AAO or variable concentrations of Ru NPs in presence of 100 μM NaA. (b) Time-dependent absorbance spectra generated upon the oxidation of 100 μM NaA in presence of 150 μg mL−1 Ru NPs. (c) Time-dependent amount of NaA remaining in different NaA dispersions containing 50 μg mL−1 Ru NPs. (d) Lineweaver–Burk plot of the activity in the presence of 50 μg mL−1 Ru NPs and different concentrations of NaA. | ||
We further demonstrated that Ru NPs can mimic the activity of AAO by using ESR spectroscopy, which is the most reliable and direct method for identification and quantification of short-lived free radicals. The intermediate AA˙ is an indicator for the oxidation of the active reducing agent NaA. AA˙ can be easily detected by ESR spectroscopy at room temperature without using any spin trap or spin label. The ESR spectra of AA˙ were shown in Fig. 6. There was no ESR signal in the pure NaA solution which indicates that NaA itself has not been oxidized within 3 min. However, significant ESR signals were detected when mixing NaA with different concentrations of Ru NPs or 1 U mL−1 AAO. The oxidation of NaA to AA˙ was dependent on the concentration of Ru NPs (Fig. 6a and b). At low NaA concentrations, the ESR signal rapidly increased. At high NaA concentrations, the ESR signal slowly increased (Fig. 6c). CTPO is a stable, water soluble nitroxide and has been widely used for ESR oximetry.57 The resolution of the super hyperfine structure of the low-field line of the ESR spectrum of CTPO depends on the oxygen concentration of the sample solution. Thus, CTPO is commonly used for detecting changes in the concentration of oxygen. As Fig. 6d showed, at the same incubation time, the ESR spectra of CTPO in the samples containing Ru NPs showed more super hyperfine splitting than the control sample, indicating more consumption of oxygen. A higher increase in super hyperfine splitting was observed for the sample containing high concentration of Ru NPs. The above results revealed that Ru NPs exhibit intrinsic AAO-like activity.
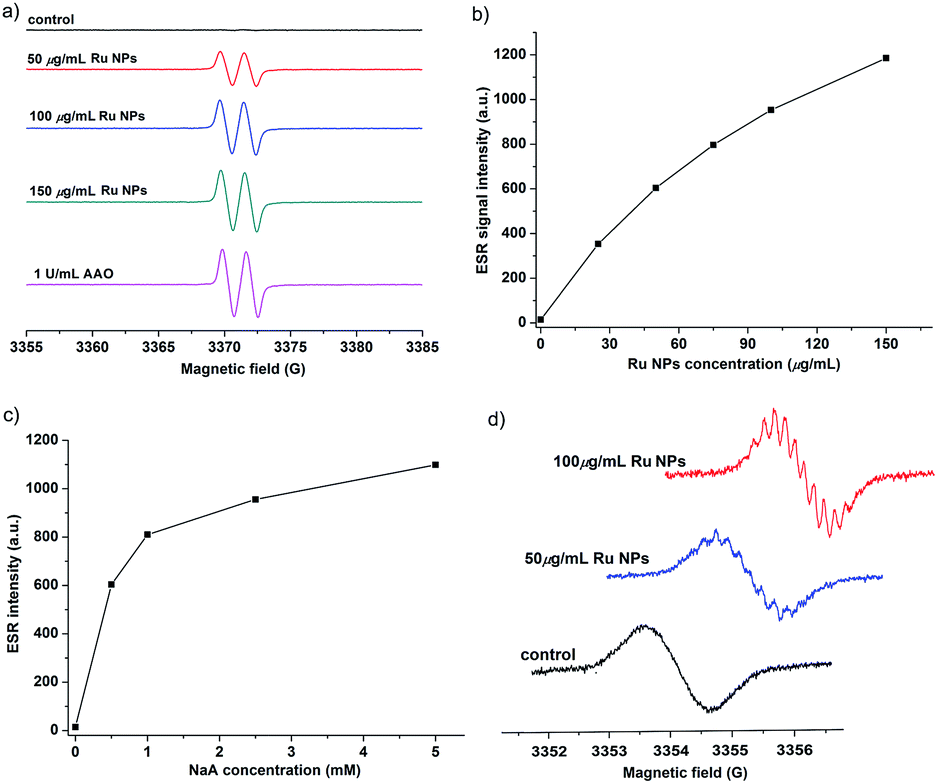 | ||
| Fig. 6 (a) ESR spectra upon oxidation of NaA using 1 U mL−1 AAO or variable concentrations of Ru NPs in presence of 5 mM NaA after 3 min incubation. (b) Effect of Ru NP concentration on NaA oxidation after incubation for 3 min. (c) Effect of NaA concentration on the oxidation in presence of 50 μg mL−1 Ru NPs after incubation for 3 min. (d) ESR spectra of samples containing 0.1 mM CTPO, 5 mM NaA, 10 mM PBS 7.4 and different concentrations of Ru NPs after mixing for 5 min. | ||
Catalytic mechanism of Ru NPs mimicking HRP and oxidase
The enzyme-like catalytic mechanisms of noble metal nanoparticles have been reported in related literature.41,42,48 Generation of hydroxyl radical and accelerating electron transfer process are two most possible mechanisms responsible for the enzyme mimic activities of Ru NPs (Scheme S1†). To test the hypothesis, we applied two models to measure the hydroxyl radical generation and electron transfer process. First, Ru/H2O2/DMPO and Ru/DMPO systems were applied to examine whether the enzyme mimicking property of Ru NPs was related to the generation of the hydroxyl radical, which could oxidize the substrate molecules. The catalytic mechanism of Ru NPs was evaluated in pH conditions of 1.12, 3.2, 7.4 and 10.96, respectively. The sample solution contains 5 mM DMPO, and different concentrations of Ru NPs (25, 50, 75, 100, 150 μg mL−1) in the absence or presence of 1 mM or 0.1 mM H2O2. All of these samples showed no ESR signal of DMPO–OH adduct (Fig. S7a†). The above results are further supported by ESR measurements using BMPO as a spin trap (Fig. S7b†). Thus, the catalytic behavior of Ru NPs can not be assumed to the ˙OH generation. It is very likely that Ru NPs act as an efficient electron transfer intermediate and facilitate the electron transfer between substrates and H2O2 or O2. Substrates were absorbed onto the surface of Ru NPs and donated electrons to Ru NPs, resulting in increased of electron density on Ru NPs. Then the electrons transferred from Ru NPs to H2O2 or O2 and facilitated the oxidation of substrates. This assumption is supported by ESR measurements using CPH as a spin probe, which is often used in ESR studies of electron transfer in biological system.58,59 CPH is an ESR silent probe, and can be oxidized to form CP-nitroxide radicals (CP˙) with a typical ESR spectrum of three lines with intensity ratios of 1:1:1. As shown in Fig. 7a, CPH itself is ESR silent. Upon addition of Ru NPs, a triplet ESR signal appeared with hyperfine splitting constant aN = 16.2 G, implying of the oxidation of CPH. The signal intensity increased in a time and dose-dependent manner (Fig. 7b).
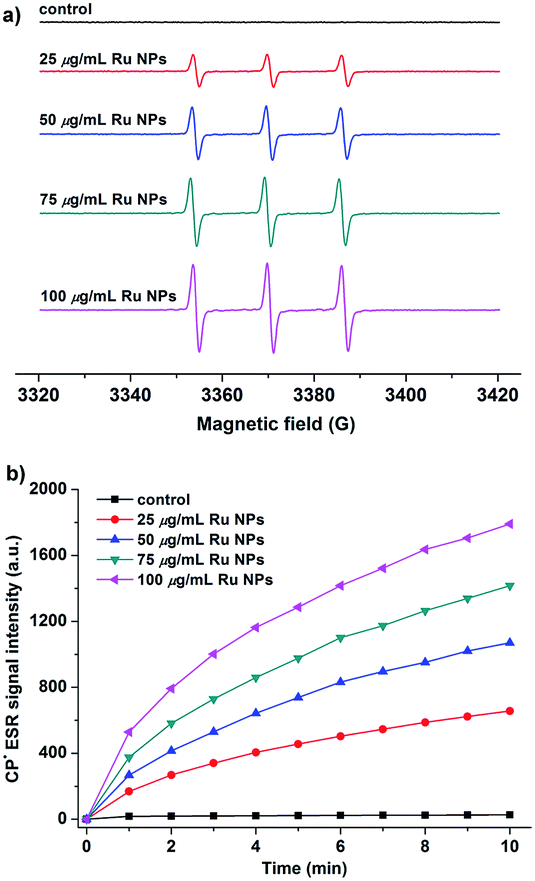 | ||
| Fig. 7 (a) ESR spectra of CP˙ obtained from samples containing 20 μM CPH and variable concentrations of Ru NPs after mixing for 10 min. (b) CP˙ ESR signal intensity vs. time in the absence and presence of Ru NPs with different concentrations. | ||
Conclusions
In summary, this study has introduced Ru NPs mimicking HRP and oxidase functionalities. Ru NPs could catalyze the oxidation of substrate TMB, OPD and DA in the presence of H2O2 to produce the color products. We also report for the first time that Ru NPs possess intrinsic oxidase-like activity, which could catalyze the oxidization of TMB and NaA by dissolved molecular oxygen. The HRP-like and oxidase-like activities of Ru NPs were found to be relevant to the concentrations of Ru NPs. The enzyme mimicking activities of the Ru NPs might originate from their characteristic of accelerating electron transfer between substrates and H2O2 or O2. Our findings offer a better understanding of enzyme-mimicking activities of Ru NPs and should provide important insights for future applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
G.-J. Cao appreciates the National Natural Science Foundation of China (Grant No. 21601035) and Natural Science Foundation of Fujian Province (Grant No. 2016J01043) for partial support. This work was also supported by a regulatory science grant under the FDA Nanotechnology CORES Program. This paper is not an official U.S. FDA guidance or policy statement. No official support or endorsement by the U.S. FDA is intended or should be inferred.Notes and references
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196–1202 CrossRef CAS PubMed.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2006, 312, 208–209 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- T. Matsuo, A. Hayashi, M. Abe, T. Matsuda, Y. Hisaeda and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 15124–15125 CrossRef CAS PubMed.
- Y. Xianyu, Y. Chen and X. Jiang, Anal. Chem., 2015, 87, 10688–10692 CrossRef CAS PubMed.
- Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef CAS PubMed.
- Y. Lin, J. Ren and X. Qu, Adv. Mater., 2014, 26, 4200–4217 CrossRef CAS PubMed.
- W. He, W. Wamer, Q. Xia, J.-J. Yin and P. P. Fu, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2014, 32, 186–211 CAS.
- J. Huang, L. Lin, D. Sun, H. Chen, D. Yang and Q. Li, Chem. Soc. Rev., 2015, 44, 6330–6374 RSC.
- L. Gao, M. Liu, G. Ma, Y. Wang, L. Zhao, Q. Yuan, F. Gao, R. Liu, J. Zhai, Z. Chai, Y. Zhao and X. Gao, ACS Nano, 2015, 9, 10979–10990 CrossRef CAS PubMed.
- Y. Liu, D. L. Purich, C. Wu, Y. Wu, T. Chen, C. Cui, L. Zhang, S. Cansiz, W. Hou, Y. Wang, S. Yang and W. Tan, J. Am. Chem. Soc., 2015, 137, 14952–14958 CrossRef CAS PubMed.
- C.-P. Liu, T.-H. Wu, Y.-L. Lin, C.-Y. Liu, S. Wang and S.-Y. Lin, Small, 2016, 12, 4127–4135 CrossRef CAS PubMed.
- Y. Hu, H. Cheng, X. Zhao, J. Wu, F. Muhammad, S. Lin, J. He, L. Zhou, C. Zhang, Y. Deng, P. Wang, Z. Zhou, S. Nie and H. Wei, ACS Nano, 2017, 11, 5558–5566 CrossRef CAS PubMed.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- X. Yan, Y. Song, X. Wu, C. Zhu, X. Su, D. Du and Y. Lin, Nanoscale, 2017, 9, 2317–2323 RSC.
- H. Jia, D. Yang, X. Han, J. Cai, H. Liu and W. He, Nanoscale, 2016, 8, 5938–5945 RSC.
- W. Zhang, S. Hu, J.-J. Yin, W. He, W. Lu, M. Ma, N. Gu and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 5860–5865 CrossRef CAS PubMed.
- T. Naganuma, Nano Res., 2017, 10, 199–217 CrossRef CAS.
- A. A. Vernekar, D. Sinha, S. Srivastava, P. U. Paramasivam, P. D'Silva and G. Mugesh, Nat. Commun., 2014, 5, 5301 CrossRef CAS PubMed.
- S. Wang, R. Cazelles, W.-C. Liao, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nano Lett., 2017, 17, 2043–2048 CrossRef CAS PubMed.
- M. Vázquez-González, W.-C. Liao, R. Cazelles, S. Wang, X. Yu, V. Gutkin and I. Willner, ACS Nano, 2017, 11, 3247–3253 CrossRef PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef CAS PubMed.
- S. Singh, K. Mitra, A. Shukla, R. Singh, R. K. Gundampati, N. Misra, P. Maiti and B. Ray, Anal. Chem., 2017, 89, 783–791 CrossRef CAS PubMed.
- Y. Wang, Y. Zhu, A. Binyam, M. Liu, Y. Wu and F. Li, Biosens. Bioelectron., 2016, 86, 432–438 CrossRef CAS PubMed.
- H.-H. Zeng, W.-B. Qiu, L. Zhang, R.-P. Liang and J.-D. Qiu, Anal. Chem., 2016, 88, 6342–6348 CrossRef CAS PubMed.
- H. Yang, J. Xiao, L. Su, T. Feng, Q. Lv and X. Zhang, Chem. Commun., 2017, 53, 3882–3885 RSC.
- S. Zhang, D. Zhang, X. Zhang, D. Shang, Z. Xue, D. Shan and X. Lu, Anal. Chem., 2017, 89, 3538–3544 CrossRef CAS PubMed.
- L. Jiang, S. Fernandez-Garcia, M. Tinoco, Z. Yan, Q. Xue, G. Blanco, J. J. Calvino, A. B. Hungria and X. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 18595–18608 CAS.
- K. Wang, N. Li, J. Zhang, Z. Zhang and F. Dang, Biosens. Bioelectron., 2017, 87, 339–344 CrossRef CAS PubMed.
- H.-B. Noh and Y.-B. Shim, J. Mater. Chem. A, 2016, 4, 2720–2728 CAS.
- J. Xie, X. Zhang, H. Wang, H. Zheng, Y. Huang and J. Xie, TrAC, Trends Anal. Chem., 2012, 39, 114–129 CrossRef CAS.
- X. Wang, Y. Hu and H. Wei, Inorg. Chem. Front., 2016, 3, 41–60 RSC.
- Y. Jv, B. Li and R. Cao, Chem. Commun., 2010, 46, 8017–8019 RSC.
- C. Wang, Y. Shi, Y.-Y. Dan, X.-G. Nie, J. Li and X.-H. Xia, Chem.– Eur. J., 2017, 23, 6717–6723 CrossRef CAS PubMed.
- M. C. Ortega-Liebana, J. L. Hueso, R. Arenal and J. Santamaria, Nanoscale, 2017, 9, 1787–1792 RSC.
- W. He, Y. Liu, J. Yuan, J.-J. Yin, X. Wu, X. Hu, K. Zhang, J. Liu, C. Chen, Y. Ji and Y. Guo, Biomaterials, 2011, 32, 1139–1147 CrossRef CAS PubMed.
- Y.-T. Zhou, W. He, W. G. Wamer, X. Hu, X. Wu, Y. M. Lo and J.-J. Yin, Nanoscale, 2013, 5, 1583–1591 RSC.
- W. He, X. Han, H. Jia, J. Cai, Y. Zhou and Z. Zheng, Sci. Rep., 2017, 7, 40103 CrossRef CAS PubMed.
- U. Carmona, L. Zhang, L. Li, W. Münchgesang, E. Pippel and M. Knez, Chem. Commun., 2014, 50, 701–703 RSC.
- C. Chen, S. Fan, C. Li, Y. Chong, X. Tian, J. Zheng, P. P. Fu, X. Jiang, W. G. Wamer and J.-J. Yin, J. Mater. Chem. B, 2016, 4, 7895–7901 RSC.
- L. Jin, Z. Meng, Y. Zhang, S. Cai, Z. Zhang, C. Li, L. Shang and Y. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 10027–10033 CAS.
- J. Wei, X. Chen, S. Shi, S. Mo and N. Zheng, Nanoscale, 2015, 7, 19018–19026 RSC.
- T. Wen, W. He, Y. Chong, Y. Liu, J.-J. Yin and X. Wu, Phys. Chem. Chem. Phys., 2015, 17, 24937–24943 RSC.
- C. Ge, G. Fang, X. Shen, Y. Chong, W. G. Wamer, X. Gao, Z. Chai, C. Chen and J.-J. Yin, ACS Nano, 2016, 10, 10436–10445 CrossRef CAS PubMed.
- Q. Wang, L. Zhang, C. Shang, Z. Zhang and S. Dong, Chem. Commun., 2016, 52, 5410–5413 RSC.
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. McKenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994–10004 CrossRef CAS PubMed.
- H. Su, D.-D. Liu, M. Zhao, W.-L. Hu, S.-S. Xue, Q. Cao, X.-Y. Le, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2015, 7, 8233–8242 CAS.
- M. Cui, J. Zhou, Y. Zhao and Q. Song, Sens. Actuators, B, 2017, 243, 203–210 CrossRef CAS.
- H. Ye, J. Mohar, Q. Wang, M. Catalano, M. J. Kim and X. Xia, Science Bulletin, 2016, 61, 1739–1745 CrossRef CAS.
- G.-J. Cao, X. Jiang, H. Zhang, J. Zheng, T. R. Croley and J.-J. Yin, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2017 DOI:10.1080/10590501.2017.1391516.
- P. J. Tarcha, V. P. Chu and D. Whittern, Anal. Biochem., 1987, 165, 230–233 CrossRef CAS PubMed.
- C. Zhao, Z. Jiang, R. Mu and Y. Li, Talanta, 2016, 159, 365–370 CrossRef CAS PubMed.
- M. Vázquez-González, R. M. Torrente-Rodríguez, A. Kozell, W.-C. Liao, A. Cecconello, S. Campuzano, J. M. Pingarrón and I. Willner, Nano Lett., 2017, 17, 4958–4963 CrossRef PubMed.
- E. Gitto, D.-X. Tan, R. J. Reiter, M. Karbownik, L. C. Manchester, S. Cuzzocrea, F. Fulia and I. Barberi, J. Pharm. Pharmacol., 2001, 53, 1393–1401 CrossRef CAS PubMed.
- S. J. Padayatty, A. Katz, Y. Wang, P. Eck, O. Kwon, J.-H. Lee, S. Chen, C. Corpe, A. Dutta, S. K. Dutta and M. Levine, J. Am. Coll. Nutr., 2003, 22, 18–35 CrossRef CAS PubMed.
- M. Karajibani, M. Hashemi, F. Montazerifar and M. Dikshit, J. Nutr. Sci. Vitaminol., 2010, 56, 436–440 CrossRef CAS PubMed.
- Y.-T. Zhou, J.-J. Yin and Y. M. Lo, Magn. Reson. Chem., 2011, 49, S105–S112 CrossRef CAS PubMed.
- S. I. Dikalov, I. A. Kirilyuk, M. Voinov and I. A. Grigor'ev, Free Radical Res., 2011, 45, 417–430 CrossRef CAS PubMed.
- W. He, H.-K. Kim, W. G. Wamer, D. Melka, J. H. Callahan and J.-J. Yin, J. Am. Chem. Soc., 2014, 136, 750–757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme, table and figures. See DOI: 10.1039/c7ra10370k |
| This journal is © The Royal Society of Chemistry 2017 |